
Abstract
Herein, we present a route to obtain bi-functional cellulose nanofibrils (CNF) by a one-pot approach using an already established functionalisation route, carboxymethylation, to which a subsequent functionalisation step, allylation or alkynation, has been added in the same reaction pot, eliminating the need of solvent exchange procedures. The total charge of the fibres and the total surface charge of the nanofibrils were determined by conductometric and polyelectrolyte titration, respectively. Furthermore, the allyl and alkyne functionalised cellulose were reacted with methyl 3-mercaptopropionate and azide-functionalised disperse red, respectively, to estimate the degree of functionalisation. The samples were further assessed by XPS and FT-IR. Physical characteristics were evaluated by CP/MAS 13C-NMR, XRD, AFM and DLS. This new approach of obtaining bi-functionalised CNF allows for a facile and rapid functionalisation of CNF where chemical handles can easily be attached and used for further modification of the fibrils.
Graphical abstract

Similar content being viewed by others

Introduction
Nanocellulosic materials, such as cellulose nanofibrils (CNF) have been given an increased interest during the past decades as components in bio-based and/or renewable composite materials (Dufresne 2012, 2013; Eichhorn et al. 2001, 2010; Klemm et al. 2011). This is mostly attributed not only to their unique combination of comparably high strength and stiffness in combination with a low density compared to for example Kevlar or steel, but also to the possibilities of producing these materials with lower consumption energy (Chauve and Bras 2014; Eriksen et al. 2008; Klemm et al. 2011). Additionally, the high aspect ratio and large interfacial surface area of CNFs contributes to the development of functional composite materials with remarkable mechanical properties (Dufresne 2012; Eichhorn et al. 2010). However, due to the inherent hydrophilic nature of cellulose, CNFs have to be surface modified in order to adequately take advantage of their principal nano-reinforcing characteristics in hydrophobic polymer matrices.
CNFs are prepared by subjecting the wood fibres to mechanical disintegration in order to fibrillate the fibres. The fibrillation process yields liberated nanofibrils aggregates/bundles or separated nanofibrils. In most cases, the fibres are chemically pre-treated, to promote the mechanical treatment and to improve the colloidal stability of the obtained fibril dispersion. Among other techniques, TEMPO-mediated oxidation or carboxymethylation are commonly used as pre-treatment methods (Dufresne 2012). Both methods introduce charges to the fibrils by substituting hydroxyl groups on the cellulose backbone with carboxylate/carboxymethylated groups.
There are a variety of surface modification approaches for CNFs, spanning from non-covalent or covalent attachment of small or large molecules to polymer grafting from or onto the surface (Daud and Lee 2017; Filpponen et al. 2012; Habibi 2013, 2014; Hatton et al. 2016; Missoum et al. 2013; Roy et al. 2009). These modifications are conducted either prior to or after the preparation of CNFs. Although there are several examples of modification routes for nanocellulosic material, it is not seldom that the modification is preceded by several (tedious) solvent exchange steps or that the nanocellulose is dried and redispersed in non-aqueous solvents prior to modification (Andresen et al. 2006; Felix and Gatenholm 1991; Goussé et al. 2004; Heinze et al. 2000; Klemm et al. 1997; Lu et al. 2008; Siqueira et al. 2008). Unfortunately, this will induce fibril aggregation that most likely will hinder re-dispersion of dried CNFs in the reaction medium/solvent due to the differences in polarity between the fibrils and the solvent, which subsequently lead to inefficient modification. In light of this, it would be advantageous to develop a sustainable functionalization protocol where the different modification steps as well as the consumption of solvent would be reduced and the usage of sustainable media (aqueous or hydroalcoholic based) is utilised. An appropriate basis for this concept is carboxymethylation, an well-established cellulose functionalisation method (Hollabaugh et al. 1945; Walecka 1956) which yields carboxymethyl cellulose (CMC), also a pre-treatment method when preparing CNF (Wågberg et al. 2008). The carboxymethylation is conducted in a mild alcoholic medium and allows for the possibility of using Williamson etherification chemistry simultaneously to add another functional moiety during the pre-treatment process, prior to mechanical disintegration.
Hence, the aim of this work was to investigate the possibility to develop a simple one-pot protocol where a reactive moiety could be attached to cellulose fibres in conjunction with the carboxymethylation process. The one-pot protocol was evaluated on the basis of two subsequent reactions, i.e. carboxymethylation and conventional etherification, in one-pot and the effect of this approach on the fibre charge and the degree of substitution. Maintained functionalisation after mechanical disintegration and the colloidal stability of the functionalised fibrils were also evaluated.
Experimental
Materials
Never-dried softwood dissolving pulp, kindly donated by Aditya Birla, Domsjö Fabriker AB, Örnsköldsvik, Sweden, was used as starting material for the preparation of CNF dispersions. The pulp was acid (HCl, pH 2) and then base (NaOH, pH 9) washed with an intermediate water wash followed by a thorough wash with deionised water and reslushed using a disintegrator at 30,000 revolutions (PTI Austria) prior to use. Ethanol (EtOH 96%), methanol (MeOH, HPLC grade), 2-propanol (IPA ≥ 99.5%), acetic acid (glacial ≥ 99.8%), dichloromethane (DCM, reagent grade), NaOH [puriss > 98%, pellets (anhydrous)], allyl bromide (99%), propargyl bromide (80% in toluene), disperse red (DR13, dye content 95%), (+)-sodium-L-ascorbate (NaAsc 98%), copper (II) sulphate (Cu(II)SO4 ≥ 99.0%, anhydrous) and 4,4′-azobis-(4-cyanovaleric acid) (ACVA ≥ 98.0%) and methyl 3-mercaptopropionate (98%) were purchased from Sigma Aldrich, Sweden. NaHCO3 (analysis grade) was purchased from Merck KGaA, Germany and used as received. 1,1′-Carbonyldiimidazole (98%) was purchased from Carbosynth Ltd., UK. Monochloroacetic acid (puriss > 99%) was purchased from Fluka. All chemicals were used as received. 6-Azidehexanoic acid was kindly donated and synthesised as previously described in literature (Montanez et al. 2011).
Preparation of carboxymethylated CNF (CM-CNF)
The CM-CNF was prepared according to a procedure described elsewhere (Wågberg et al. 2008). Briefly, fibres (3 g) were solvent-exchanged to ethanol by immersing the fibres in 150 mL ethanol (15 min) followed by a filtration step. This procedure was conducted four times. The fibres were then added to a solution of 0.3 g monochloroacetic acid in 15 mL isopropanol and impregnated for 30 min. After impregnation the fibres were added to a heated solution (82 °C) of 0.49 g NaOH in 15 mL MeOH and 60 mL isopropanol in a three-necked round-bottom flask. The carboxymethylation was allowed to proceed for 60 min under reflux. The carboxymethylation reaction was followed by a work-up procedure consisting of three washing steps and intermediate filtration steps: 1 L deionised water, 100 mL acetic acid (0.1 M) and finally 1 L deionised water. In order to convert the fibres to their sodium form the fibres were impregnated with NaHCO3 (4 wt%) for 30 min. After impregnation the carboxymethylated fibres (CM-fibres) were washed and filtered with 1 L of deionised water. The fibres were diluted with Milli-Q water (0.2 wt%) and then mechanically disintegrated in a high pressure microfluidizer (M-110EH, Microfluidics Corp, US) by a single pass through two chambers (400 and 200 μm) in series and then four times through two chambers (200 and 100 μm) in series. The homogenized dispersion was further diluted with Milli-Q water (0.15 wt%) and then ultra-sonicated for 10 min at 30% amplitude with a microtip probe (6 mm in diameter) on a Vibracell VCX750 (Sonics & Materials Inc., US). Thereafter, the sonicated dispersions were centrifuged for 1 h at a gravitational force of 4100g using a Rotina 420 bench-top centrifuge (Andreas Hettich GmbH & Co.KG, Germany). The supernatant was collected and a stock dispersion of CM-CNF (0.1 wt%) was prepared by dilution with Milli-Q water.
One-pot preparation of carboxymethylated allyl-CNF (CM-ene-CNF)
The one-pot preparation of CM-ene-CNF followed the same procedure as for the preparation of the CM-CNF except for an added reaction step after the carboxymethylation. After undergoing 60 min of carboxymethylation, allyl bromide (11 g, 5:1 molar eq. to the anhydroglucose units (AGUs)) was added to the reaction vessel and the allylation was allowed to proceed for another 60 min. Then the work-up of CM-ene-CNF followed the exact same procedure as described for CM-CNF.
One-pot preparation of carboxymethylated alkyne-CNF (CM-yne-CNF)
The one-pot preparation of CM-yne-CNF followed the same procedure as for the preparation of the CM-ene-CNF except for using propargyl bromide (14 g, 5:1 molar eq. to the AGUs) as the added reagent in the second functionalisation step. Then the work-up of CM-yne-CNF followed the exact same procedure as described for CM-CNF.
Thiol-ene coupling of methyl 3-mercaptopropionate on CM-ene-fibres (CM-ene-fibre-T)
To demonstrate the presence of enes, CM-ene-fibres (50 mg) were subjected to a solvent-exchange procedure: EtOH (100 mL, 99%) and then MeOH (100 mL). The fibres were then mixed with MeOH (10 mL) in a glass vial and stirred for 60 min, thereafter methyl 3-mercaptopropionate (2 μL) and ACVA (200 μg) were added followed by a purging step with argon for 30 min in an ice bath. The vial was then immersed in a heated oil bath (set to 60 °C). The reaction was allowed to proceed for 18 h. After the reaction was stopped the fibres were filtered and then thoroughly washed and filtered intermittently with MeOH (500 mL) to remove unreacted reagents. The fibres were dried in a vacuum oven at 50 °C and stored at room temperature. Carboxymethylated cellulose fibres (CM-fibres) were also subjected to the same procedure and used as reference. Blank samples were conducted using exactly the same synthetic procedure but without the addition of ACVA (radical initiator) and are denoted CM-ene-fibre-T-blank and CM-fibre-T-blank, respectively.
Thiol-ene coupling of methyl 3-mercaptopropionate to CM-ene-CNF (CM-ene-CNF-T)
To confirm the preservation of enes after homogenisation aerogels from CM-ene-CNF dispersion (0.5 wt%) were prepared by instantly freezing samples, in aluminium pans, using liquid nitrogen and then freeze drying the samples for 4 days. The CM-ene-CNF aerogels were then subjected to the same thiol-ene coupling procedure as described for CM-ene-fibres and CM-CNF aerogels were used as reference. Blank samples were conducted using exactly the same synthetic procedure but without the addition of ACVA (radical initiator) and are denoted CM-ene-CNF-T-blank and CM-CNF-T-blank, respectively.
Azide-alkyne Huisgen cycloaddition of DR13-N3 on CM-yne-fibres
To demonstrate the presence of alkyne groups, CM-yne-fibres (100 mg) were subjected to a solvent-exchange procedure: EtOH (100 mL, 99%) and then MeOH (100 mL). The fibres were then mixed with MeOH (44 mL) in a round bottom-flask and stirred for 60 min, thereafter NaAsc (15 mg) and Cu(II)SO4 (6 mg), dissolved in a small amount of deionised water, were added to the reaction vessel, followed by the addition of DR13-N3 (19 mg) dissolved in DCM (7 mL). The reaction vessel was covered in aluminium foil and the reaction was allowed to proceed overnight. After the reaction was stopped the fibres were filtered and thoroughly washed with a mixed solution of MeOH and DCM (7:1). The fibres were then soxhlet extracted using DCM for 24 h to remove the last traces of unreacted reagents. The fibres were dried in a vacuum oven at 50 °C and stored at room temperature. Carboxymethylated cellulose fibres (CM-fibres) were also subjected to the same procedure and used as reference.
Azide-alkyne Huisgen cycloaddition of DR13-N3 on CM-yne-CNF
To confirm the preservation of alkyne groups after homogenisation aerogels from CM-yne-CNF dispersion (0.5 wt%) were prepared by instantly freezing samples of the dispersion, in aluminium pans, using liquid nitrogen and then freeze drying the samples for 4 days. The obtained aerogels underwent the same cycloaddition procedure as described for the CM-yne-fibres. CM-CNF-aerogels were also subjected to the same procedure and used as reference.
Characterisation methods
Fourier transform infrared spectroscopy (FT-IR) was performed on a Perkin-Elmer Spectrum 100 FT-IR equipped with a MKII Golden Gate, heat controlled single reflection attenuated total reflection (ATR) accessory from Specac Ltd. (Kent, England). Each spectrum was based on 16 scans averaged at 4.0 cm−1 resolution in the range of 600–4000 cm−1. Transmission was also carried out using KBr-pellets.
Cross-polarization magic angle spinning 13C nuclear magnetic resonance (CP/MAS 13C-NMR) spectra were recorded in a Bruker Avance III AQS 400 SB instrument operating at 9.4 T. All measurements were carried out at 295 (± 1) K with a magic angle spinning (MAS) rate of 10 kHz. A 4-mm double air-bearing probe was used. All samples were packed uniformly in a zirconium oxide rotor. Water swollen samples had a water content of 40–60%. Data acquisition was performed using a cross-polarization (CP) pulse sequence, i.e., a 2.95 ms proton 90°-pulse and an 800 ms ramped (100–50%) falling contact pulse, with a 2.5 s delay between repetitions. A SPINAL64 pulse sequence was used for 1H decoupling. The Hartmann-Hahn matching procedure was based on glycine. The chemical shift scale was calibrated to the TMS-scale (tetramethylsilane, (CH3)4Si) by assigning the data point of maximum intensity in the alpha-glycine carbonyl signal to a shift of 176.03 ppm. 4096 transients were recorded on each sample leading to an acquisition time of about 3 h. The software for spectral fitting was developed at Innventia AB and is based on a Levenberg–Marquardt algorithm (Larsson et al. 1997). All computations were based on integrated signal intensities obtained from spectral fitting (Wickholm et al. 1998). The errors given for parameters obtained from the fitting procedure are the standard error with respect to the quality of the fit.
Conductometric titration according to SCAN-CM 65:02 was used to determine the total charge density of the fibres using a Metrohm 702SM Titrino titrator. Briefly, the fibres were dispersed in deionised water at a concentration of 0.6–0.7 g/L with a pH of 4 and a salt concentration of 2 mM in the suspension and then titrated with 0.1 M NaOH.
Polyelectrolyte titration (PET) was carried out to determine the surface charge of the CNF dispersions, at pH 9, by titration with a poly(diallyldimethylammonium chloride) solution (PDADMAC) with the aid of a Stabino Particle Charge Mapping unit (Particle Metrix GmbH, Germany).
Dynamic light scattering (DLS) measurements were performed with a Zetasizer ZEN3600 (Malvern Instruments Ltd., UK) to study the dynamics of the dispersions by monitoring the correlation decay rate and the intercept of the intensity correlation function (ICF). For each sample, five measurements were made, each measurement consisting of ten runs.
X-ray diffraction (XRD) measurements were carried out, to study the changes in crystallinity, by ARL™ X’TRA Powder Diffractometer (Thermo Fisher Scientific Inc., USA) using Cu Kα radiation generated at 45 kV and 40 mA. Scans were obtained from 5 to 50 degrees 2θ in 0.02° steps for one second per step. The crystallinity index was calculated according to:
where I200 is the maximum intensity of the (200) lattice peak and IAm is the corresponding intensity of the amorphous part at 18°.
X-ray photon spectroscopy (XPS) was performed at Umeå University and collected with Axis Ultra DLD electron spectrometer (Kratos analytical Ltd., UK) using monochromatised Al Kα (1486.6 eV) source operated at 150 W. Survey spectra where collected from 1100 to 0 eV at pass energy of 160 eV. Kratos software was used for spectra processing and high resolution XPS spectra were fitted using linear combinations of 70:30 Gauss–Lorentzian functions on Shirley background-subtracted spectra. Binding energy (BE) scale was calibrated using aliphatic C1s component, set at 285.0 eV. The degree of substitution (DS) was calculated according to Eq. (2). For the CM-ene-fibres the calculations were based on the content of sulphur and for the CM-yne-fibres the calculations were based on the content of nitrogen.
where x is the weight fraction, obtained by conversion from the atomic percentage, of an element Y (S or N) in the analysed sample. MY and MR are the molecular weights of the element Y and the functional group F, respectively, and NY(AGU) and NY(F) are the number of atoms of element Y in the repeating D-anhydroglucopyranose unit (AGU) and in the functional group (F).
Contact angle measurements were conducted on pressed CNF aerogels using a CAM200 contact angle meter (KSV Instruments Ltd, Finland) and images were recorded and analysed by the software OneAttension (Biolin Scientific, Sweden). Measurements were performed at room temperature at 50% RH. A 10 µL droplet was deposited on the aerogel surface using Milli-Q water.
Results and discussion
Carboxymethylated and functionalised fibres
In this work, carboxymethylation of cellulose fibres has been successfully combined with a subsequent functionalisation step (Williamson etherification), yielding a one-pot preparation method of functionalised carboxymethylated cellulose fibres (CM-F-fibre) and fibrils (CM-F-CNF). As proof of concept, two different functionalities were targeted, allyl and alkyne functional groups (Scheme 1).

Carboxymethylation and subsequent allylation or alkynation of cellulose fibres
Initially, it was investigated if allyl bromide or propargyl bromide could be added, concurrently with the monochloroacetic acid, however, extremely low values of the total fibre charge were determined, indicating that the reactions of monochloroacetic acid and the alkyl halides were competing reactions. Hence, these attempts were discarded. Nonetheless, to the best knowledge of the authors this is the first reported procedure where cellulose fibres have been both charge stabilised and functionalised in a subsequent one-pot approach. The synthetic approach is robust and repetitive reactions renders reproducible results.
Characterisation of functionalised CM-fibres
Successful carboxymethylation and preserved carboxylation after functionalisation was confirmed by conductometric titration. The targeted total charge was 600 μeq g−1, and as seen in Table 1, the functionalised samples, CM-ene-fibres and CM-yne-fibres, maintained their total charge density after the functionalisation step. X-ray diffraction and CP/MAS 13C-NMR spectroscopy were utilised to evaluate any changes in crystallinity between the different samples (Table 1 and Figure S3). There is a discrepancy between the estimates of the degree of crystallinity obtained by CP/MAS 13C-NMR and XRD (Park et al. 2010), both in absolute values and in trends. The degree of crystallinity by XRD is systematically higher than corresponding results obtained by NMR. Both techniques indicate that there is no significant change in the degree of crystallinity as a result of the chemical modifications. Differences in the absolute values of crystallinity assessments obtained by XRD and NMR results is not uncommon, however the absence of common trends in XRD and NMR results deserves some attention. The chemical modification performed in this study primarily attach substituents to polymers that constitute the cellulose fibril surface, the cellulose particle phase boundary. NMR estimates of the degree of crystallinity are calculated based on the surface-to-volume ratio of the cellulose fibrils (Nocanda et al. 2007; Wickholm et al. 1998). The surface-to-volume ratio is sensitive to any changes in the signal intensity originating from polymers located at the cellulose fibril surfaces, why it is inherently sensitive to chemical changes occurring at the cellulose fibril surfaces. It can be argued that the fibril surface polymers, located at the cellulose particle phase boundary, are not part of a complete unit cell, why they could be considered a contribution to the amorphous (non-crystalline) signal in XRD, a situation that would most likely prevail also after a partial chemical modification of such polymers. This could explain the lack of consistent trends in the NMR and XRD data on crystallinity. Nonetheless, both NMR and XRD results indicate that no remarkable change in crystallinity is seen due to the functionalisation.
In the case of alkynation, successful functionalisation was confirmed by FT-IR in transmission mode utilizing KBr-pellets, by detecting the sharp but weak peak around 2120 cm−1, ascribed to the carbon–carbon alkyne bond stretch vibration (Socrates 2001). Although moderately sharp, the peak was apparent in the CM-yne-fibre sample (Fig. 1a), confirming a successful functionalisation.

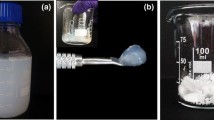
FT-IR spectra of CM- and CM-yne fibres (a) and images of CM-fibre (b) and CM-yne-fibre (c) that has been subjected to azide-alkyne Husigen cycloaddition with DR13-N3. The slight green colour of the neat sample (1b) is due to the remaining copper in the fibres after the washing steps
Azide-alkyne Husigen cycloaddition employing azide-functionalised disperse red 13 (DR13-N3) was also conducted to further establish alkynation (Fig. 1b, c) and the strong red colour of the alkynated sample (Fig. 1c) and the absence of colour in the neat sample (Fig. 1b) affirm a successful alkynation.
Attempts to directly verify allylation were ineffective since the characteristic alkene bond stretch or bending vibrations (3010–3100 cm−1, 1620–1680 cm−1 or 675–1000 cm−1) (Socrates 2001) were all hidden in the characteristic peaks attained from the carboxymethylated cellulose fibres (Fig. 2). However, when looking in detail at the region 1800–1500 cm−1 (Fig. 2) it is evident that a new peak has appeared at 1739 cm−1, anticipated to be attributed to a carbonyl bond stretch of an ester (Socrates 2001). The appearance of this new peak is possibly a result of the formation of an allyl ester at the carboxylate group. This may occur via an SN1-reaction due to the dissociation of the bromide from the allyl bromide, allowing the carboxylate group to react with the allyl (Sykes 1986). However, the formation of allyl esters during the etherification is a side-reaction, and is not considered to be a competing reaction since no significant difference was noted in the total charge density between CM-fibres and CN-ene-fibres (Table 1), which would have been the inevitable result of a competing reaction.

FT-IR spectrum of CM-fibres and CM-ene-fibres (left) and detailed spectrum in the region 1800–1500 cm−1 (right)
Alternately, a thiol-ene coupling reaction using methyl 3-mercaptopropionate was employed to determine the presence of alkenes since a new peak attributed the carbonyl bond stretch vibration for the ester should appear approximately around 1730 cm−1 (Socrates 2001) after successful coupling, distinguishable from the carboxylate group bond stretch vibration of the carboxymethylated cellulose fibres at 1580 cm−1. It was, however, anticipated that the ester would also physically adsorb to the reference sample (CM-fibre) and not only covalently couple with the allylated samples. Hence, a blank reaction with only methyl-3-mercaptopropionate was conducted to estimate the amount of adsorption (Fig. 3a). The results show that 3-mercaptopropionate is adsorbed to both CM-fibres and CM-ene-fibres in small amount and that the adsorption is seemingly equivalent in both samples. In Fig. 3b, it is observed that a new peak has appeared at 1725 cm−1 in both samples but the ratio between the ester carbonyl (1725 cm−1) and the cellulose carbonyl (1580 cm−1) is greater for the CM-ene-fibre sample indicating that this sample comprise of both adsorbed and covalently coupled ester.

FT-IR spectra of CM-fibres and CM-ene-fibres, after blank reactions (a) and thiol-ene reactions (b)
The samples were analysed by XPS to determine the degree of substitution (DS) by analysing the samples after coupling with methyl 3-mercaptopropionate or DR13-N3 and calculating the content of coupled sulphur onto the CM-ene-fibres (Figure S5) and the nitrogen content, originating from DR13-N3, onto the CM-yne-fibres (Figure S6), respectively. The data obtained were given as atomic percentage and hence, these were converted to weight percentage. Using Eq. (2), the DS of allyl and alkyne-functionality were calculated to be 0.016 and 0.0018, respectively. These degrees of substitution may seem low but the mild conditions need to be taken into account when evaluating the effect of the etherification. As previously studied and discussed (Mangiante et al. 2012), the reaction parameters are governed by the reaction medium, the NaOH weight ratio, the ratio between the functional reagent and the anhydroglucose units (F/AGU) and the reaction time. However, a prerequisite for our study was to employ identical reaction conditions as commonly used for carboxymethylation of cellulose fibres targeting a fibre charge of 600 µeq g−1, i.e. a purely alcoholic medium, approximately 0.7 wt% NaOH and not more than 60 min for the subsequent reaction in order to not implicate the targeted charge. Hence, the discussion is hinged on these postulated conditions.
The rationale behind the reaction time was the incomplete understanding how the reaction time would influence the targeted charge, hence a subsequent reaction time of 60 min was added for the functionalisation and studied. In this regard, our choice of reaction time proved to be reasonable since the apparent charge was not affected by the extended reaction time. Further, the carboxymethylation protocol is conducted in a purely alcoholic medium. On the contrary to the previous work (Mangiante et al. 2012) the absence of water as a co-solvent in our system did not give rise to de-mixing issues for NaOH, which in turn was reported as a probable cause for the major alterations of the crystallinity when a high alcohol/water ratio was used. Instead, our analyses of the changes in crystallinity showed no major alterations of the crystallinity. The F/AGU ratio was chosen with a sensible environmental approach, and a higher DS would reasonably have been achieved with a higher F/AGU ratio (Mangiante et al. 2012). A DS of approximately 0.02 for the CM-F-fibres should be considered as a good value considering the restrictions in reaction parameters.
Characterisation of CM-F-CNFs
The neat and functionalised fibres were further treated mechanically to disintegrate the fibres into fibrils, followed by sonication and centrifugation to obtain stable aqueous dispersions. The surface charge of the fibrils was determined by polyelectrolyte titration (Table 2). The surface charges are lower compared to the total charge density of the fibres (Table 1), however, this is expected since it is assumed that although stable dispersions are obtained not all fibrils are completely disintegrated and small aggregates remaining after centrifugation will lower the surface charge value compared to the apparent total charge density value.
AFM was used to analyse the morphology of the nanofibrillar samples (Figure S2), and the height images do not indicate any noticeable changes of the morphology when comparing the neat and functionalised nanofibrils.
Films from the cellulose nanofibrillar dispersions were made in order to evaluate if the hydrophobic functionalities would increase the contact angle (CA) compared to the neat CNF film. As seen in Table 3, the initial CA measured (10 s) for the functionalised samples had increased with roughly 20%, indicating a more hydrophobic surface compared to the neat sample. Moreover, the water droplet prevailed after 30 and 60 s for both the functionalised samples, although with a decreasing angle, while the water droplet on the neat sample was adsorbed in less than 30 s.
To confirm maintained functionalisation after mechanical treatment aerogels of CM-yne-CNF were made and then subjected to the azide-alkyne Husigen cycloaddition employing DR13-N3. As seen in Fig. 4, the reference sample (CM-CNF) has not been stained with the dye, whilst the CM-yne-CNF sample has been coloured. The colour is not as uniform and solid as for the fibre sample but this is considered more a consequence by the difficulty to homogeneously soak the dry aerogel with solvents and reagents and not due to less functionality sites compared to the fibres.

Images of CM-CNF (a) and CM-yne-CNF (b) that has been subjected to azide-alkyne Husigen cycloaddition using DR13-N3
The maintained presence of alkenes in CM-ene-CNF aerogels was indirectly confirmed by employing the same thiol-ene reaction used for the CM-ene-fibre samples, and blank reactions were also conducted to display the non-covalent adsorption of the methyl-3-mercaptopropionate (Fig. 5a). Similarly, as with the fibre coupling the bond stretch vibration attributed to the carbonyl ester at 1725 cm−1 is present in both samples, displaying yet again the physical adsorption of the ester but as with the fibre sample the ratio between the ester carbonyl (1725 cm−1) and the CM-cellulose carbonyl (1580 cm−1) is greater for CM-ene-fibril, which indirectly demonstrates the presence of alkenes (Fig. 5b).

FT-IR spectra of CM-CNF and CM-ene-CNF, after blank reactions (a) and thiol-ene reactions (b)
The stability of the CM-F-CNF compared to CM-CNF was further evaluated by DLS since the mobility of the dispersed fibrils can be expressed by the decay rate of the normalized intensity function (Tables S7 and S2). It is observed that the added functionality had not significantly changed the mobility, i.e. dispersion stability, nor the size of the fibrils, ensuring that stable dispersions are achievable with an added functionality.
Conclusions
In this work, a one-pot protocol for preparing bi-functional cellulose nanofibrils has been developed using the combination of both carboxymethylation of fibres in order to introduce charge and the concurrent conventional etherification incorporating an additional functionality, in this case alkenes or alkynes, which can be used for further modification of the cellulose nanofibrils. The developed one-pot protocol eliminates the need of tedious solvent exchange procedure and it is conducted in a mild alcoholic medium. Polyelectrolyte titration and FT-IR analysis of the mechanically treated fibres into fibrils corroborated that both the total surface charge and the additional functionalisation are preserved after homogenisation. Moreover, the added functionality had no impact on the morphology or colloidal stability of the fibrils. The developed one-pot protocol is a simple but powerful procedure which can provide a good toolbox to further build on new materials requiring nanocellulosic material with a chemical handle.
References
Andresen M, Johansson L-S, Tanem BS, Stenius P (2006) Properties and characterization of hydrophobized microfibrillated cellulose. Cellulose 13(6):665–677
Chauve G, Bras J (2014) Industrial point of view of nanocellulose materials and their possible applications. In: Handbook of green materials: 1 bio-nanomaterials: separation processes, characterization and properties, World Scientific, Singapore, pp 233–252
Daud JB, Lee KY (2017) Surface modification of nanocellulose. Handb Nanocellul Cellul Nanocompos 1:101–122
Dufresne A (2012) Nanocellulose, from nature to high performance tailored materials. Walter de Gruyter GmbH & Co KG, Berlin
Dufresne A (2013) Nanocellulose: a new ageless bionanomaterial. Mater Today 16(6):220–227. https://doi.org/10.1016/j.mattod.2013.06.004
Eichhorn S, Baillie C, Zafeiropoulos N, Mwaikambo L, Ansell M, Dufresne A, Groom L (2001) Review: current international research into cellulosic fibres and composites. J Mater Sci 36(9):2107–2131
Eichhorn SJ, Dufresne A, Aranguren M, Marcovich N, Capadona J, Rowan SJ, Renneckar S (2010) Review: current international research into cellulose nanofibres and nanocomposites. J Mater Sci 45(1):1–33
Eriksen O, Syverud K, Gregersen O (2008) The use of microfibrillated cellulose produced from kraft pulp as strength enhancer in TMP paper. Nord Pulp Pap Res J 23(3):299–304
Felix JM, Gatenholm P (1991) The nature of adhesion in composites of modified cellulose fibers and polypropylene. J Appl Polym Sci 42(3):609–620
Filpponen I, Kontturi E, Nummelin S, Rosilo H, Kolehmainen E, Ikkala O, Laine J (2012) Generic method for modular surface modification of cellulosic materials in aqueous medium by sequential “click” reaction and adsorption. Biomacromol 13(3):736–742
Goussé C, Chanzy H, Cerrada M, Fleury E (2004) Surface silylation of cellulose microfibrils: preparation and rheological properties. Polymer 45(5):1569–1575
Habibi Y (2013) Chemical modification of nanocelluloses. Biopolym Nanocompos Process Prop Appl 8:367–390
Habibi Y (2014) Key advances in the chemical modification of nanocelluloses. Chem Soc Rev 43(5):1519–1542
Hatton FL, Ruda M, Lansalot M, D’Agosto F, Malmström E, Carlmark A (2016) Xyloglucan-functional latex particles via RAFT-mediated emulsion polymerization for the biomimetic modification of cellulose. Biomacromol 17(4):1414–1424
Heinze T, Dicke R, Koschella A, Kull AH, Klohr EA, Koch W (2000) Effective preparation of cellulose derivatives in a new simple cellulose solvent. Macromol Chem Phys 201(6):627–631
Hollabaugh C, Burt LH, Walsh AP (1945) Carboxymethylcellulose. Uses and applications. Ind Eng Chem 37(10):943–947
Klemm D, Heinze T, Philipp B, Wagenknecht W (1997) New approaches to advanced polymers by selective cellulose functionalization. Acta Polym 48(8):277–297
Klemm D, Kramer F, Moritz S, Lindström T, Ankerfors M, Gray D, Dorris A (2011) Nanocelluloses: a new family of nature-based materials. Angew Chem Int Ed 50(24):5438–5466
Larsson PT, Wickholm K, Iversen T (1997) A CP/MAS13C NMR investigation of molecular ordering in celluloses. Carbohyd Res 302(1–2):19–25
Lu J, Askeland P, Drzal LT (2008) Surface modification of microfibrillated cellulose for epoxy composite applications. Polymer 49(5):1285–1296
Mangiante G, Alcouffe P, Burdin B, Gaborieau M, Zeno E, Petit-Conil M, Fleury E (2012) Green nondegrading approach to alkyne-functionalized cellulose fibers and biohybrids thereof: synthesis and mapping of the derivatization. Biomacromol 14(1):254–263
Missoum K, Belgacem MN, Bras J (2013) Nanofibrillated cellulose surface modification: a review. Materials 6(5):1745–1766
Montanez MI, Hed Y, Utsel S, Ropponen J, Malmstrom E, Wagberg L, Malkoch M (2011) Bifunctional dendronized cellulose surfaces as biosensors. Biomacromol 12(6):2114–2125
Nocanda X, Larsson PT, Spark A, Bush T, Olsson A, Madikane M, Iversen T (2007) Cross polarisation/magic angle spinning 13C-NMR spectroscopic studies of cellulose structural changes in hardwood dissolving pulp process. Holzforschung 61(6):675–679
Park S, Baker JO, Himmel ME, Parilla PA, Johnson DK (2010) Cellulose crystallinity index: measurement techniques and their impact on interpreting cellulase performance. Biotechnol Biofuels 3(1):10
Roy D, Semsarilar M, Guthrie JT, Perrier S (2009) Cellulose modification by polymer grafting: a review. Chem Soc Rev 38(7):2046–2064
Siqueira G, Bras J, Dufresne A (2008) Cellulose whiskers versus microfibrils: influence of the nature of the nanoparticle and its surface functionalization on the thermal and mechanical properties of nanocomposites. Biomacromol 10(2):425–432
Socrates G (2001) Infrared and Raman characteristic group frequencies: tables and charts. Wiley, New York
Sykes P (1986) A guidebook to mechanism in organic chemistry. Pearson Education India, Bengaluru
Wågberg L, Decher G, Norgren M, Lindström T, Ankerfors M, Axnäs K (2008) The build-up of polyelectrolyte multilayers of microfibrillated cellulose and cationic polyelectrolytes. Langmuir 24(3):784–795
Walecka JA (1956) An investigation of low degree of substitution carboxymethylcelluloses. Georgia Institute of Technology, Atlanta
Wickholm K, Larsson PT, Iversen T (1998) Assignment of non-crystalline forms in cellulose I by CP/MAS 13 C NMR spectroscopy. Carbohyd Res 312(3):123–129
Acknowledgments
TK would like to thank the Wallenberg Wood Science Center (WWSC) for financial support. Sandra Garcia-Gallego is thanked for synthesising the 6-azidehexanoic acid.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Kaldéus, T., Larsson, P.T., Boujemaoui, A. et al. One-pot preparation of bi-functional cellulose nanofibrils. Cellulose 25, 7031–7042 (2018). https://doi.org/10.1007/s10570-018-2066-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10570-018-2066-y