
Abstract
Objective
To investigate baseline fat intake and the risk of colon and rectal tumors lacking MLH1 (mutL homolog 1, colon cancer, nonpolyposis type 2) repair gene expression and harboring mutations in the APC (adenomatous polyposis coli) tumor suppressor gene and in the KRAS (v-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog) oncogene.
Methods
After 7.3 years of follow-up of the Netherlands Cohort Study (n = 120,852), adjusted incidence rate ratios (RR) and 95% confidence intervals (CI) were computed, based on 401 colon and 130 rectal cancer patients.
Results
Total, saturated and monounsaturated fat were not associated with the risk of colon or rectal cancer, or different molecular subgroups. There was also no association between polyunsaturated fat and the risk of overall or subgroups of rectal cancer. Linoleic acid, the most abundant polyunsaturated fatty acid in the diet, was associated with increased risk of colon tumors with only a KRAS mutation and no additional truncating APC mutation or lack of MLH1 expression (RR = 1.41, 95% CI 1.18–1.69 for one standard deviation (i.e., 7.5 g/day) increase in intake, p-trend over the quartiles of intake <0.001). Linoleic acid intake was not associated with risk of colon tumors without any of the gene defects, or with tumors harboring aberrations in either MLH1 or APC.
Conclusion
Linoleic acid intake is associated with colon tumors with an aberrant KRAS gene, but an intact APC gene and MLH1 expression, suggesting a unique etiology of tumors with specific genetic aberrations.
Similar content being viewed by others

Introduction
Although dietary fat has been implicated in the etiology of colorectal cancer [1], results from epidemiological studies are inconsistent [2, 3] and often do not support an association, as observed recently in the Women’s Health Study [4]. Fortunately, current molecular techniques to detect DNA alterations on a large scale allow studying molecular endpoints for colorectal cancer, characterized by acquired (epi) genetic defects in tumor DNA [5]. This approach may improve our ability to observe associations between dietary factors and cancer that may otherwise remain undetected.
A multistep model linking sporadic colorectal carcinogenesis to molecular aberrations has been proposed [6–8], with DNA repair genes, tumor suppressor genes and oncogenes, operating in multiple genetic pathways. About 10–20% of sporadic colon carcinomas are characterized by microsatellite instability, predominantly due to promoter methylation of the MLH1 (mutL homolog 1, colon cancer, nonpolyposis type 2) DNA mismatch repair gene, which prevents expression of the enzyme [9]. Up to 90% of colon and rectum carcinomas are chromosomally instable [10, 11] and are associated with mutations in the APC (adenomatous polyposis coli) and TP53 (tumor protein 53) tumor suppressor genes and in the KRAS (v-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog) oncogene [12]. However, simultaneous occurrence of mutations in these three genes is rare suggesting that, even within this group of chromosomally instable tumors, different genetic pathways to colorectal cancer exist [13, 14]. Mutations in the APC gene are found to occur relatively early in colorectal tumorigenesis and are observed in up to 80% of both adenomas and carcinomas [8, 15]. Mutations in the KRAS gene are observed in approximately 10–20% of small adenomas and 40–50% of larger adenomas and carcinomas, suggesting it to be an important event in the progression of adenoma to carcinoma [15]. Mutations in the TP53 gene are postulated to affect relatively late stages of colorectal carcinogenesis [15].
Breivik et al. proposed that the type of genetic instability in cancer cells reflects the selection pressures exerted by specific carcinogens [16]. Bardelli et al. subsequently tested this hypothesis in immortal genetically stable human cells and concluded that exposure to specific carcinogens can indeed select for tumor cells with distinct forms of genetic instability and vice versa [17]. Therefore, DNA adducts derived from dietary fat metabolism could also be associated with colorectal tumors exhibiting chromosomal instability. This is supported by the observations that malondialdehyde (MDA), generated during lipid peroxidation and arachidonic acid metabolism, can form DNA adducts and induce G→T transversions and G→A transitions in DNA [18, 19]. In addition, higher levels of MDA-DNA adducts have been observed in colorectal tissue of adenoma patients than in tissue of controls [20]. MDA levels are modulated by dietary factors, with polyunsaturated fatty acids, and specifically ω-6 polyunsaturated fatty acids, presumably increasing MDA levels [21]. This is in line with our previous report of a significant association between the intake of linoleic acid, the most abundant ω-6 polyunsaturated fatty acid in the diet, and increased risk of colon carcinomas with a mutated KRAS gene within the Netherlands Cohort Study (NLCS) on diet and cancer [22].
These observations and hypotheses prompted us to investigate the associations between the intake of total fat and different types of fat and the risk of colon and rectal tumors lacking MLH1 expression and with and without APC gene mutations, two early events in colorectal tumorigenesis, independent of tumors harboring KRAS gene mutations.
Materials and methods
Study population
The prospective NLCS was initiated in The Netherlands in September 1986. The study design has been described in detail elsewhere [23]. Briefly, at baseline a total of 58,279 men and 62,573 women, between the ages of 55 and 69 years, completed a self-administered food frequency and lifestyle questionnaire. Incident cancer cases are identified by monitoring of the entire cohort for cancer occurrence through annual record linkage to the National Cancer Registry (NCR), consisting of nine regional cancer registries throughout The Netherlands, and to PALGA, a nationwide network and registry of histo- and cytopathology [24]. The NCR and PALGA together provide a near 100% coverage of the 204 municipalities included in the NLCS.
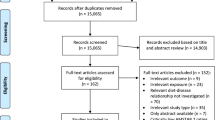
Accumulation of person-time in the cohort was estimated through biennial vital status follow-up of a subcohort of 3,500 men and women who were randomly selected after baseline exposure measurement [24]. Cases with prevalent cancer other than non-melanoma skin cancer were excluded from the subcohort, which left 3,346 men and women for analysis next to all colorectal cancer cases from the entire cohort. No subcohort members were lost to follow-up. A flow diagram of subcohort members and patients on whom the analyses are based is given in Fig. 1.

Flow diagram of the number of subjects on whom the final statistical analyses were based. aNetherlands Cancer Registry. bPathologisch Anatomisch Landelijk Geautomatiseerd Archief. cPatients with rectosigmoid tumors were not included in the analyses. dmutL homolog 1, colon cancer, nonpolyposis type 2. eAdenomatous polyposis coli. fMutation cluster region. gv-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog. hPatients with rectal tumors were not included in the analysis according to MLH1 expression
The first 2.3 years of follow up were excluded because of possible preclinical disease affecting exposure status and because of incomplete nationwide coverage of PALGA alone (i.e., not in combination with the NCR) in some of the municipalities included in the NLCS. Within this period, 83 subcohort members deceased or were diagnosed with cancer other than non-melanoma skin cancer, leaving 3,263 subcohort members for analysis. From 1989 to 1994, 929 incident cases with histologically confirmed colorectal cancer were identified within the entire cohort, of whom 819 could also be linked to a PALGA report of the lesion. The PALGA reports were used to identify and locate tumor tissues from eligible colorectal cancer patients in 54 pathology laboratories throughout the Netherlands. Cancers were classified according to site as follows, colon: cecum through sigmoid colon (ICD-O codes 153:0, 153.1, 153.2, 153.3, 153.4, 153.5, 153.6, 153.7), proximal colon (ICD-O codes 153.0, 153.1, 153.4, 153.5, 153.6), distal colon (ICD-O codes 153.2, 153,3, `53.7), rectosigmoid (ICD-O code 154.0), and rectum (ICD-O code 154.1).
Tissue samples
Approval for collection of archival tissue samples from colorectal cancer patients was obtained from the Ethical Review Board of University Maastricht, the NCR and PALGA. The tissue specimen collection started in August 1999 and was completed in December of 2001. For five percent of patients, tissue samples could not be retrieved (44/819) due to administrative inconsistencies. Of 775 available tissue samples, 737 (95%) contained sufficient tumor material for molecular analyses of MLH1 expression and mutations in APC and KRAS genes.
Since the rectosigmoid can be considered as a clinically applied term rather than an anatomically defined transitional zone between the colon and rectum, the 85 patients with a rectosigmoid tumor were excluded from data analysis. Moreover, the group of patients with a rectosigmoid tumor was too small for adequate stratified analysis.
MLH1 expression analysis
Formalin-fixed, paraffin-embedded tissues were sectioned at 4 μm and contained tumor tissue and normal adjacent mucosa. Endogeneous peroxidase activity was blocked with 3% H2O2. Slides were submitted to microwave antigen retrieval in 1mM EDTA buffer (pH 8.0) and incubated with 10% normal horse serum for 10 min at room temperature. Then, sections were incubated overnight at 4°C with mouse monoclonal antibodies against MLH1 protein (clone G168–15, PharMingen, San Diego, CA) at a 1:100 dilution. Antibody binding was detected by incubating the sections at room temperature with the peroxidase-labeled DAKO Envision System (DAKO, Carpinteris, CA) and using DAB as a chromogen. Sections were counterstained with haematoxylin.
Lesions were considered to lack MLH1 protein expression when unequivocal absence of nuclear staining of the tumor epithelial cells was observed. Nuclear staining of normal epithelial and stromal cells or lymphocytes served as internal positive control. Two investigators reviewed the immunohistochemical staining independently and discrepancies were re-examined and discussed with a pathologist until consensus was reached. MLH1 expression status was determined successfully in 98% of samples, i.e., 468 colon tumors and 173 rectum tumors (Fig. 1).
APC mutation analysis
The majority of somatic mutations in APC occur within the mutation cluster region. Mutation analysis of the mutation cluster region (codons 1,286–1,520), was performed on archival adenocarcinoma specimens, using macrodissection followed by extraction of tumor DNA. Then nested PCR was used to amplify the mutation cluster region in four overlapping DNA fragments and the purified fragments were sequenced. This procedure has been described in detail elsewhere [25]. In brief, in a first round of PCR, two overlapping fragments were generated, that served as templates for a second round of PCR to amplify four overlapping biotin-labeled PCR fragments that were subsequently used for direct sequencing. The sequence profile was analyzed on ALFexpress DNA Analysis System using ALFwin software (Amersham Biosciences, Roosendaal, The Netherlands). Evaluation of the sequence patterns and data entry were independently performed by two observers. Sensitivity and specificity was assessed by analyzing the mutational status of APC in six colorectal cancer cell lines. Both sensitivity and specificity were regarded to be satisfactory since specific mutations in the mutation cluster region of APC were confirmed in CaCo2 cells, SW480 cells and LOVO cells, as previously described [25, 26], and wild type sequences were confirmed in HCT116, Colo205 and HT29, for the mutation cluster region of APC [25]. In addition, the detection limit was 5% of mutated DNA [25]. Reproducibility of mutation analysis was regarded to be satisfactory since 85% of duplicate analyses, from flank PCR of genomic DNA to sequencing of the four fragments (i.e., 61 out of 72 fragments), revealed identical mutation status of APC [25].
From 47 colon cancer patients and 25 rectum cancer patients, one or more fragments of the APC gene mutation cluster region could not be amplified and these patients were not included in this study, leaving 429 colon and 151 rectum cancer cases with successful analysis of the mutation cluster region of the APC gene (Fig. 1).
KRAS mutation analysis
Mutation analysis of the exon1 fragment of the KRAS oncogene, spanning codons 8–29, was performed on archival adenocarcinoma specimens, using nested PCR, followed by direct sequencing of purified fragments [27]. The detection limit was 5% mutated DNA. Reproducibility was regarded to be satisfactory, since 88% of duplicate analyses, from tissue sectioning to DNA sequencing (i.e., 28 out of 32), revealed identical mutation status of KRAS [27]. Mutation analysis was performed with success on 476 colon and 176 rectum cancer cases (Fig. 1).
Exposure assessment
The 150–item semi-quantitative food frequency questionnaire concentrated on habitual consumption of food and beverages during the year preceding the start of the study. Mean individual nutrient intakes per day were computed using the computerized Dutch food composition table of 1986 [28]. The questionnaire was validated against a 9-day diet record [29]. Crude and energy-gender-adjusted (in parentheses) correlation coefficients were 0.72 (0.52) for total fat, 0.73 (0.58) for saturated fat and 0.73 (0.75) for polyunsaturated fat [29]. For energy intake the correlation coefficient was 0.74. On average, the questionnaire covered 91% of the energy intake assessed by the record intake. Questionnaire data were key-entered twice and processed for all incident cases in the cohort and for all subcohort members in a manner blinded with respect to case/subcohort status.
For 7% of subjects (either cases or subcohort members), dietary data were incomplete or inconsistent, and they were excluded from the analyses. Questionnaires were considered incomplete when either: (1) more than 60 items were left blank and less than 35 items were eaten at least once a month; or (2) one or more item blocks (groups of items, e.g., beverages) were left blank. Additional details are given elsewhere [29]. This resulted in the availability of 3,048 subcohort members, 441 colon cancer cases for whom MLH1 expression status was known, and 414 colon and 136 rectal cancer cases for whom APC and KRAS mutation status was known. No data-analyses were conducted for lack of MLH1 expression in rectal cancer cases since there were only two such cases in the cohort (Fig. 1).
Intake of specific fatty acids was based on a food composition database with specific fatty acids derived from the TRANSFAIR study [30]. For this database, the hundred foods that contributed most to fat intake in the Dutch dietary pattern were sampled and analyzed as methyl esters of the fatty acids present in the foods. In the database, total fat includes triglycerides and other lipids such as phospholipids and sterols. The percentage of triglycerides in total fat is assumed to be 93% on average, but varies across food sources. Daily intakes of total fat (g/day), saturated fat (g/day), monounsaturated fat (g/day), polyunsaturated fat (g/day), and linoleic acid (C18:2, C9, 12) (g/day) and linolenic acid (C18:3, C9, 12, 15) (g/day) as the main constituents of polyunsaturated fat, were used as exposure variables. Linoleic and linolenic acid were used as the most abundant sources of ω-6 polyunsaturated fatty acids and ω-3 polyunsaturated fatty acids in the diet. In all analyses, the values for fat intake variables are adjusted for energy intake by the residual method [31]. For data analyses, quartiles of the intake of fat and different types of fats were computed based on the distribution of subcohort members. Daily intake of dietary fiber (g/day), alcohol (g/day), fruit (g/day), vegetables (g/day) and total energy (kJ/day) and age at baseline (years), sex (men/women), body mass index (kg/m2), non-occupational physical activity (<30 min/day, 30–60 min/day, 60–90 min/day, >90 min/day), family history of colorectal cancer (yes/no) and smoking status (never/ex/current) were regarded as potential confounders.
Statistical analysis
Data analyses were based on study participants for whom data on fat intake and confounding variables were complete, i.e., 2,948 subcohort members, 428 colon cancer patients for whom MLH1 expression status was known, and 401 colon cancer and 130 rectal cancer patients for whom APC and KRAS mutation status was known (Fig. 1).
Data analyses were conducted separately for overall colon and rectal cancer, colon cancer lacking MLH1 expression, colon and rectal cancer with or without a truncating APC mutation, described here as APC + and APC − tumors respectively. Truncating APC mutations lead to the introduction of a stop codon and result in a truncated and therefore, inactive APC protein. The analyses with truncating APC mutations were also conducted separately for the most common point mutations resulting in the introduction of a stop codon, i.e., C:G → T:A or G:C → T:A point mutations. As indicated previously, associations between fat intake and KRAS mutated tumors have been described in this population previously, and a positive association between the intake of linoleic acid and KRAS mutated colon tumors was observed [22]. Therefore, when (borderline) significant associations were observed with any of the colon tumor endpoints, analyses were repeated excluding tumors harboring mutations in KRAS.
Since tumors may harbor multiple mutations it is difficult to assess whether observed associations are specific for tumors with a particular gene defect. We therefore, conducted additional analyses when (borderline) significant associations were observed. In these analyses subgroups of tumors were formed characterized by either the absence of the three gene defects, or by defects in a single gene, i.e., either only lack of MLH1 expression, only a truncating APC mutation or only an activating KRAS mutations. Activating KRAS mutations are defined as mutations in codons 12 and 13 of the KRAS gene leading to an altered amino acid.
Mean values of the intake of fat variables (g/day), and possible confounding variables including age at baseline (years), dietary fiber (g/day), alcohol (g/day), intake of fruit (g/day), vegetable (g/day), energy (kJ/day), and BMI (kg/m2), as well as distributions of the variables sex, family history of colorectal cancer (yes/no), smoking status (never/ex/current smoker) and physical activity in leisure time (<30, 30–60, 60–90, >90 min/day) were evaluated for subcohort members, colon and rectal cancer patients with or without a truncating APC mutation and colon cancer patients lacking MLH1 expression. Differences in mean values of the continuous variables between patients with or without truncating nonsense or frameshift mutations in the mutation cluster region of the APC gene, and between patients with or without MLH1 expression, were tested using the Mann–Whitney-U-test since the variables were not normally distributed among cases. The distributions of the categorical variables between patients with and without truncating APC mutations were tested with the χ2–test.
Incidence rate ratios (RR) and corresponding 95% confidence intervals (CI) for colon and rectal cancer patients were estimated according to quartiles of intake of fat variables, and one standard deviation increment of intake, using Cox proportional hazards regression models. In addition, associations were estimated for specific molecular endpoints of the tumors.
Standard errors were estimated using the robust Huber–White sandwich estimator to account for additional variance introduced by sampling the subcohort from the entire cohort [32, 33]. The proportional hazards assumption was tested using the scaled Schoenfeld residuals [34]. Tests for dose response trends over the different quartiles and categories of fat intake were estimated by fitting the ordinal exposure variables as continuous variables and evaluated using the Wald test.
The covariates included in the multivariate analyses were those found to significantly (p < 0.05) contribute to the multivariate model for colon and/or rectal cancer (age at baseline, sex, body mass index, family history of colorectal cancer, and smoking status) or to influence the RR by more than ten percent, as well as energy intake.
Results
Lack of expression in MLH1 was observed in 13% (54 out of 428) of tumors from colon cancer patients (Table 1). APC truncating mutations were observed in tumors from 32% of colon cancer patients (127 out of 401) and 44% of rectal cancer patients (57 out of 130) (Table 1). C:G→T:A transitions or G:C→T:A transversions that would result in a stop codon were observed in 10% and 5% of colon cancer patients and 12% and 5% of rectal cancer patients, respectively. These figures are similar to the percentages reported for the total group of colon and rectal cancer patients for whom APC mutation status was available, but for whom dietary intake data were not always complete [25].
Colon and rectal cancer patients were generally older and more frequently men than subcohort members (Table 1). Colon cancer patients lacking MLH1 expression in their tumor were significantly less often men than patients with expression of the gene (41% vs. 56%). There were no striking differences in fat intake between patients and subcohort members or between patients with or without MLH1 expression or APC mutations in their tumors. Only rectal cancer patients with a tumor harboring a truncating APC mutation had a higher intake of saturated fat than rectal cancer patients without a truncating APC mutation (p = 0.05).
Neither total fat nor different types of fat appeared to be associated with overall colon cancer risk in this population (Table 2). For different subgroups of colon cancer based on absence of MLH1 expression or absence or presence of APC truncating mutations in their tumors, total fat intake and most of the specific types of fat intake variables were also not associated with risk. However, polyunsaturated fat intake, and especially linoleic acid intake, appeared to be associated with an increased risk of colon tumors without MLH1 expression and with colon tumors without APC truncating mutations, but not with colon tumors with APC truncating mutations (Table 2). For colon tumors without MLH1 expression, the RR according to the quartiles of linoleic acid intake were increased, though not significantly, for all the categories of intake above the reference (lowest quartile of intake), i.e., 1.66 (95% CI 0.69–3.98), 2.14 (95% CI 0.91–5.00) and 2.02 (95% CI 0.86–4.76) for the second through the fourth quartiles respectively, and the test for linear trend was borderline significant (p = 0.08). A similar trend was observed for the risk of colon tumors without APC truncating mutations (RR over the quartiles of linoleic acid intake: 1.50 (95% CI 1.02–2.21), 1.68 (95% CI 1.15–2.45) and 1.44 (95% CI 0.99–2.11) respectively, p-trend = 0.05 (Table 2). Additional analyses for subgroups of colon tumors with specific truncating point mutations in APC did not show any associations with the intake of fat or different types of fat (results not shown).
For overall rectal cancer, associations with the intake of total fat or different types of fat were not observed (Table 3). Also after taking account of truncating APC mutations in rectal tumors, none of the fat intake variables were significantly associated with risk of rectal cancer. Only the intake of saturated fat appeared to be inversely associated with rectal tumors without APC truncating mutations (RR for the highest versus the lowest quartile of intake: 0.46 (95% CI 0.22–0.97), p-trend = 0.07) (Table 3). For rectal tumors with specific types of APC truncating mutations no associations were observed with any of the fat intake variables (results not shown).
Additional analyses were conducted to assess whether the observed associations of polyunsaturated fat intake, and especially linoleic acid intake, with the increased risk of colon tumors lacking MLH1 expression and with the increased risk of colon tumors without APC truncating mutations, were observed because of an underlying association with colon tumors harboring a KRAS mutation, as previously observed [22]. Excluding tumors with a KRAS mutation resulted in the absence of a statistically significant association of polyunsaturated fat intake and linoleic acid intake with colon tumors lacking MLH1 expression (p-trend = 0.34 and 0.12, respectively) and those lacking APC tuncating mutations (p-trend = 0.77 and 0.99, respectively). Intake of polyunsaturated fat or linoleic acid was neither associated with the risk of colon cancer without any of the three gene defects, nor with the risk of colon cancer only lacking MLH1 expression, nor with the risk of colon cancer with only truncating APC mutations (Table 4). With increasing intake of polyunsaturated fat and of linoleic acid, a strongly increased risk of colon cancer with only activating KRAS mutations was observed (Table 4) (p-trend ≤ 0.001 for both polyunsaturated fat and linoleic acid intake). The RRs for one standard deviation increase in intake were 1.40 (95% CI 1.17–1.68) and 1.41 (95% CI 1.18–1.69), respectively. The RRs for polyunsaturated fat (not shown) and linoleic acid intake were of similar size when estimated separately for men (1.41, 95% CI 1.15–1.72 for 1 standard deviation increase in linoleic acid intake) and women (1.42, 95% CI 0.96–2.10), and were elevated for proximal (1.23, 95% CI 0.99–1.53) and distal colon cancer (1.53, 95% CI 1.21–1.95). Likewise, a positive association was observed for all colorectal cancers (1.24, 95% CI 1.06–1.47 and p-trend = 0.01), based on a total of 87 cases (i.e., including the rectosigmoid).
Likewise, additional analyses were conducted for saturated fat intake in relation to risk of rectal cancer without APC truncating mutations, also excluding individuals with a KRAS mutation and lack of MLH1 expression. The association did not change substantially. Again, only the highest level of intake showed a significant reduced risk of cancer compared to the reference category (RR 0.40 95% CI 0.14–1.15, p-trend = 0.09).
Discussion
In this prospective study, we observed that the intake of total, saturated and monounsaturated fat was not associated with the risk of colon cancer, rectal cancer, or the different molecular subgroups of cancer based on lack of MLH1 expression or truncating mutations in the APC gene. This was also found for polyunsaturated fat intake and rectal cancer. However, linoleic acid showed an association with increased risk of colon tumors with only an activated KRAS mutation and no additional truncating APC mutation or lack of MLH1 expression.
None of the other epidemiological studies report on specific fatty acids and the risk of molecular surrogate end-points for colon or rectal cancer or adenomas [35–43]. Some of these studies report on various types of fat depending on saturation level, but the results are inconsistent across the studies [35, 38, 41, 42] including the current study.
Diergaarde et al. observed unsaturated fat intake to be associated with increased colon carcinomas with a truncating APC mutation [38]. No distinction was made between mono- and polyunsaturated fats. In our study, we did not observe any association between various types of fat intake and risk of colon or rectal cancer with or without truncating APC mutations after patients also harboring a KRAS mutation in their tumor were excluded from the analyses. We observed a possible inverse association between saturated fat intake and risk of rectal tumors without a truncating APC mutation. However, the association was weak, did not increase gradually according to the quartiles of intake and was only a result of the reduced risk in the highest category of intake. Furthermore, in absence of a biological explanation for this finding and considering the large number of associations investigated, the observation may best be attributed to chance.
Slattery et al. observed saturated and monounsaturated fats, but not polyunsaturated fat, to be associated with increased risk of colon tumors with specific KRAS mutations, i.e., a G→T transversion at codon 12 [41]. No distinction was made between ω-6 and ω-3 fatty acids. We observed an association between polyunsaturated fat intake, especially linoleic acid, and increased risk of colon tumors with a KRAS mutation, regardless of the type of mutations [22].
Finally, Bautista et al. observed an inverse association between monounsaturated fats, mostly derived from olive oil in the Spanish diet, and risk of colon cancer without KRAS mutations [35]. Since olive oil was rarely consumed by this elderly Dutch population in the years preceding 1986 (the cohort baseline), this could explain the lack of association for this type of fat in our study. However, a recent Dutch case–control study on risk factors for colorectal adenomas showed a significant positive association between monounsaturated fats and adenoma risk [42].
Several factors hamper comparisons between our findings and those of other epidemiological studies and may in part explain observed inconsistencies. First, our study is the first large prospective cohort study incorporating molecular end-points for colon and rectal cancer. One of the other studies was a cross-sectional case–case comparison study [40], and the others were case–control studies of varying size (ranging from 108 to 1,510 cases) [35–39, 41–43]. Second, varying end-points were considered. Three of the other studies focused on adenomas instead of carcinomas [37, 40, 42], and three studies also incorporated rectal tumors but did not distinguish between colon and rectum [35, 37, 42].
Previously, we reported the association between linoleic acid intake and increased risk of colon tumors with KRAS mutations (adjusted RR for one standard deviation of increase 1.22 (95% CI 1.05–1.42)) [22]. Now, we report that the association appears to be confined to those colon tumors with activating KRAS mutations and an otherwise intact APC gene and with MLH1 expression. In addition, the association appears to be robust since RRs clearly increase over the quartiles of linoleic acid intake and the RRs for one standard deviation increase in linoleic acid intake are similar for men and women, and is increased for proximal and distal colon cancer as well as for overall colorectal cancer, including the rectosigmoid. The activating KRAS mutations at codons 12 and 13 are predominantly G→T and G→A mutations [27] which could be a result of MDA DNA adduct formation [18, 19] associated with increased ω-6 polyunsaturated fat intake [21]. Therefore, even though chance cannot be ruled out and verification by others is warranted, the association seems quite plausible.
Still several issues remain puzzling. First, why is the observed association for linoleic acid confined to colon cancer and not rectum cancer with only a KRAS mutation? The multistep model for molecular aberrations underlying colorectal carcinogenesis is likely to apply equally for both tumor subsites [7, 8, 15]. However, lack of MLH1 expression in our study is rare among rectum cancer patients and there is growing evidence for differences in the etiology of colon and rectal tumors [33].
Second, why is the association with linoleic acid observed for KRAS and not for truncating APC gene mutations? It is unlikely that adduct formation selectively occurs in one gene but not in the other. However, KRAS is an oncogene requiring only one mutation for the gene to be activated, whereas APC is a tumor suppressor gene requiring an additional aberration in the other allele for loss of function. In addition, more than half of the patients with an APC mutation had multiple mutations in this gene [25], complicating data analyses and interpretation. Additional analyses for subgroups of colon tumors with specific truncating point mutations in APC did not show any associations with the intake of fat or different types of fat.
This still does not satisfy our third query, i.e., why are the associations specifically confined to this subgroup of colon cancer patients whose tumors are characterized by activating KRAS mutations, and not truncating APC mutations or lack of MLH1 expression? It is speculative, but plausible, that when KRAS is the only one of the three genes affected, the mutation may more likely be the result of exogenous exposure, for example a relatively high linoleic acid intake. In contrast, when a KRAS mutation co-occurs with a mutation in APC or, although more rarely, in addition to a defective MLH1, these other early gene defects also had a role in tumor formation and may have resulted in a mutator phenotype leading to mutations in other genes (such as the KRAS gene) irrespective of exogenous factors. Since there is no information on the timing of genetic aberrations in this type of human studies, we cannot verify this with our data. Aberrations in other genes, not available for this study, but possibly involved in early tumorigenesis of colorectal cancer, could not be accounted for in analyses and may have influenced results. However, a recent systematic sequence analysis of 13,023 exons in individual colorectal cancers showed that the prevalence of mutations other than in APC, KRAS and TP53 is rather low [44], and mutations in TP53 is not an early event in colorectal carcinogenesis. Finally, results are based on relatively small numbers of patients, especially in the reference group of polyunsaturated fat or linoleic acid intake (four patients, see Table 4), and point estimates of RRs for quartiles of intake of polyunstaturated fat or linoleic acid should therefore, be interpreted cautiously. Nevertheless, as discussed previously, the association appears to be robust when regarding the results for one standard deviation increase in linoleic acid intake (based on a total of 65 patients). Therefore, Breivik and Glaudernack’s hypothesis for distinct carcinogens to exert their effect on two proposed types of genetic instability, i.e., microsatellite instability and chromosomal instability [16], may be extended to the potential effect of carcinogens on more specific genetic pathways to colorectal tumorigenesis, as for example the KRAS mutated pathway.
The data from this large prospective cohort study suggest that linoleic acid intake is strongly associated with colon tumors with an aberrant KRAS gene, but an intact APC gene and MLH1 expression. Verification in other studies is warranted. Possibly, tumors revealing the involvement of distinct genetic pathways on the basis of specific genetic aberrations, may have a unique etiology.
References
Woutersen RA, Appel MJ, van Garderen-Hoetmer A, Wijnands MV (1999) Dietary fat and carcinogenesis. Mutat Res 443(1–2):111–127
Potter JD (1996) Nutrition and colorectal cancer. Cancer Causes Control 7(1):127–146
Willett WC (2000) Diet and cancer. Oncologist 5(5):393–404
Lin J, Zhang SM, Cook NR, Lee IM, Buring JE (2004) Dietary fat and fatty acids and risk of colorectal cancer in women. Am J Epidemiol 160(10):1011–1022
Sanderson P, Johnson IT, Mathers JC, Powers HJ, Downes CS, McGlynn AP, et al (2004) Emerging diet-related surrogate end points for colorectal cancer: UK Food Standards Agency diet and colonic health workshop report. Br J Nutr 91(2):315–323
Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M et al (1988) Genetic alterations during colorectal-tumor development. N Engl J Med 319(9):525–532
Gryfe R, Swallow C, Bapat B, Redston M, Gallinger S, Couture J (1997) Molecular biology of colorectal cancer. Curr Probl Cancer 21(5):233–300
Fodde R, Smits R, Clevers H (2001) APC, signal transduction and genetic instability in colorectal cancer. Nat Rev Cancer 1(1):55–67
Kuismanen SA, Holmberg MT, Salovaara R, de la Chapelle A, Peltomaki P (2000) Genetic and epigenetic modification of MLH1 accounts for a major share of microsatellite-unstable colorectal cancers. Am J Pathol 156(5):1773–1779
Breivik J, Gaudernack G (1999) Carcinogenesis and natural selection: a new perspective to the genetics and epigenetics of colorectal cancer. Adv Cancer Res 76:187–212
Lengauer C, Kinzler KW, Vogelstein B (1997) Genetic instability in colorectal cancers. Nature 386(6625):623–627
Shih IM, Zhou W, Goodman SN, Lengauer C, Kinzler KW, Vogelstein B (2001) Evidence that genetic instability occurs at an early stage of colorectal tumorigenesis. Cancer Res 61(3):818–822
Smith G, Carey FA, Beattie J, Wilkie MJ, Lightfoot TJ, Coxhead J et al (2002) Mutations in APC, Kirsten-ras, and p53-alternative genetic pathways to colorectal cancer. Proc Natl Acad Sci U S A 99(14):9433–9438
Luchtenborg M, Weijenberg MP, Wark PA, Saritas AM, Roemen GM, van Muijen GN et al (2005) Mutations in APC, CTNNB1 and K-ras genes and expression of hMLH1 in sporadic colorectal carcinomas from the Netherlands Cohort Study. BMC Cancer 5:160
Fearon ER, Vogelstein B (1990) A genetic model for colorectal tumorigenesis. Cell 61(5):759–767
Breivik J, Gaudernack G (1999) Genomic instability, DNA methylation, and natural selection in colorectal carcinogenesis. Semin Cancer Biol 9(4):245–254
Bardelli A, Cahill DP, Lederer G, Speicher MR, Kinzler KW, Vogelstein B et al (2001) Carcinogen-specific induction of genetic instability. Proc Natl Acad Sci USA 98(10):5770–5775
Marnett LJ (2002) Oxy radicals, lipid peroxidation and DNA damage. Toxicology 181–182:219–222
Niedernhofer LJ, Daniels JS, Rouzer CA, Greene RE, Marnett LJ (2003) Malondialdehyde, a product of lipid peroxidation, is mutagenic in human cells. J Biol Chem 278(33):31426–31433
Leuratti C, Watson MA, Deag EJ, Welch A, Singh R, Gottschalg E, et al (2002) Detection of malondialdehyde DNA adducts in human colorectal mucosa: relationship with diet and the presence of adenomas. Cancer Epidemiol Biomarkers Prev 11(3):267–273
Hendrickse CW, Kelly RW, Radley S, Donovan IA, Keighley MR, Neoptolemos JP (1994) Lipid peroxidation and prostaglandins in colorectal cancer. Br J Surg 81(8):1219–1223
Brink M, Weijenberg MP, De Goeij AF, Schouten LJ, Koedijk FD, Roemen GM et al (2004) Fat and K-ras mutations in sporadic colorectal cancer in The Netherlands Cohort Study. Carcinogenesis 25(9):1619–1628
van den Brandt PA, Goldbohm RA, van’t Veer P, Volovics A, Hermus RJ, Sturmans F (1990) A large-scale prospective cohort study on diet and cancer in The Netherlands. J Clin Epidemiol 43(3):285–295
Van den Brandt PA, Schouten LJ, Goldbohm RA, Dorant E, Hunen PM (1990) Development of a record linkage protocol for use in the Dutch cancer registry for epidemiological research. Int J Epidemiol 19(3):553–558
Luchtenborg M, Weijenberg MP, Roemen GM, de Bruine AP, van den Brandt PA, Lentjes MH et al (2004) APC mutations in sporadic colorectal carcinomas from The Netherlands Cohort Study. Carcinogenesis 25(7):1219–1226
Rowan AJ, Lamlum H, Ilyas M, Wheeler J, Straub J, Papadopoulou A et al (2000) APC mutations in sporadic colorectal tumors: A mutational “hotspot” and interdependence of the “two hits”. Proc Natl Acad Sci USA 97(7):3352–3357
Brink M, de Goeij AF, Weijenberg MP, Roemen GM, Lentjes MH, Pachen MM et al (2003) K-ras oncogene mutations in sporadic colorectal cancer in The Netherlands Cohort Study. Carcinogenesis 24(4):703–710
Nevo table (1987) Dutch food composition table 1986–1986
Goldbohm RA, van den Brandt PA, Brants HA, van’t Veer P, Al M, Sturmans F et al (1994) Validation of a dietary questionnaire used in a large-scale prospective cohort study on diet and cancer. Eur J Clin Nutr 48(4):253–265
van Poppel G (1998) Intake of trans fatty acids in western Europe: the TRANSFAIR study. Lancet 351(9109):1099
Willett W (1998) Nutritional epidemiology. Oxford University Press, New York
Lin DY, Wei LJ (1989) The robust inference for the Cox proportional hazards model. JASA 84(408):1074–1078
Wei EK, Giovannucci E, Wu K, Rosner B, Fuchs CS, Willett WC et al (2004) Comparison of risk factors for colon and rectal cancer. Int J Cancer 108(3):433–442
Schoenfeld D (1982) Partial residuals for the proportional hazards regression models. Biometrika 69(1):239–241
Bautista D, Obrador A, Moreno V, Cabeza E, Canet R, Benito E et al (1997) Ki-ras mutation modifies the protective effect of dietary monounsaturated fat and calcium on sporadic colorectal cancer. Cancer Epidemiol Biomarkers Prev 6(1):57–61
Diergaarde B, Braam H, van Muijen GN, Ligtenberg MJ, Kok FJ, Kampman E (2003) Dietary factors and microsatellite instability in sporadic colon carcinomas. Cancer Epidemiol Biomarkers Prev 12(11 Pt 1):1130–1136
Diergaarde B, Tiemersma EW, Braam H, Van Muijen GN, Nagengast FM, Kok FJ et al (2005) Dietary factors and truncating APC mutations in sporadic colorectal adenomas. Int J Cancer 113(1):126–132
Diergaarde B, van Geloof WL, van Muijen GN, Kok FJ, Kampman E (2003) Dietary factors and the occurrence of truncating APC mutations in sporadic colon carcinomas: a Dutch population-based study. Carcinogenesis 24(2):283–290
Kampman E, Voskuil DW, van Kraats AA, Balder HF, van Muijen GN, Goldbohm RA et al (2000) Animal products and K-ras codon 12 and 13 mutations in colon carcinomas. Carcinogenesis 21(2):307–309
Martinez ME, Maltzman T, Marshall JR, Einspahr J, Reid ME, Sampliner R et al (1999) Risk factors for Ki-ras protooncogene mutation in sporadic colorectal adenomas. Cancer Res 59(20):5181–5185
Slattery ML, Curtin K, Anderson K, Ma KN, Edwards S, Leppert M et al (2000) Associations between dietary intake and Ki-ras mutations in colon tumors: a population-based study. Cancer Res 60(24):6935–6941
Wark PA, Van der Kuil W, Ploemacher J, Van Muijen GN, Mulder CJ, Weijenberg MP et al (2006) Diet, lifestyle and risk of K-ras mutation-positive and -negative colorectal adenomas. Int J Cancer 119(2):398–405
Slattery ML, Anderson K, Curtin K, Ma KN, Schaffer D, Samowitz W (2001) Dietary intake and microsatellite instability in colon tumors. Int J Cancer 93(4):601–607
Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD et al (2006) The consensus coding sequences of human breast and colorectal cancers. Science 314(5797):268–274
Acknowledgments
We thank Dr. P. Wark and G. van Wijhe for assessing MLH1 expression. We are grateful to Prof. J.W. Arends and Dr. M. van Engeland for their participation in the initiation of the study. In addition, we thank Drs. A. Volovics and A. Kester for statistical advice; Dr. L. Schouten, S. van de Crommert, H. Brants, J. Nelissen, C. de Zwart, M. Moll, W. van Dijk, M. Jansen, and A. Pisters for assistance; and H. van Montfort, T. van Moergastel, L. van den Bosch, and R. Schmeitz for programming assistance. We also thank the participants of this study and wish to thank the regional cancer registries (IKA, IKL, IKMN, IKN, IKO, IKR, IKST, IKW, IKZ), and the Dutch nationwide network and registry of histo- and cytopathology (PALGA). Finally, we are grateful to the departments of Pathology of the following hospitals for providing the tissue blocks: Academisch Ziekenhuis Nijmegen Sint Radboud, Academisch Ziekenhuis Groningen, Rijnland Ziekenhuis, Antoni van Leeuwenhoek Ziekenhuis, Academisch Ziekenhuis Rotterdam, Stichting Laboratorium Pathologie Oost Nederland, Pathologisch Instituut Utrecht, Ziekenhuis Rijnstate Arnhem, Laboratorium Volksgezondheid Leeuwarden, Ziekenhuis Bethesda, Stichting Samenwerkend Ziekenhuizen Oost Groningen, Martini Ziekenhuis Groningen, Samenwerkend Stichting Delftse Ziekenhuizen, Leyenburg Ziekenhuis, Academisch Ziekenhuis Vrije Universiteit, Academisch Medisch Centrum, Sint Franciscus Ziekenhuis, Dr. Daniel den Hoed Kliniek, Academisch Ziekenhuis Maastricht, Goudse Ziekenhuizen Stichting Laboratorium, Canisius Wilhelmina Ziekenhuis, Slootervaart Ziekenhuis, Maaslandziekenhuis, Atrium Heerlen, Atrium Kerkrade and Brunssum, Microbiologie St. Medische Stedendriehoek, IJsselmeer Ziekenhuizen, Ziekenhuis Centrum Apeldoorn, Isala Klinieken, Elkeriekziekenhuis, Groot Ziekengasthuis, Ziekenhuis Gooi Noord, Medisch Centrum Alkmaar, Regionaal Pathologisch en Cytologisch Laboratorium voor Eemland en Noord-West Veluwe, Diakonesse Ziekenhuis, Sint Antonius Ziekenhuis, Onze Lieve Vrouwe Gasthuis, St. Lucas Andreas Ziekenhuis, Pathologisch Anatomisch Laboratorium SPALK, Ziekenhuis de Heel, Diakonessenhuis, Rode Kruis Ziekenhuis, Ziekenhuis Bronovo, Laurentius Ziekenhuis Roermond, Pathologisch Anatomisch Laboratorium Dordrecht, Zuiderziekenhuis, Sint Clara Ziekenhuis, Medisch Centrum Haaglanden, St. Streeklaboratorium Zeeland, Sint Elisabeth Ziekenhuis, Catharinaziekenhuis, Sint Maartensgasthuis and Spaarne Ziekenhuis. This work was supported by The Netherlands Organisation for Scientific Research (980–10–26) for Margreet Lüchtenborg and The Dutch Cancer Society (UM 99–1980) for Mirian Brink.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Weijenberg, M.P., Lüchtenborg, M., de Goeij, A.F.P.M. et al. Dietary fat and risk of colon and rectal cancer with aberrant MLH1 expression, APC or KRAS genes. Cancer Causes Control 18, 865–879 (2007). https://doi.org/10.1007/s10552-007-9032-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10552-007-9032-6