
Abstract
Background
Hepatocellular carcinoma (HCC) is a common malignancy worldwide with poor outcomes. Therefore, it is important to identify a valuable prognostic biomarker for HCC. The present study aimed to identify novel prognostic biomarkers for HCC and evaluate the potential role of hub genes in HCC.
Methods
Weighted gene co-expression network analysis and protein–protein interaction analysis were performed to identify important potential prognostic genes. The expression of hub genes was confirmed by the GEPIA, Oncomine, UALCAN, and HPA database. Furthermore, survival analysis of hub genes was performed using the Kaplan–Meier plotter database. Finally, we investigated the association between hub genes and immune factors in HCC through GSEA, the TIMER, and TISIDB database.
Results
HSD17B6 expression was significantly lower in HCC than in normal tissues. Low HSD17B6 expression is associated with poorer overall survival and progression-free survival in HCC patients, particularly at medium disease stages (stage II and III or grade III). HSD17B6 showed a strong correlation with tumor-infiltrating B cells, CD4 + and CD8 + T cells, macrophages, neutrophils, and dendritic cells. Somatic copy number alteration might be the main cause of the negative correlation between HSD17B6 expression and immune infiltration. HSD17B6 expression in HCC negatively correlated with the expression of several immune cell markers, including exhausted T cell markers, PD-1 and CTLA-4, suggesting its role in regulating tumor immunity.
Conclusions
HSD17B6 is a potential prognostic biomarker that determines cancer progression and is correlated with tumor immune cells infiltration in HCC.
Similar content being viewed by others


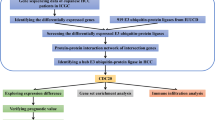
Introduction
Hepatocellular carcinoma (HCC) is one of the most common lethal cancers in the world [1]. Statistically, about 700,000 people die from liver cancer worldwide each year [2], and most liver cancers occur in cirrhosis [3]. Although the survival rate of HCC patients has improved in the last decade because of improved surgical techniques, novel chemotherapies, and targeted therapies, the prognosis of HCC patients remains unsatisfactory. Early recurrence and metastasis are the leading causes of poor patient outcome [4]; therefore, to improve HCC outcome, the investigation of novel molecular mechanisms underlying HCC recurrence and metastasis is urgently required.
Traditional cancer therapeutic regimens focus more on the tumor cells themselves, but HCCs, due to the chronic inflammation and cirrhosis, develop in a highly complex and dynamic environment [5, 6]. The tumor microenvironment (TME) consists of tumor cells and tumor-associated stroma. The tumor stroma consists of tumor-infiltrating lymphocytes (TILs), fibroblasts, and the extracellular matrix surrounding the tumor [7]. Tumor-infiltrating lymphocytes are heterogeneous lymphocytes in the TME and are involved in the antitumor immune response, mainly including T lymphocytes (T cells), B lymphocytes (B cells), natural killer (NK) cells, macrophages, and dendritic cells (DCs) [8]. In the microenvironment, the interaction between tumor tissues and the immune system often has a bidirectional effect. For example, the immune system can positively identify and kill tumors, while tumor cells can form an immunosuppressive microenvironment through the expression of inhibitory molecules and secretion of tumor-related cytokines on the surface to mediate the body’s immune tolerance to tumors, resulting in immune escape. The interaction between tumor cells and related stroma not only influences the occurrence and progression of the disease but also is closely related to patient prognosis [9]. In recent years, the emergence of nivolumab and pembrolizumab has led HCC therapy into the immune era [10,11,12]. Besides, the expression of programmed cell death protein-1 (PD-1), programmed cell death ligand 1 (PD-L1), tumor mutational load (TMB), and TILs have shown some correlation with patients’ response to immunotherapy [13]. However, less than 30% of patients currently have a clinical response to immunotherapy [14, 15]. Therefore, the search for new HCC markers and their interaction mechanisms with immune cells is of great scientific importance. In this way, researchers can remodel TME to improve the body’s antitumor effect.
In this study, we comprehensively studied the expression of HSD17B6 and its correlation with the patient’s clinical outcome in HCC. We also shed light on the role of HSD17B6 in the HCC microenvironment.
Materials and Methods
Data Source and Preprocessing
We downloaded the standardized expression profile of GSE14520 (GPL3921, n = 445) and associated clinical data from the Gene Expression Omnibus database (GEO, http://www.ncbi.nlm.nih.gov/geo/). All probes were converted to Entrez IDs. But if a probe corresponded to multiple Entrez IDs, we removed it. If multiple probes corresponded to the same Entrez ID, we selected the probe with the highest expression. Finally, we performed the weighted gene co-expression network analysis (WGCNA) for the tumor samples (n = 221).
Identifying Key Genes by WGCNA Analysis and PPI Analysis
Unlike previous studies (pre-filtering some genes by differential expression analysis), we performed WGCNA on all genes via the WGCNA R package. Since WGCNA is an unsupervised analysis method for clustering genes based on gene expression, this means that differential expression filtering may lose some important data or even invalidate the WGCNA analysis.
In this analysis, we first converted the gene expression profile data into the Pearson correlation matrices for different gene pairs. For gene k and gene j, their Pearson’s correlation was Skj = | cor (k, j) |. Then, we transformed the Pearson’s correlation matrix into the connection strength matrix through the power function Ukj = power (Skj, β) = | Skj|β. By doing so, we emphasized strong correlations between genes also exponentially reduced the influence of weak correlations. Through network topology analysis (1–20), we obtained the best soft threshold power (β). We then identified gene co-expression modules by hierarchically clustering the average connectivity between genes, with a minimum module size of 50 genes. Next, we correlate the gene modules with clinicopathological information. Here, all clinicopathological characteristics, including age, sex, HBV status, ALT, tumor size, multinodular, cirrhosis, AFP, TNM stage, BCLC stage, survival, and recurrence, were included in the analysis.
After module identification, we selected key genes based on the gene significance (GS) and module membership (MM). GS is the absolute value of the correlation between genes and traits; MM is the correlation of the module eigengenes (the first principal component of a module) and gene expression profiles. First, gene modules that were highly correlated with survival information were identified as target modules. Then, we screened the genes with GS > 0.3 and MM > 0.8 in the target modules as key genes. By using the STRING database (https://string-db.org/), we constructed a protein–protein interaction (PPI) network for the key genes. Finally, we screened highly related but unreported genes in the PPI network as potential biomarkers.
Functional Enrichment Analysis of Potential Biomarkers
Relying on the “Metascape” online tool, we further investigated the pathway and process enrichment situation of key genes. Terms with P < 0.01, a minimum count of three, and an enrichment factor > 1.5 were considered to be important.
Expression Levels of Biomarkers in HCC
To understand whether potential marker expression was associated with HCC, we analyzed marker expression levels across databases. At the transcriptional level, we counted the expression levels of key genes in the GSE14520 cohort by R package limma (log2FC > |1.5|, P < 0.001), based on the GEPIA (http://gepia2.cancer-pku.cn/) database (log2FC > |1|, P < 0.001) and the Oncomine (https://www.oncomine.org/) database (log2FC > |1|, P < 0.05). We also confirmed the expression levels of key genes on the TCGA-LIHC cohort and other cohorts. At the translational level, based on the Human Protein Atlas (HPA, https://www.proteinatlas.org/) database, we compared immunohistochemistry images of key genes in normal liver and HCC tissues.
To further understand whether biomarkers could influence the progression of hepatocellular carcinoma, we compared their expression levels on various clinicopathological groups with the UALCAN (http://ualcan.path.uab.edu/) database. Besides, we explored the mRNA levels of biomarkers in different immune subtypes using the TISIDB (http://cis.hku.hk/TISIDB/) database.
Survival Analysis for Biomarkers
The Kaplan–Meier plotter (http://kmplot.com/analysis/) database is a web tool that can assess the effects of specific genes on cancer prognosis. Here, we tested the ability of biomarkers to predict OS and progression-free survival (PFS) at 1, 3, and 5 years using the Kaplan–Meier plotter. We also assessed biomarkers’ ability in predicting prognosis for different clinicopathological subgroups. We divided the TCGA-LIHC cohort into two groups based on the biomarkers’ median expression. The Kaplan–Meier plotter automatically chooses the log-rank test in the analysis.
Gene Set Enrichment Analysis (GSEA)
We download the GSEA-3.0.jar software and gene sets (c7.all.v6.2.symbols.gmt [immunologic signatures]) from the website of Broad Institute and run under the support of Java 8.0. We divided HCC patients in the GSE14520-GPL3921 cohort into high and low groups according to the median of the biomarker. Enriched pathways with |NES| > 1.8, NOM p value < 0.01 FDR < 0.25 were considered to be significant terms.
Evaluation of the Immunological Infiltrate
Tumor Immune Estimation Resource (TIMER) is a publicly available resource for the systematic analysis of immune infiltrates across different types of cancer (https://cistrome.shinyapps.io/timer/). It contains seven modules, including gene, survival, mutation, somatic copy numbers alteration (SCNA), differential gene expression, correlation, and estimation. We collected immunologic infiltration data from the TIMER platform to explore their relationships with biomarkers. The abundances of six immunes infiltrate (CD4 + T cells, CD8 + T cells, B cells, neutrophils, macrophages, and dendritic cells) are estimated by statistical method mining sequencing data retrieved from TCGA. TIMER displays the purity-corrected partial Spearman’s correlation with scatterplots. Based on TIMER, we further conducted the univariate Cox analysis and multivariate Cox analysis of HSD17B6 and immune infiltration cells. Cox regression analysis was fitted by function coxph() from R package survival, which was built in TIMER. The hazard ratio (HR) and 95% confidence interval (CI) were presented. Copy number alteration is one of the causes of the inactivation of cancer suppressor genes. To explore the possible mechanism of down-regulation of HSD17B6 expression in hepatocellular carcinoma, we used the cBioPortal (https://www.cbioportal.org/) website to explore the relationship between the mRNA expression level of HSD17B6 and copy number alteration in hepatocellular carcinoma. Next, we analyzed the relationship between HSD17B6 copy number variation and immune cell infiltration using the SCNA module in the TIMER database. SCNAs are divided into five types, namely deep deletion (−2), arm-level deletion (−1), diploid/normal (0), arm-level gain (1), and high amplification (2). Boxplots show the distribution of each immune subset at each copy number status in selected types of cancer. The level of infiltration for each SCNA category is compared with the normal using the Kruskal–Wallis test.
Immunomodulators and Chemokines Analysis Through the TISIDB Database
TISIDB database (http://cis.hku.hk/TISIDB/) integrates plentiful immunological-related source retrieved from databases. For probing the hidden function of biomarkers in immune therapy, we investigated the relationship between biomarkers and multiple immunomodulators, and chemokines.
Statistical Analysis
P value < 0.05 was considered statistically significant, if not especially noted.
Results
Identification of Key Genes Via WGCNA and PPI Analysis
β = 5 was first chosen to construct the scale-free network (Fig. 1a). Then, the hierarchical clustering tree was built under the dynamic hybrid cutting. Each leaf represents a single gene, and genes with similar expression were clustered into a tree branch, representing a gene module. According to Zhou et al. [16] and Qi et al. [17], we selected the blue module consisting of 1291 genes as the target module since it had a higher correlation with clinicopathological traits like AFP (R = −0.43, P = 1e−11), TNM stage (R = −0.33, P = 7e−07), and survival time (R = 0.31, P = 2e−06). Based on GS > 0.3 and MM > 0.8, we sorted nineteen highly linked genes from the blue module as candidate biomarkers (Fig. 1d). Then, relying on the PPI network analysis (connectivity > 5 nodes), we identified eight potential key genes (Fig. 1e), including ALDH2, ETNPPL (AGXT2L1), CAT (Catalase), DAO (diamine oxidase), HSD17B6, SULT2A1, SLC27A5, and SLC10A1 (Table 1).

Choosing hub genes through WGCNA and PPI analysis. a Choosing a fit soft threshold power (β) from 1 to 20. b Clustering dendrogram of genome-wide genes in hepatocellular carcinoma samples. c Heatmap shows correlations of module eigengenes with clinicopathological traits. d A scatter plot of co-expression genes in the blue module. e PPI network of candidate hub genes
Function and Disease Enrichment Analyses
Next, we analyzed 19 key genes in terms of pathways, processes, and disease enrichment. The results indicate that most of these genes are involved in drug metabolism and influence the development of cirrhosis (Table 2).
Expression Level of HSD17B6 in HCC
All key genes were down-regulated in GSE14520-GPL3921 tumor tissues (Table S2). By November 2019, searching with “gene symbol,” “HCC,” and “survival,” we found that only SULT2A1, DAO, and HSD17B6 were not reported before. In the next expression analysis, we found that HSD17B6 showed significant down-regulation not only on the GEPIA database but also on the Oncomine and HPA databases. In the two immunohistochemistry images, we could see that there were many deeply eosinophilic cells, thick fibrous bands, and large pseudogland on the HCC. These are all features of cirrhosis that develops from hepatitis. HSD17B6 was highly stained in normal hepatocytes, especially in the cytoplasm and membranes; however, HSD17B6 was negative in HCC. This suggests that HSD17B6 might be a potential biomarker for HCC.
To investigate the relationship between HSD17B6 mRNA expression and the clinicopathological characteristics of HCC, we analyzed TCGA data using the UALCAN database. We found that the normal tissue data were concentrated and tended to be normally distributed, but the tumor subgroup data were discrete and tended to be skewed. However, the expression of HSD17B6 in each tumor subgroup was generally lower than its expression in normal liver tissue. Moreover, the expression of HSD17B6 gradually decreased with tumor progression. Besides, down-regulation of HSD17B6 was also associated with tumor lymph node metastasis. Among the different immune subgroups, we noted that HSD17B6 expression was highest in C3 (inflammatory type) and lowest in C1 (wound healing type) (Fig. 2e). In contrast, in molecular typing, HSD17B6 was highly expressed in the iCluster 2 subtype and least expressed in iCluster 1 (Fig. 2f). This evidence suggests that HSD17B6 is a potential HCC biomarker, which may be involved in immune responses and thus influence tumor progression.

The expression level of HSD17B6 in HCC. a Validation of hub genes by LIHC data in GEPIA. b Validation of HSD17B6 at transcriptional level via Oncomine. c Validation of HSD17B6 at translational level via HPA. d Clinicopathological subgroups analysis for HSD17B6 in LIHC. e The expression level of HSD17B6 in different immune subtypes. f The expression level of HSD17B6 in different molecular subtypes (Student’s t test, *, P < 0.05, **, P < 0.01, ***, P < 0.001, ****, P < 0.0001)
HSD17B6 Down-Regulation Suggested the Poor Survival for HCC Patients
To understand the impact of HSD17B6 expression levels on the prognosis of HCC patients, we explored the prognostic value of HSD17B6 in the TCGA-LIHC cohort using the Kaplan–Meier Plotter database. The 1, 3, and 5-year OS analysis and PFS analysis consistently showed that increased HSD17B6 was a favorable factor for HCC patients (Fig. 3a, b). Not only that, patients with high HSD17B6 expression also showed higher prognostic levels in different clinicopathological subgroups (Fig. 3c). In terms of overall survival, HSD17B6 played a beneficial role in patients with the following characteristics: male (HR 0.58 [95% CI 0.37–0.92], P = 0.018), Asian (HR 0.35 [95% CI 0.19–0.67], P = 0.00082), yes-alcohol consumption (HR 0.53 [95% CI 0.28–1], P = 0.047), stage 3 (HR 0. 53 [95% CI 0.29–0.97], P = 0.035), stage 2 and 3 (HR 0.63 [95% CI 0.39–1], P = 0.049), grade 3 (HR 0.49 [95% CI 0.26–0.9], P = 0.018), AJCC T3 (HR 0.49 [95% CI 0.26–0.89], P = 0.018). In terms of progression-free survival, HSD17B6 high expression was still protective to male patients (HR 0.36 [95% CI 0.2–0.65], P = 0.0004), Asian (HR 0.44 [95% CI 0.2–0.98], P = 0.04), yes-alcohol consumption (HR 0.51 [95% CI 0.3–0.86], P = 0.0098), but was newly discovered to be beneficial to patients who with no-alcohol consumption (HR 0.65 [95% CI 0.34–0.97], P = 0.036), no-hepatitis virus (HR 0.63 [95% CI 0.4–0.97], P = 0.034), stage 1 and 2 (HR 0.64 [95% CI 0.44–0.95], P = 0.024) and grade 2 (HR 0.6 [95% CI 0.39–0.94], P = 0.023). In conclusion, patients with high HSD17B6 usually had better survival outcomes. This suggested that HSD17B6 might act as a protective factor against HCC.

The prognostic value of HSD17B6 in HCC. a 1-year, 3-year and 5-year OS in LIHC cohort. b 1-year, 3-year and 5-year PFS in LIHC cohort. c Subgroup analysis for OS and PFS in the LIHC cohort (log-rank test, *, P < 0.05, **, P < 0.01, ***, P < 0.001, ****, P < 0.0001)
Evaluation of Associations Among HSD17B6, Immune Cells, and Survival
The results of the expression analysis in Fig. 2 suggested that HSD17B6 might be immune-related (Fig. 2e, f). Therefore, it was necessary to further explore the association between immune status and HSD17B6 expression in the HCC microenvironment. Our GSEA results showed that high HSD17B6 expression was associated with down-regulation of macrophages, CD4 + T cells and DC, but with the upregulation of NK (Fig. 4a). We then validated the results using the TIMER database, which disclosed that the expression of HSD17B6 was not significantly correlated with tumor purity in HCC (R = 0.073, P = 0.175). However, its expression negative correlated (Fig. 4b) with the infiltration levels of B cells (R = −0.253, P value = 2.07e−06), CD8 + T cells (R = −0.213, P value = 7.19e−05), CD4 + T cells (R = −0.137, P value = 1.08e−02), macrophages (R = −0.251, P value = 2.69e−06), neutrophils (R = −0.183, P value = 6.15e−04), and dendritic cells (R = −0.202, P value = 1.81e−04) in HCC. Our results suggested that HSD17B6 might affect the infiltration of immune cells, especially on macrophages, CD4 + T cells, and DC.

Correlation between HSD17B6 mRNA expression and immune cell infiltration. a GSEA analysis for high vs. low HSD17B6 group in immunologic signature gene sets (GSE14520-GPL3921 cohort). b Correlations of HSD17B6 expression with immune infiltration level in HCC. c Correlation between the SCNA and HSD17B6 mRNA expression. d The SCNA of HSD17B6 affects the infiltrating levels in HCC
To explore immune infiltrates and HSD17B6 expression on HCC prognosis, we performed univariate and multivariate cox analysis via the TIMER database. Univariate cox analysis showed that the infiltration level of macrophages and neutrophils were unfavorable factors (HR > 1, P < 0.05) for HCC patients, while HSD17B6 expression was favorable (HR < 1, P < 0.05). Multivariate analysis further confirmed that HSD17B6 and macrophages were independent prognostic factors. Furthermore, tumor purity, macrophages, and dendritic cells were unfavorable prognostic factors (HR > 1, P < 0.05), whereas B cells, CD8 + T cells, and HSD17B6 were found to be favorable prognostic factors (HR < 1, P < 0.05). Thus, these data suggested that HSD17B6 and macrophages were independent prognostic markers in immune infiltration.
Compared with the diploid group, the gain group showed lower HSD17B6 expression levels (Fig. 4c). We then compared tumor infiltration levels between different SCNAs of HSD17B6 and tumors using TIMER. We found that HSD17B6 mainly mutated into the arm-level-gain class and the infiltration level of immune cells (except CD8 + T cells and neutrophils) was significantly elevated in the arm-level gain group (Fig. 4d). These results indicated that the SCNAs of HSD17B6 were closely related to the degree of immune infiltration of HCC.
Regulation of Immune Molecules by HSD17B6
The Spearman’s correlations between HSD17B6 expression and immunomodulators, and chemokines were analyzed using the TISIDB database (Fig. 5). Figure 5a shows correlations between HSD17B6 expression levels and immuneinhibitors. The top six immuneinhibitors included KDR (R = 0.445, P < 2.2e−16), TGFB1 (R = −0.343, P = 1.29e−11), CTLA4 (R = −0.326, P = 1.52e−10), LGALS9 (R = −0.323, P = 1.99e−10), PDCD1 (R = −0.321, 2.74e−10), and VTCN1 (R = −0.26, P = 3.92e−07). Figure 5b shows correlations between HSD17B6 expression and immunostimulators, and the top six immunostimulators included CD276 (R = −0.367, P = 3.58e−13), TNFSF15 (R = −0.35, P = 4.41e−12), IL6R (R = 0.349, P = 5.07e−12), TNFRSF18 (R = −0.338, P = 2.56e−11), TNFSF9 (R = −0.302, P = 1.3e−09), and ULBP1 (R = −0.294, P = 8.3e−09). Figure 5c shows correlations between HSD17B6 expression and chemokines, and the top six chemokines included CCL16 (R = 0.526, P < 2.2e−16), CCL14 (R = 0.497, P < 2.2e−16), CCL20 (R = −0.355, P = 2.2e−12), CCL26 (R = −0.324, P = 1.97e−10), CXCL5 (R = −0.312, P = 9.18e−10), CCL15 (R = −0.295, P = 7.05e−09). Figure 5c shows correlations between HSD17B6 expression and receptors, and the top six receptors included CCR6 (R = −0.343, P = 1.24e−11), CXCR4 (R = −0.278, P = 5.2e−08), CCR10 (R = −0.222, P = 1.56e−05), CXCR5 (R = −0.189, P = 0.000255), CXCR3 (R = −0.164, P = 0.00154), and CCR5 (R = −0.16, P = 0.00197). Therefore, HSD17B6 might regulate the above immune molecules.

Correlation between HSD17B6 mRNA expression and immune-related molecules. a Spearman’s correlation of HSD17B6 with immunoinhibitors in HCC. b Spearman’s correlation of HSD17B6 with immunostimulators in HCC. c Spearman’s correlation of HSD17B6 with chemokines in HCC. d Spearman’s correlation of HSD17B6 with receptors
Discussion
HSD17B6 encoded protein functions as oxidoreductase and epimerase and metabolizes androgen. In disease studies, the single-nucleotide polymorphism (SNP) of HSD17B6 was first reported being associated with polycystic ovary syndrome (PCOS) [18]. In cancer, HSD17B6 has turned out to be down-regulated in prostate cancer and was associated with tumor growth [19] and metastasis [20]. A recent study also showed that HSD17B6 is a potential prognostic marker in non-small cell lung cancer and may contribute to the development of non-small cell lung cancer and drive tumor progression [21]. HSD17B6 is specifically expressed in liver tissue, but little is known about the role of HSD17B6 in liver disease, and the SNP of HSD17B6 is significantly associated with the risk of hepatic fibrosis [22]. In HCC, Yang et al. [23] had suggested that HSD17B6 was a potential tumor suppressor—both CpG hypomethylation and down-regulation of mRNA expression are associated with HCC recurrence. However, they did not investigate the detailed mechanism by which HSD17B6 may affect the prognosis of liver cancer. Role of HSD17B6 in HCC requires further clarification. Thus, our study was performed.
We analyzed HSD17B6 expression levels in HCC samples from the TCGA dataset and found that low levels of HSD17B6 positively correlated with the higher histological grade, TNM stage of the tumor (Fig. 2d), which are indicators of greater tumor malignancy. HSD17B6 down-regulation was also associated with worse survival and poor recurrence (Fig. 3). Moreover, multivariate Cox regression analysis further confirmed HSD17B6 as an independent favorable prognostic factor of HCC (Table 2). Together, these results demonstrated that HSD17B6 might inhibit the progression of HCC and be a potential biomarker.
Then, GO and KEGG analysis for HSD17B6 and its co-expression gene showed that HSD17B6 participated in the peroxisome pathway (Table 1). And peroxisome-localized hepatitis Bx protein increases the invasion property of hepatocellular carcinoma cells [24]. Also, disruption of peroxisome function leads to metabolic stress, mTOR inhibition, and lethality in liver cancer cells [25]. Therefore, we speculate that HSD17B6 dysfunction promotes HCC development by regulating the peroxisome signaling pathway. The overall metabolic state of the organism is now considered to be an important and long-neglected immune mediator. The peroxisome, on the other hand, has recently been identified as a key regulator of immune function and inflammation during development and infection [26]. HCC is a highly heterogeneous cancer, and tumor heterogeneity is instrumental in immune evasion [27,28,29]. Numerous studies have shown that tumor-infiltrating immune cells are prognostic markers for cancer progression [30,31,32,33]. In contrast, our study showed that HSD17B6 was significantly differentially expressed among different immune subgroups (Fig. 2e). The above evidence suggests that HSD17B6 is likely to be involved in immune regulation.
Various state-of-the-art algorithms, including TIMER [34], xCell [35], CIBERSORT [36], MCP-counter [37], EPIC [38] and quanTIseq [39], have been developed to estimate immune infiltration. But TIMER is the only method that takes tissue specificity into account when estimating immune cell populations. Though to date, it only estimates six immune cell types. Other algorithms, like xCell, can make estimations on the higher number of different immune cell types but may fail to detect signals from homogeneous samples [34]. Therefore, for this study, we used the TIMER to make a reliable immune infiltration estimation. Accordingly, we found that HSD17B6 expression was negatively correlated with the degree of infiltration of B cells, CD8 + T cells, CD4 + T cells, macrophages, neutrophils, and dendritic cells (Fig. 4b). GSEA results (Fig. 4a) also corroborated the negative correlation between HSD17B6 and CD4 + T cells, macrophages, and DCs. Copy number alteration (CNA) is genetically important and may interfere with genes, altering the genetic content leading to phenotypic differences [40]. Our results revealed that the predominant CNA type of HSD17B6 was arm-level gain, and this variation resulted in a down-regulation of the mRNA expression level of HSD17B6 (Fig. 4c). However, arm-level gain led to an increase in infiltration of most immune cells (Fig. 4d).
Immune responses to primary and secondary tumor sites rely on the different types of immune cells that infiltrate into the tumor microenvironment. The infiltration of different types of immune cells is closely regulated by the various chemokines, which modulate tumor immunity and the biological phenotype of the tumors, and also influence tumor progression, therapy, and prognosis [41]. This study also shows that HSD17B6 expression correlated with the infiltration status of immune cells in HCC. In HCC, there was a strong negative correlation between HSD17B6 expression with infiltration of B cells, CD4 + and CD8 + T cells, macrophages, neutrophils, and DCs. This suggests that HSD17B6 plays a major role in regulating tumor immunity, and therefore influences HCC prognosis. Moreover, expression of exhausted T cells markers, PDCD1 (PD-1) and CTLA-4, which are critical inhibitory immune checkpoint proteins negatively correlates with HSD17B6 expression. Most cancers, including HCC, overexpress inhibitory ligands to evade immune response by dampening T cell function, thus contributing to cancer progression [42]. The expression of inhibitory immune checkpoint proteins is altered in the tumor microenvironment [43]. HSD17B6 may bind to chemokine receptors, such as CCR5 and CCR6 (Fig. 5), and regulate activation and migration of different leukocytes by mobilizing Ca2 + influx [44, 45]. Altered Ca2 + flux in the T cell subsets promotes cytokine production and down-regulates CTLA-4 and PD-1 expression [46]. We postulate that low HSD17B6 expression in the tumor microenvironment diminishes the Ca2 + influx and upregulates the expression of inhibitory immune checkpoint proteins, PD-1 and CTLA-4 on the exhausted T cells. These mechanistic changes can alter antitumor function of T cells and result in the poorer prognosis of HCC. However, this hypothesis needs to be further investigated. Taken together, our findings indicate that HSD17B6 plays an important role in regulating tumor-infiltration of immune cells in HCC. Besides, the latest research showed the dual anti-PD-1/VEGFR-2 therapy has a durable vessel fortification effect in HCC and can overcome treatment resistance to either treatment alone and increase overall survival in both anti-PD-1 therapy-resistant and anti-PD-1 therapy-responsive HCC models [47]. Here, we found that HSD17B6 was highly correlated with both PDCD1 (PD-1) and KDR (VEGFR2). Therefore, we reasoned that if HSD17B6 expression was induced in tumors, it might further enhance the therapeutic effect of HCC. In short, HSD17B6, which is associated with these immune molecules, may provide a new target for studying the immune evasion of HCC cells and can potentially serve as an immunotherapeutic target for liver cancer.
Our study has a few limitations. Firstly, our investigations into the role of HSD17B6 in tumors were based on data that were already reported in the ONCOMINE, Kaplan–Meier plotter, GEPIA, and TIMER databases. However, we did not verify these outcomes by testing our own clinical samples. Secondly, we did not conduct in vitro and animal experiments to confirm the role of HSD17B6 in the growth and progression of HCC, and its relationship with the infiltration of immune cells into the tumor microenvironment. Hence, further studies are needed to verify the role played by HSD17B6 in HCC.
Conclusions
In summary, our results suggest that HSD17B6 is an independent potential prognostic biomarker for HCC that can be used to evaluate the levels of immune cell infiltration in the tumor tissues. SCNA may cause the down-regulation of HSD17B6 expression. Relatively low levels of HSD17B6 in HCC tissues may indicate greater risk of tumor relapse after treatment, and close medical supervision will be necessary for such patients.
Abbreviations
- HCC:
-
Hepatocellular carcinoma
- WGCNA:
-
Weighted gene co-expression network analysis
- PPI:
-
Protein–protein interaction
- GEPIA:
-
Gene expression profiling interactive analysis
- HPA:
-
Human protein atlas
- TIMER:
-
Tumor immune estimation resource
- SCNA:
-
Somatic copy numbers alteration
- TIL:
-
Tumor infiltration lymphocyte
- TMB:
-
Tumor mutational load
- GEO:
-
Gene expression omnibus database
- GS:
-
Gene significance
- MM:
-
Module membership
- TCGA:
-
The cancer genome atlas
- OS:
-
Overall survival
- PFS:
-
Progression-free survival
- HR:
-
Hazard ratio
- 95% CI:
-
95% confidence interval
- GSEA:
-
Gene set enrichment analysis
- NES:
-
Normalized enrichment score
- STRING:
-
Search tool for the retrieval of interacting genes
References
Lurje I, Czigany Z, Bednarsch J, et al. Treatment strategies for hepatocellular carcinoma (-) a multidisciplinary approach. Int J Mol Sci. 2019. https://doi.org/10.3390/ijms20061465.
Bruix J, Reig M, Sherman M. Evidence-based diagnosis, staging, and treatment of patients with hepatocellular carcinoma. Gastroenterology. 2016;150:835–853. https://doi.org/10.1053/j.gastro.2015.12.041.
Llovet JM, Montal R, Villanueva A. Randomized trials and endpoints in advanced HCC: Role of PFS as a surrogate of survival. J Hepatol. 2019;70:1262–1277. https://doi.org/10.1016/j.jhep.2019.01.028.
Grandhi MS, Kim AK, Ronnekleiv-Kelly SM, Kamel IR, Ghasebeh MA, Pawlik TM. Hepatocellular carcinoma: From diagnosis to treatment. Surg Oncol. 2016;25:74–85. https://doi.org/10.1016/j.suronc.2016.03.002.
Lu C, Rong D, Zhang B, et al. Current perspectives on the immunosuppressive tumor microenvironment in hepatocellular carcinoma: Challenges and opportunities. Mol Cancer. 2019;18:130. https://doi.org/10.1186/s12943-019-1047-6.
Cariani E, Missale G. Immune landscape of hepatocellular carcinoma microenvironment: Implications for prognosis and therapeutic applications. Liver Int Off J Int Assoc Study Liver. 2019;39:1608–1621. https://doi.org/10.1111/liv.14192.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. https://doi.org/10.1016/j.cell.2011.02.013.
Sasada T, Suekane S. Variation of tumor-infiltrating lymphocytes in human cancers: Controversy on clinical significance. Immunotherapy. 2011;3:1235–1251. https://doi.org/10.2217/imt.11.106.
Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer. 2009;9:239–252. https://doi.org/10.1038/nrc2618.
Prieto J, Melero I, Sangro B. Immunological landscape and immunotherapy of hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2015;12:681–700. https://doi.org/10.1038/nrgastro.2015.173.
Liu X, Qin S. Immune checkpoint inhibitors in hepatocellular carcinoma: Opportunities and challenges. Oncologist. 2019;24:S3–S10. https://doi.org/10.1634/theoncologist.2019-io-s1-s01.
Tovoli F, Casadei-Gardini A, Benevento F, Piscaglia F. Immunotherapy for hepatocellular carcinoma: A review of potential new drugs based on ongoing clinical studies as of 2019. Digest Liver Dis Off J Italian Soc Gastroenterol Italian Assoc Study Liver. 2019;51:1067–1073. https://doi.org/10.1016/j.dld.2019.05.006.
Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer. 2016;16:275–287. https://doi.org/10.1038/nrc.2016.36.
Kudo M. Immune checkpoint inhibition in hepatocellular carcinoma: Basics and ongoing clinical trials. Oncology. 2017;92:50–62. https://doi.org/10.1159/000451016.
Lu LC, Hsu C, Shao YY, et al. Differential organ-specific tumor response to immune checkpoint inhibitors in hepatocellular carcinoma. Liver Cancer. 2019;8:480–490. https://doi.org/10.1159/000501275.
Zhou Z, Cheng Y, Jiang Y, et al. Ten hub genes associated with progression and prognosis of pancreatic carcinoma identified by co-expression analysis. Int J Biol Sci. 2018;14:124–136. https://doi.org/10.7150/ijbs.22619.
Qi YF, Sun JN, Ren LF, et al. Intestinal microbiota is altered in patients with gastric cancer from Shanxi Province, China. Dig Dis Sci. 2019;64:1193–1203. https://doi.org/10.1007/s10620-018-5411-y.
Jones MR, Italiano L, Wilson SG, et al. Polymorphism in HSD17B6 is associated with key features of polycystic ovary syndrome. Fertil Steril. 2006;86:1438–1446. https://doi.org/10.1016/j.fertnstert.2006.04.027.
Zhang A, Zhang J, Plymate S, Mostaghel EA. Classical and non-classical roles for pre-receptor control of DHT metabolism in prostate cancer progression. Hormones Cancer. 2016;7:104–113. https://doi.org/10.1007/s12672-016-0250-9.
Jernberg E, Thysell E, Bovinder Ylitalo E, et al. Characterization of prostate cancer bone metastases according to expression levels of steroidogenic enzymes and androgen receptor splice variants. PLoS ONE. 2013;8:e77407. https://doi.org/10.1371/journal.pone.0077407.
Ma Q, Xu Y, Liao H, et al. Identification and validation of key genes associated with non-small-cell lung cancer. J Cell Physiol. 2019;234:22742–22752. https://doi.org/10.1002/jcp.28839.
White DL, Liu Y, Garcia J, et al. Sex hormone pathway gene polymorphisms are associated with risk of advanced hepatitis C-related liver disease in males. Int J Mol Epidemiol Genet. 2014;5:164–176.
Yang JD, Seol SY, Leem SH, et al. Genes associated with recurrence of hepatocellular carcinoma: integrated analysis by gene expression and methylation profiling. J Korean Med Sci. 2011;26:1428–1438. https://doi.org/10.3346/jkms.2011.26.11.1428.
Han JM, Kang JA, Han MH, et al. Peroxisome-localized hepatitis Bx protein increases the invasion property of hepatocellular carcinoma cells. Adv Virol. 2014;159:2549–2557. https://doi.org/10.1007/s00705-014-2105-4.
Cai M, Sun X, Wang W, et al. Disruption of peroxisome function leads to metabolic stress, mTOR inhibition, and lethality in liver cancer cells. Cancer Lett. 2018;421:82–93. https://doi.org/10.1016/j.canlet.2018.02.021.
Di Cara F, Andreoletti P, Trompier D, et al. Peroxisomes in immune response and inflammation. Int J Mol Sci. 2019;20:3877. https://doi.org/10.3390/ijms20163877.
Jeng KS, Chang CF, Jeng WJ, Sheen IS, Jeng CJ. Heterogeneity of hepatocellular carcinoma contributes to cancer progression. Crit Rev Oncol Hematol. 2015;94:337–347. https://doi.org/10.1016/j.critrevonc.2015.01.009.
Fu Y, Liu S, Zeng S, Shen H. From bench to bed: the tumor immune microenvironment and current immunotherapeutic strategies for hepatocellular carcinoma. J Exp Clin Cancer Res. 2019;38:396. https://doi.org/10.1186/s13046-019-1396-4.
Sachdeva M, Chawla YK, Arora SK. Immunology of hepatocellular carcinoma. World J Hepatol. 2015;7:2080–2090. https://doi.org/10.4254/wjh.v7.i17.2080.
Zhang S-C, Hu Z-Q, Long J-H, et al. Clinical implications of tumor-infiltrating immune cells in breast cancer. J Cancer.. 2019;10:6175–6184. https://doi.org/10.7150/jca.35901.
Ye L, Zhang T, Kang Z, et al. Tumor-infiltrating immune cells act as a marker for prognosis in colorectal cancer. Front Immunol. 2019. https://doi.org/10.3389/fimmu.2019.02368.
Pagès F, Galon J, Dieu-Nosjean MC, Tartour E, Sautès-Fridman C, Fridman WH. Immune infiltration in human tumors: a prognostic factor that should not be ignored. Oncogene. 2010;29:1093–1102. https://doi.org/10.1038/onc.2009.416.
Guo F, Dong Y, Tan Q, Kong J, Yu B. Tissue infiltrating immune cells as prognostic biomarkers in endometrial cancer: A meta-analysis. Dis Mark. 2020;2020:1805764. https://doi.org/10.1155/2020/1805764.
Li T, Fu J, Zeng Z, et al. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucl Acids Res. 2020;48:W509–W514. https://doi.org/10.1093/nar/gkaa407.
Aran D, Hu Z, Butte AJ. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017;18:220. https://doi.org/10.1186/s13059-017-1349-1.
Newman AM, Liu CL, Green MR, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12:453–457. https://doi.org/10.1038/nmeth.3337.
Becht E, Giraldo NA, Lacroix L, et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016;17:218. https://doi.org/10.1186/s13059-016-1070-5.
Racle J, de Jonge K, Baumgaertner P, Speiser DE, Gfeller D. Simultaneous enumeration of cancer and immune cell types from bulk tumor gene expression data. eLife. 2017;6:e26476. https://doi.org/10.7554/elife.26476.
Finotello F, Mayer C, Plattner C, et al. Molecular and pharmacological modulators of the tumor immune contexture revealed by deconvolution of RNA-seq data. Genome Med. 2019;11:34. https://doi.org/10.1186/s13073-019-0638-6.
Urrutia E, Chen H, Zhou Z, Zhang NR, Jiang Y. Integrative pipeline for profiling DNA copy number and inferring tumor phylogeny. Bioinformatics. 2018;34:2126–2128. https://doi.org/10.1093/bioinformatics/bty057.
Balkwill F. Cancer and the chemokine network. Nat Rev Cancer. 2004;4:540–550. https://doi.org/10.1038/nrc1388.
Granier C, De Guillebon E, Blanc C, et al. Mechanisms of action and rationale for the use of checkpoint inhibitors in cancer. ESMO Open. 2017;2:e000213. https://doi.org/10.1136/esmoopen-2017-000213.
Philips GK, Atkins M. Therapeutic uses of anti-PD-1 and anti-PD-L1 antibodies. Int Immunol. 2015;27:39–46. https://doi.org/10.1093/intimm/dxu095.
Vongsa RA, Zimmerman NP, Dwinell MB. CCR6 regulation of the actin cytoskeleton orchestrates human beta defensin-2- and CCL20-mediated restitution of colonic epithelial cells. J Biol Chem. 2009;284:10034–10045. https://doi.org/10.1074/jbc.m805289200.
Al-Alem L, Puttabyatappa M, Rosewell K, et al. Chemokine ligand 20: A signal for leukocyte recruitment during human ovulation? Endocrinology. 2015;156:3358–3369. https://doi.org/10.1210/en.2014-1874.
Rodríguez-Perea AL, Rojas M, Velilla-Hernández PA. High concentrations of atorvastatin reduce in vitro function of conventional T and regulatory T cells. Clin Exp Immunol. 2019;196:237–248. https://doi.org/10.1111/cei.13260.
Shigeta K, Datta M, Hato T, et al. Dual programmed death Receptor-1 and vascular endothelial growth factor Receptor-2 blockade promotes vascular normalization and enhances antitumor immune responses in hepatocellular carcinoma. Hepatology (Baltimore, MD). 2020;71:1247–1261. https://doi.org/10.1002/hep.30889.
Acknowledgments
2020 is a very unusual year. The outbreak of COVID-19 has disrupted our lives. First of all I would like to thank all the scientists for their contribution to the world. It is your selfless dedication that has saved the world. This year, along with COVID-19, I had some amazing things happen to me. Thanks to my family, friends, and elders for their support. Thanks especially to Dr. Li Jiang and Dr. Yuan Xuejia. Without their help, I could not have been reborn. Finally, I would like to extend my special thanks to the editorial board of Digestive Diseases and Sciences and all the reviewers. I would like to thank you for your valuable comments and allowing enough time for revision. This means a lot to me. Thank you all very much.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Wu, M., Jiang, L. Hydroxysteroid 17-Beta Dehydrogenase 6 Is a Prognostic Biomarker and Correlates with Immune Infiltrates in Hepatocellular Carcinoma. Dig Dis Sci 67, 146–158 (2022). https://doi.org/10.1007/s10620-021-06832-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10620-021-06832-7