
Abstract
Glucocorticoids (GCs) regulate cell homeostasis and can affect carcinogenesis. An inherited germline mutation in the BRCA1 gene, a tumor suppressor gene, confers a predisposition to breast and ovarian cancers. BRCA1 participates in the maintenance of genome stability through DNA repair, in cellular homeostasis through gene transcription, and in signaling regulation. The interaction between BRCA1 and the glucocorticoid receptor (GR) signaling pathway was studied in normal breast tissues and triple-negative breast cancers from BRCA1 mutation carriers. A loss of the active Ser211 phosphorylated form of GR was found in the mutant as compared to the non-mutant. In in vitro studies, the BRCA1 status in breast cancer cell lines regulates GC-dependent proliferation/apoptosis and impacts GC-dependent gene expression. The lack of BRCA1 inhibited dexamethasone actions on its target genes’ expression and the opposite effect was seen with BRCA1 overexpression. BRCA1 overexpression enhances MAPK p38 phosphorylation, resulting in an amplification of GR phosphorylation on Ser 211 and GR basal expression. Our results indicate that BRCA1 is essential to develop an efficient GC signalization. GR P-Ser211 levels may constitute an important diagnostic factor for screening BRCA1 loss of expression in tumors from BRCA1 mutation carriers as well as in sporadic BRCAness tumors. This marker may help to optimize therapeutic strategies.
Similar content being viewed by others


Introduction
Breast cancer is the leading cause of cancer-related deaths in women worldwide. In the recent years, an estimated 234,000 and 425,000 annual new cases of breast cancer occur in the United States and in Europe, respectively (http://globocan.iarc.fr).
BRCA1 is a tumor suppressor gene involved in the maintenance of genomic stability through DNA repair and gene transcription. Inherited germline mutations in BRCA1 confer a 40 and 80 % increased risk to develop for ovarian and breast cancer, respectively [1, 2]. While these cancers occur in hormonal responsive tissues, 90 % of breast cancers observed in BRCA1 carriers are “triple-negative” types, lacking estrogen and progesterone expression and HER2 amplification [3, 4].
The link between BRCA1 and steroid hormone receptors has been widely investigated during the last decade. Using preclinical models of total or partial abolition of BRCA1 expression in the murine mammary gland or in breast cancer cells, a relationship among BRCA1, estrogen receptor (ER), progesterone receptor (PR), and androgen receptor (AR) was reported [5–8]. Physical interactions between BRCA1 and these receptors have been demonstrated. BRCA1 was also found to limit transactivation of target gene transcription of ER and PR and enhance those of the AR [5–8]. In normal human breast tissue from mutation carriers, little data have been published. Bramley et al. reported that the estradiol (E2) increased proliferation in breast epithelium, while Mote et al. reported an altered expression of PR and pS2, both estrogen-responsive proteins in normal breast tissue from BRCA1 carriers [9, 10].
The relationship between BRCA1 and the glucocorticoid receptor (GR) has not been extensively studied. It has been shown that in the absence of hydrocortisone, unliganded GR increases BRCA1 expression, whereas the addition of hydrocortisone downregulates BRCA1 expression in non-malignant mouse mammary cells [11, 12].
Our investigation concentrated on the interaction between BRCA1 and GR because of the effect of glucocorticoids (GCs) on survival of normal and breast cancer cells [13, 14]. Our group has recently demonstrated that the human luminal breast cells were highly responsive to the proliferative and anti-apoptotic effects of GCs [15]. In breast cancer cells, GCs induced anti-apoptotic and anti-proliferative effects, but tended to improve survival when cells were exposed to chemotoxic treatments [15–18]. The expression of GR and its potential prognostic value in breast cancer remain controversial and poorly evaluated [13]. In most of the studies, GR expression appears to be lost during tumor progression; a recent publication suggested that its expression was correlated with a better outcome in ERpos breast cancers and carried a negative prognostic correlation in ERneg tumors [14]. GCs regulate cellular response by positively controlling the GR mRNA and protein levels at their transcriptional level [19–21]. GR, like other steroid receptors, constantly shuttles between the cytoplasm and the nucleus [22]. Binding of hormones to GR forces conformational changes and modifies the composition of the hsp90 complex. Once translocated into the nucleus, GR dimerizes and acts as a transcription factor that binds to the GC response elements (GREs) in the promoter regions of GC-inducible genes. GR undergoes phosphorylation upon GC stimulation which determines the GR activity in the cell. Of the GR, the phosphorylated amino acid P-Ser211 is crucial for maximal transactivation of GC signaling [21, 23, 24]. GR P-Ser211 expression in breast cancer cells has not been previously reported.
As compared to controls, a substantial decrease of GR P-Ser211 expression in BRCA1 mutation carriers and in triple-negative breast cancers arising in patients with BRCA1 mutations is observed. BRCA1 extinction using RNA-silencing technology in breast cells leads us to establish a link between BRCA1 expression and GR expression/activation.
Materials and methods
Reagents and steroids
Dexamethasone, insulin, epidermal growth factor (EGF), transferrin, choleratoxin, cortisol, hyaluronidase, thymidine, mevinolin, and ribonuclease A were purchased from Sigma-Aldrich. The GR antagonist ORG34116 (AG) was kindly provided by Dr HJ Kloosterboer, Organon (Oss, The Netherlands).
Immunohistochemistry (IHC)
Normal breast tissue samples were obtained between 2009 and 2012 from 23 consecutive women (median age of 32.5 years) with non-mutated (BRCA1 +/+) undergoing surgery for mammoplasty reduction and from 20 consecutive women (median age of 42.5 years) with confirmed BRCA1 +/− mutation undergoing prophylactic mastectomies. Additional breast cancer samples were obtained from 14 women with non-mutated BRCA1 +/+ (median age of 56.5 years) and from 13 BRCA1 mutation carriers (median age of 47 years) with triple-negative [ERα(−), PR(−), and HER2(−)] breast cancer .
Tissues were processed for IHC (10 % formalin fixed and paraffin embedded). Paraffin sections were then dewaxed and rehydrated, and antigens sites were retrieved by treating sections in sodium citrate buffer overnight in the PickCell Retriever 2100 apparatus. Sections were incubated with a primary polyclonal antibody against human total GR at 1/75 (H300 sc-8992, Santa Cruz), a monoclonal antibody against human total GR at 1/20 (NCL-GCR, Novocastra), or a polyclonal antibody against GR P-Ser211 at 1/200 (#4161, Cell Signaling). A streptavidin–biotin–peroxidase method was used in the IHC processing (Vectastain kit, Abcys). A negative control (omitting the first antibody) and a positive control (normal appendix) were included for quality purposes.
The first semi-quantitative quantification was done by three investigators blinded to the type of samples (AG, JH, and NM). A quantification using the Allred score commonly used to quantify ER expression in breast cancer was then systematically performed by JH. The proportion of positive cells is expressed as follows: 0, none; 1 < 1/100; 2, 1/100–1/10; 3, 1/10–1/3; 4, 1/3–2/3; and 5, >2/3. Next, an intensity score is assigned, which represents the average intensity of positive tumor cells (0, none; 1, weak, 2, intermediate; and 3, strong). The proportion and intensity scores were then added to obtain a total score, which ranged from 0 to 8 [25].
Cell culture procedures
MCF-7 and MDA-MB-231 cell lines were, respectively, maintained in DMEM and in RPMI 1640 media (PAA Laboratories) supplemented with 10 % fetal calf serum. Human breast epithelial (HBE) cell cultures were established from breast specimens obtained from five BRCA1 +/+ women with the median age of 31.5 years. The procedure used for the HBE culture has been described elsewhere [26]. HBE cells were obtained from the same patients used for the IHC studies and maintained in HAM F10 medium (PAA Laboratories) containing NaHCO3 (0.24 %), penicillin–streptomycin (1 %), cortisol (5 ng/ml), T3 (6.5 ng/ml), choleratoxin (10 ng/ml), transferrin (5 mg/ml), insulin (0.016 U/ml), EGF (10 ng/ml), and 5 % human serum.
Steroid treatments
After seeding and transfection, cells were treated with dexamethasone (10 nM) alone or in combination with AG (1 μM). Control cells were treated with ethanol as vehicle (1:1,000). Treatments were carried out in a phenol red-free medium containing 5 % dextran-charcoal-stripped serum for a period of 24 h in MCF-7 and MDA-MB-231 or 48 h in HBE cells.
BRCA1 siRNA transient transfections
Human breast epithelial, MCF-7, and MDA-MB-231 cells were transiently transfected with 10 nM of either siRNA control or siRNA BRCA1 using the HiPerFect Reagent (Qiagen) in MCF-7 and MDA-MB-231, or the lipofectamine LTX reagent (Invitrogen) in HBE cells according to the manufacturer instructions. SiRNA BRCA1 transfection consisted of a transfection of four siRNA BRCA1 sequences from Qiagen Flexitube Gene Solution GS672 (Hs_BRCA1_9, Hs_BRCA1_13, Hs_BRCA1_14, and Hs_BRCA1_15), which recognized BRCA1 transcripts at four different positions. For gene or protein expression analyses, cells were washed 48 h after siRNA and hormone treatments were performed for 24 h in MCF-7 and MDA-MB-231 or 48 h in HBE cells.
BRCA1 vector and transient transfections
MCF-7 and MDA-MB-231 cells were transiently transfected with 5 μg of either pcDNA3 empty vector or pcDNA3 expressing wt-BRCA1 using the lipofectamine reagent according to the manufacturer instructions (Invitrogen). For gene or protein expression analyses, cells were washed 24 h after vector transfection and hormone treatments were performed for 24 h in MCF-7 and MDA-MB-231.
Luciferase reporter assays
Cells were washed either 48 h after siRNA transfection or 24 h after BRCA1 vector transfection, and then transfected with the GRE-Luc, human GR promoters 1A, or 1B/C plasmid reporter, using lipofectamine reagent for MCF-7 and MDA-MB-231 cells or lipofectamine LTX reagent for HBE cells. For each sample, the DNA mix was adjusted to 1/10th of the total content with a plasmid expressing Rous sarcoma virus beta-galactosidase as an internal control. Luciferase and beta-galactosidase assays were performed 24 h after the reporter transfection, and 24 or 48 h of hormone treatments in MCF-7 and MDA-MB-231 cells, or HBE cells, respectively. At the end of the experiment, cells were lysed and luciferase activity was determined using the luciferase assay system (Promega). A different lysate aliquot was also taken from each sample and analyzed for beta-galactosidase activity using the beta-galactosidase enzyme assay system (Promega).
Immunocytochemistry (ICC)
MDA-MB-231 cells were grown on coverslips and transfected by 5 μg of pcDNA3 empty vector or pcDNA3 expressing wt-BRCA1 for 24 h in the presence or absence of dexamethasone (100 nM). They were fixated using 4 % formaldehyde and stained with a rabbit polyclonal antibody against BRCA1 (C-20, Santa Cruz Biotechnology) at 1/50, the polyclonal antibody against human total GR at 1/100, or the polyclonal antibody against GR P-Ser211 at 1/500, and revealed by fluorescein-conjugated anti-Rabbit IgG (611-6202, Rockland, INC) at 1/1,000.
Coimmunoprecipitation
MDA-MB-231 cells were transiently transfected with 5 μg of either pcDNA3 empty vector or pcDNA3 expressing wt-BRCA1. Twenty-four hours after vector transfection, cells were harvested and immediately lysed in a 1 % Triton X-100 lysis buffer.
Immunoprecipitation was carried out in WCE (500 μg or 1 mg) with 1 μg of either anti-BRCA1 (C20) (sc-642, Santa Cruz) or GR (H300) (sc-8992, Santa Cruz) coupled with 50 μl of A/G Plus-agarose beads (Santa Cruz Biotechnology, Santa Cruz, CA) overnight at 4 °C with constant agitation. Following five washes with supplemented lysis buffer, samples were denatured in 2 % SDS loading dye, separated by SDS-PAGE, and transferred to a nitrocellulose membrane (BioRad).
Coimmunoprecipitated BRCA1 or GR was detected, respectively, with either anti-GR (H300) (sc-8992, Santa Cruz) or anti-BRCA1 (C20) (sc-642, Santa Cruz).
For endogenous protein interactions, 24 h after seeding, MDA-MB-231 cells were harvested and lysed as described above. Immunoprecipitation was also carried out as described above.
Western blots
Cell lysates (50 or 100 μg) were subjected to a combined lower 12 % section and upper 6 % section polyacrylamide gel electrophoresis (PAGE), and proteins were transferred and detected using a chemoluminescence procedure. The proteins of interest were analyzed with following antibodies directed against BRCA1 (C20) (sc-642, Santa Cruz), GR (H300) (sc-8992, Santa Cruz), GR P-Ser211 (#4161, cell signaling), phosphorylated Thr180/Tyr182 MAPK p38 (p-p38, sc-17852, Santa Cruz), MAPK p38 (MAPK p38 #9212, cell signaling), and actin (α-actin, AC-15; #A5441, Sigma-Aldrich). Quantification of Western blot bands of phosphorylated GR and p38 or total GR and p38 or actin was performed using Morpho Expert software (Explora Nova, France).
Flow cytometry analysis
BRCA1 overexpression or silencing was performed in MCF-7 and MDA-MB-231 cells. Twenty-four hours or 48 h after transfection, cells were treated with dexamethasone (100 nM) alone or in combination with AG (1 μM). Twenty-four hours after treatment, cells were washed in PBS, trypsinized, and centrifuged 5 min at 1,350 rpm. Cells were fixed with 70 % ethanol overnight, and before analysis cells were washed in PBS and stained with 10 μg/ml propidium iodide in PBS (containing 0.835 U/ml ribonuclease A). For each sample, at least 10,000 cells were counted on a BD LSR II flow cytometer (BD Biosciences). Cycle distribution and subG1 phase were analyzed using the ModFit LT software (Verity Software House).
Real-time quantitative reverse transcription (qt-PCR)
Total RNA was extracted using TriZOL reagent. Two micrograms of total RNA was subjected to RT using random primers. As described previously by Courtin et al. [15], cDNA product was subjected to qt-PCR using sequence-specific primers (300 nM) and Brilliant SYBR GREEN QPCR master mix on an Mx3000P apparatus (Agilent Technologies). Primer sequences are available in Supplementary Table S1. The data shown are mRNA expression levels normalized to the housekeeping gene 36B4.
Ethical permissions
The normal breast samples were obtained from the women with their informed consent according to the French law on clinical experimentation (L. 1243-3 and L. 1243-4) as a part of a biomedical project including collection and conservation of breast tissues obtained from women performing plastic surgery or prophylactic mastectomies. The triple-negative tumors were obtained from the Department of Pathology of Centre Jean Perrin; patients had previously provided informed consent regarding the use of their tissues. Ethical committee review and authorization were obtained (“Comité de Protection des Personnes” Number: 11826).
Statistical analysis
To determine the statistical significance of treatments, one-way ANOVA and Tukey–Kramer multiple comparisons tests were performed to compare the relative efficiency of each treatment with the Instat 3 software (GraphPad, USA). p < 0.05 was considered significant.
Results
Loss of GR P-Ser211 expression in normal and tumoral breast tissues from BRCA1-mutated and non-mutated carriers
We first analyzed GR staining in normal breast tissues from 20 BRCA1 mutation carriers and 23 women with non-mutated BRCA1, who underwent prophylactic mastectomy or plastic surgery. In mutated and non-mutated breast tissues, GR staining was equivalent and found to be more strongly expressed in the nuclei and the cytoplasm of myoepithelial cells than luminal cells, as shown in Fig. 1A-a, b (red and black arrows, respectively). This was observed with two different antibodies directed against total GR.

Total and P-Ser211 Glucocorticoid receptor expressions in normal and cancerous breast tissues. A, Immunolabelling of total GR (a, b) and GR P-Ser211(c–f) in luminal (black arrows) and myoepithelial cells (red arrows) in alveolar/lobular structures of 23 BRCA1 +/+ (a, c, e) and 20 BRCA1 +/− (b, d, f) normal human breast tissues (original magnification ×400). B Immunodetection of total GR (a, c, e, g) and GR P-Ser211 (b, d, f, h) in 14 BRCA1 +/+ (a–d) and 13 BRCA1 −/− (e–h) triple-negative (TN) human breast tissues (original magnification ×400). C Representation of the percentage of positive cells for GR P-Ser211 staining in tumor cells from BRCA1 +/+ (TN N) and BRCA1 −/− (TN M) triple-negative patients. (***p < 0.0001)
In contrast to total GR labeling, in non-mutated patients, the staining intensity of GR phosphorylated Ser 211 form (GR P-Ser211) was stronger in the nuclei of luminal cells than myoepithelial cells (Fig. 1A-c, e). But, GR P-Ser211 staining was weaker in the nuclei of the myoepithelial and luminal cells from the BRCA1 mutation carriers (Fig. 1A-d, f) compared to the non-mutated BRCA1.
Using the Allred score [25], we quantified the intensity and proportion of positive cells of the GR P-Ser211 in the two groups of women in luminal and myoepithelial cells. The proportion of GR P-Ser211-positive cells was not significantly different within the normal tissues from both groups of patients (p = 0.09 for myoepithelial cells and p = 0.07 for luminal cells); the mean GR P-Ser211 intensity score was significantly different in luminal and myoepithelial cells within the two groups (respectively, p = 0.004 and 0.001). The Allred score (proportion + intensity scores) reached the level of significance of p < 0.05 for myoepithelial cells and p < 0.01 for the luminal cells.
Because a majority of breast cancers occurring in women with germline BRCA1 mutations are “triple-negative” type (TN), the expression of total GR and GR P-Ser211 was evaluated in 14 TN tumors from women with non-mutated BRCA1 and in 13 TN-type tumors from BRCA1 mutation carriers. In non-mutated TN, total GR labeling was positive in 50 % of the samples. An example of positive or negative staining is shown in Fig. 1B-a, c. In TN with mutation, 10/13 (73 %) were negative, but the difference between the two groups of TN was not significant (Fig. 1B-e, g). GR P-Ser211 labeling from TN breast cancers associated with the BRCA1 mutation was much weaker or negative as compared to sporadic TN breast cancers (Fig. 1B-b, d compared to Fig 1B-f, h). Using the Allred score, BRCA1-associated tumors showed that an average of 28.8 ± 6 % of the cells expressed GR P-Ser211, whereas in sporadic TN breast cancers, a mean of 67.14 ± 2.7 % of the cells expressed GR P-Ser211 (p < 0.0001) (Fig. 1C).
The weaker GR P-Ser211 expression in healthy and tumoral tissues in BRCA1 mutation carrier suggested a contribution of BRCA1 on GR activation that is impaired in the mutated tissue. The following studies attempt to confirm this hypothesis.
BRCA1 extinction inhibits the GR gene regulation in HBE cells
We first examined the inhibition of BRCA1 expression, on the GC signaling pathway, in normal human primary breast cells (HBE cells) (Fig. 2a). When BRCA1 mRNA and protein contents were abolished by BRCA1-specific siRNA (siBRCA1), GR protein level was moderately decreased, whereas GR P-Ser211 remained at similar levels (Fig. 2a). The sustained GR activation can be explained by the cortisol contained in HBE cells’ medium.

Abolition of BRCA1 expression inhibits the glucocorticoid receptor activity in HBE cells. a BRCA1 mRNA level was determined by quantitative RT-PCR in HBE cells after 96 h of BRCA1 silencing (mean ± SEM, n = 3; ***p < 0.001). BRCA1, GR, and GR P-SER211 protein contents were also evaluated in these same conditions. b HBE cells silenced for BRCA1 were transfected with the GRE-luciferase reporter and then treated with dexamethasone (DEX) (100 nM) (mean ± SEM, n = 5; ***p < 0.001). c G0S8, MKP-1, IEX-1, and tPa mRNA levels were determined by quantitative RT-PCR in HBE cells silenced for BRCA1 (mean ± SEM, n = 3; ***p < 0.001; **p < 0.01). d GR mRNA level was determined by quantitative RT-PCR in HBE cells (mean ± SEM, n = 3; ***p < 0.001)
The transcriptional activity of dexamethasone was monitored on a reporter vector containing six GR-responsive elements, GRE-Luc. The level of GRE-Luc induction by dexamethasone in the presence of siBRCA1 was reduced by 50 % compared to cells transfected with siControl (p < 0.001) (Fig. 2b), suggesting that the level of BRCA1 expression affects GR transcriptional activity. This effect was confirmed on specific GC-responsive genes. Dexamethasone-induced upregulation of the regulator of G-protein signaling (G0S8/RGS2) and the mitogen-activated protein kinase phosphatase-1 (MKP-1) genes were strongly reduced when cells were transfected with siBRCA1 as compared to siControl (Fig. 2c) [16, 27]. In the same manner, the dexamethasone-induced downregulation observed for the immediate early response gene X-1 (IEX-1) and tissue plasminogen activator (tPa) was abolished in the presence of siBRCA1 as compared to siControl (Fig. 2c) [16, 27]. Supporting the interrelation between GR and BRCA1 in these cells, a significant decrease of GR mRNA in the presence of siBRCA1 (Fig. 2d) was observed.
These results illustrate that the BRCA1 silencing drastically abrogated the dexamethasone-mediated responses, indicating that BRCA1 is involved in GC-dependent signaling pathways.
BRCA1 status in breast cancer cell lines regulates GC-dependent proliferation and apoptosis
Using BRCA1 silencing or overexpression approaches, the contribution of BRCA1 expression to GC signaling pathway was explored in two breast cancer cell lines with differentiated and undifferentiated phenotypes, MCF-7 (ER+, PR+, GR+) and MDA-MB-231 (ER−, PR− and GR+), respectively. As GCs are known to exert anti-proliferative and anti-apoptotic actions in breast cancer cell lines, we investigated the role of BRCA1 on cellular homeostasis and responses to GCs [17, 28, 29].
Dexamethasone significantly decreased cell proliferation in MCF-7 and MDA-MB-231 cells, and this effect was abolished by treating cells with the anti-GC compound (AG) ORG34116 (Figs. 3a, S1a). The dexamethasone anti-proliferative effect was not observed when cell lines were exposed to siBRCA1 (Figs. 3a and S1a, left graphs). In contrast, BRCA1 overexpression enhanced the ability of dexamethasone to induce cell proliferation inhibition (Figs. 3a, S1A, right graphs).

BRCA1 regulates GC-dependent proliferation and apoptosis in MCF-7 cells. a Cellular proliferation and b apoptosis were measured by flow cytometry in MCF-7 cells silenced or overexpressing BRCA1. Cells were treated with 100 nM DEX alone or in combination with 1 μM AG. Result is expressed as percentage of control (untreated cells) (mean ± SEM, n = 4, ***p < 0.001, **p < 0.01, *p < 0.05)
A significant decrease in cell apoptosis by dexamethasone in MCF-7 and MDA-MB-231 cells was observed as compared to the control. The anti-GC AG also impeded this effect (Figs. 3b, S1B, left graphs). Inhibition of BRCA1 expression abrogated dexamethasone-dependent anti-apoptotic effect, whereas BRCA1 overexpression enhanced the dexamethasone effect in both cell lines (Figs. 3b, S1B, right graphs). Nevertheless, apoptosis concerned a smaller amount of cells (11 ± 0.5 % of the MCF-7 cells), whereas proliferation expressed as the percent of MCF-7 cells in S/G2 + M reaches 56.9 ± 2.8 % in the condition of dexamethasone antagonism.
BRCA1 status in breast cancer cell impacts GC-dependent gene expression
The transcription efficacy of dexamethasone on GRE-Luc and endogenous GR target genes was studied in MCF-7 and MDA-MB-231 cells following BRCA1 silencing or overexpression (Figs. 4, S2). BRCA1 extinction reduced GRE-Luc constitutive activity twofold (Figs. 4a, S2A, left graphs). BRCA1 overexpression significantly (p < 0.05) enhanced the basal promoter GRE-Luc activity in both cells, but to a lower extent than in the presence of the ligand (p < 0.001) (Figs. 4a, S2A, right graphs). As expected, dexamethasone induced a strong GRE-Luc transactivation in both cell types. This effect was drastically reduced by four–sixfold in cells silenced for BRCA1 and enhanced in cells overexpressing BRCA1 (Figs. 4a, S2A).

BRCA1 impacts GC-dependent gene expression in MCF-7 cells. a Cells silenced for (left) or overexpressing (right) BRCA1 were transfected with the GRE-Luciferase reporter. Cells were treated with DEX (100 nM). Results are expressed as fold induction compared to untreated cells transfected with siControl or pcDNA3, respectively (mean ± SEM, n = 3; ***p < 0.001). b G0S8 and MKP-1; c IEX-1 and tPa mRNA levels were determined by quantitative RT-PCR in MCF-7 cells silenced for or overexpressing BRCA1 and treated with 100 nM DEX or not. Results are expressed as fold induction compared to untreated cells transfected with siControl or pcDNA3, respectively (mean ± SEM, n = 3; ***p < 0.001, **p < 0.01, *p < 0.05)
Dexamethasone stimulation of GC target genes G0S8, MKP-1, IEX-1, and tPa expression was altered accordingly to BRCA1 status in MCF-7 and MDA-MB-231 cells (Figs. 4b, c, S2B, C). The lack of BRCA1 was associated with an inhibition of G0S8 and MKP-1 dexamethasone-mediated induction (Figs. 4b, S2B, left graphs), and abrogated the dexamethasone-induced inhibition of IEX-1 and tPa mRNA expression (Figs. 4c, S2C).
In conditions of BRCA1 overexpression, a mirror effect on genes induced by GCs was observed. GS08 and MKP-1 mRNA dexamethasone-induced expressions were substantially enhanced (Figs. 4b, S2B, right graphs). However, no modification was observed on the mRNA levels of dexamethasone-repressed genes, IEX-1 and tPa (Figs. 4c, S2C, right graphs). In the absence of dexamethasone, BRCA1 overexpression did not significantly increase the expression of these genes (in MCF-7 cells, 1.2- and 1.3-fold for GOS8 and MKP1, respectively; in MDA-MB231, 1- and 1.1-fold for GOS8 and MKP1, respectively) (Figs. 4b, S2B, right graphs).
These series of experiments confirmed that the BRCA1 modulates GCs target gene activities as well as biological key functions mediated by GCs such as proliferation and apoptosis.
BRCA1 regulates GR expression and activity
The potential mechanisms interconnecting BRCA1 and GR were investigated by analysis of GR expression modulation by BRCA1. It has to be noted that the activated GR protein regulates GR transcriptional machinery [30]. In MCF-7 cells, the decrease in GR mRNA level was shown to correlate with the inhibition of BRCA1 expression over time (Fig. 5a). In MDA-MB-231 cells, GR mRNA also dramatically decreased in the presence of siBRCA1, as compared to siControl (Fig. S3A, left graphs). The GR mRNA decrease was also correlated with a smaller quantity of total GR protein content in the absence ( ) or in the presence of dexamethasone (Figs. 5b, S3B).
) or in the presence of dexamethasone (Figs. 5b, S3B).

BRCA1 regulates GR expression and activation in MCF-7 cells. a Kinetic experiments to monitor BRCA1 and GR mRNA levels by quantitative RT-PCR from 0 h to 72 h in cells silenced for BRCA1 (mean ± SEM, n = 3; ***p < 0.001). b Representative immunoblot for BRCA1, GR P-Ser211, total GR, and actin, following BRCA1 silencing and 100 nM DEX treatment. c Luciferase assay on cells silenced for BRCA1 or with siControl and transfected with the human GR promoters 1A or 1B/C reporter plasmids. Cells were treated with DEX (100 nM) or not (mean ± SEM, n = 3; ***p < 0.001, **p < 0.01, *p < 0.05). d Kinetic experiments to monitor BRCA1 and GR mRNA levels by quantitative RT-PCR from 0 h to 72 h in cells overexpressing BRCA1 (mean ± SEM, n = 3; ***p < 0.001, **p < 0.01). e Representative immunoblot for BRCA1, GR P-Ser211, total GR, and actin, following BRCA1 overexpression and 100 nM DEX treatment. f Cells overexpressing BRCA1 were transfected with the human GR promoters 1A or 1B/C-Luciferase reporters, and then treated with DEX (100 nM) (mean ± SEM, n = 3; ***p < 0.001, **p < 0.01)
The ability of BRCA1 to impact GR transcription regulation was confirmed in both cell lines. In the absence of dexamethasone, basal GR promoter activity was slightly inhibited by siBRCA1 (Figs. 5c, S3C). In the presence of dexamethasone, siBRCA1 totally abolished GR promoter activation on the two main GR promoters (hGR 1A and hGR 1B) (Figs. 5c, S3C).
As expected, when BRCA1 was overexpressed, GR mRNA and protein were increased in MCF-7 and MDA-MB-231 (Figs. 5d, S3D). GR promoter basal activity was increased by 1.5- and 1.7-fold on promoter hGR 1A and hGR 1B, respectively. In the presence of dexamethasone, the promoters’ activity was drastically increased threefold (Figs. 5f, S3F). Therefore, BRCA1 expression impacts basal and ligand-activated GR promoter transactivation.
BRCA1-induced GR protein activation
Under dexamethasone exposure, GR P-Ser211 was strongly increased in cell extracts from MCF-7 and MDA-MB 231 cells, testifying that the GR P-Ser211 is a proper reporter of GR protein activation (Figs. 5b, S3B). Interestingly, we observed a strong reduction of the basal GR Ser211 phosphorylated form with the decrease of BRCA1 expression (Figs. 5b, S3B) and a constitutive phosphorylation of GR Ser211 with BRCA1 overexpression in both breast cancer cell lines (Figs. 5e, S3E). To evaluate the impact of BRCA1 on GR activation, we calculated the ratios of GR P-Ser211 BRCA1 overexpression/GR P-Ser211 wild type by total GR under BRCA1 overexpression/total GR wild type. The median increase of GR P-Ser211 form was 2- and 3.9-fold in MCF-7 and MDA-MB231 overexpressing BRCA1, respectively. These results suggested that the BRCA1 participates in determining the GR basal activity by increasing the activated form of GR in a ligand-independent manner.
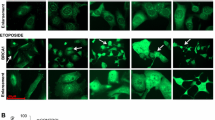
Nuclear receptors are known to move into nuclear aggregates or foci, in the presence of ligand, which represent their sites of transcription [31]. To further investigate the influence of BRCA1 on GR activation, we performed ICC studies in the presence and the absence of dexamethasone in MDA-MB231 cells under conditions of BRCA1 overexpression. In cells transfected by the control vector, both total GR and GR P-Ser211 were located in the cytoplasm (Fig. 6a, b). As expected, with dexamethasone, total GR and GR P-Ser211 were translocated into the nuclei with a subnuclear distribution mostly in foci (Fig. 6d, e). In BRCA1-transfected cells, total GR levels increased, but they remained perinuclear (Fig. 6a), whereas GR P-Ser211 was clearly located in the nuclei of transfected cells (Fig. 6b). The subnuclear distribution was distributed in foci (Fig. 6b). Under dexamethasone treatment, total GR translocated to the nuclei and the GR P-Ser211 staining remained nuclear, but with a decreased intensity, very likely because of recycling of the GR (Fig. 6d, e). These experiments confirmed that upon BRCA1 overexpression and in the absence of ligand, total GR and GR P-Ser211 levels are increased. We concluded that the BRCA1 induced GR activation in a ligand-independent mechanism. Nevertheless, the GR BRCA1-induced activation does not interfere with the transcription of GR-targeted genes since GS08 and MKP-1 basal transcription levels were was not modified (Figs. 4B, S2).

BRCA1 overexpression is associated with increased GR P-Ser211 and total GR. MDA-MB-231 cells were transfected by the control plasmid (pDNA3) or by BRCA1 for 24 h. a total GR staining without treatment, b GR P-Ser211 staining, c BRCA1 staining. d Total GR staining under dexamethasone (100 nM), e GR P-Ser211 staining under dexamethasone. Original magnification ×630
The interaction between BRCA1 and various MAPK has been reported previously [32]. To further evaluate this mode of action, we used some MAP kinase inhibitors. While ERK inhibitor (PD98059) did not display any significant effects on GR P-211 phosphorylation status, a MAPK phosphorylated p38 (P-p38) highly specific inactivator and stabilizer, the SB 202190 (SB) was able to inhibit GR P-Ser211 phosphorylation. This inhibitor binds both active and inactive forms of p38 and has no effect on other MAP kinases up to 100 μM (Sigma datasheet). As MAPK p38 is involved in the GC-induced phosphorylation of GR on Ser211 in lymphoid cells [23] and BRCA1 can induce MAPK p38 phosphorylation [33], it was hypothesized that the BRCA1 could modulate GR P-Ser211 level through the activation of MAPK p38. In the MCF-7 cellular model, the decrease in p38 levels of twofold under basal conditions when BRCA1 was silenced supports this hypothesis (Fig. 7a). Levels of P-p38 were increased in the presence of SB, supporting the stabilization effect of SB on P-p38 (Fig. 7a). Under basal conditions ( ), in the absence of the inhibitor SB, GR P-Ser211 levels were, respectively, either decreased or enhanced following BRCA1 silencing or overexpression. In comparison to basal levels, in cells exposed to SB, a constant decrease of GR P-Ser211 occurred, even in conditions of BRCA1 overexpression (Fig. 7a). This was also observed in cells treated by dexamethasone + SB compared to dexamethasone treatment alone (Fig. 7a). These results support the hypothesis that p38 plays a critical role in GR P-Ser211 phosphorylation and argue for the contribution of BRCA1 in the regulation of GR phosphorylation at the Ser211 by MAPK p38.
), in the absence of the inhibitor SB, GR P-Ser211 levels were, respectively, either decreased or enhanced following BRCA1 silencing or overexpression. In comparison to basal levels, in cells exposed to SB, a constant decrease of GR P-Ser211 occurred, even in conditions of BRCA1 overexpression (Fig. 7a). This was also observed in cells treated by dexamethasone + SB compared to dexamethasone treatment alone (Fig. 7a). These results support the hypothesis that p38 plays a critical role in GR P-Ser211 phosphorylation and argue for the contribution of BRCA1 in the regulation of GR phosphorylation at the Ser211 by MAPK p38.

p38 pathway is involved in BRCA1-induced GR activity. a Representative immunoblot for BRCA1, GR P-Ser211, total GR, phospho-MAPK p38, and total MAPK p38 from MCF-7 cells silenced or overexpressing BRCA1. Cells were treated with 100 nM DEX in combination or not with 10 μg/ml SB 202190, a p38 inhibitor. b Potential mechanism as BRCA1 regulates GR expression and activity
BRCA1 does not interact with GR
Further experiments were designed to address the question of an interaction between BRCA1 and GR at the GR promoter and then a potential recruitment of p38 in the transcriptional complex. BRCA1 was immunoprecipitated with GR antibody, but no BRCA1 protein was coimmunoprecipitated and detected by Western blot. Attempts to immunoprecipitate GR with BRCA1 antibody were undertaken, but no GR was coimmunoprecipitated.
wt-BRCA1 expression plasmid was also used in coimmunoprecipitation experiments. Brca1 or GR were immunoprecipitated by either a GR antibody or a BRCA1 antibody respectively; neither GR nor BRCA1 were, respectively, coimmunoprecipitated and detected by Western blots. These results demonstrated that GR and BRCA1 did not interact in our experimental conditions.
Our results establish a link between BRCA1 and GR expression and activation. Our data suggest that BRCA1 regulates GR activity through GR phosphorylation on Ser211 by modulating MAPK p38. GR requires BRCA1 action to activate its auto-transcription and its target genes’ transcription (Fig. 7b); thus, the alteration of BRCA1 expression impairs this pathway for both GR and GR P-Ser211.
Discussion
In this study, we showed that the wild-type expressed BRCA1 is crucial for GCs to exert efficient signaling through GR P-Ser211 phosphorylation. This evidence is provided by a set of data and observations from biopsies of normal human breast tissues from BRCA1 mutation carriers, triple-negative breast cancers with documented BRCA1 mutations, and in vitro studies using BRCA1 cellular engineering. We conclude that in cells bearing mutated BRCA1, GR signaling pathway would be expected to be attenuated. These observations suggest a potential influence of GR deregulation in BRCA1 mutation carriers at two levels: the modulation of the risk to develop a breast cancer and an impact on cancer progression by modulation of breast cell proliferation and apoptosis.
A DNA repair deficiency has been associated with BRCA1 haplo-insufficiency in mutated heterozygous cells [34], which could promote the mammary cells into an early transformation process and result in a higher and earlier risk of breast cancer development for a BRCA1 mutation carrier. As per the data generated in this series of experiments, this risk may also be modulated by a weaker GR activity in normal breast cells. Physiologically, GCs are continuously secreted by adrenal glands, and this release is enhanced under stress conditions. Recently, clinical and experimental studies suggested that “stress” contributes to cancer emergence and progression and could alter the intracellular GR distribution [13, 35–39]. In two recent publications, the inhibition of GR-dependent activity decreased the proliferative effect of dexamethasone in normal cells, suggesting a protective effect against breast cancer in BRCA1 +/− cells [15, 40]. Thus, the relation observed between BRCA1 and GCs signalization pathway may have complex influence on the risk of BRCA1 +/− cells transformation. This observation requires additional investigation in order to determine if a preventive treatment impacting GCs pathway could be considered as a potential future treatment option.
In addition, the failure of GR activation may also influence breast carcinogenesis progression in women with BRCA1 mutations. We have shown that the BRCA1 suppression in MCF-7 and MDA-MB-231 tumoral cells inhibits anti-proliferative and anti-apoptotic effects of dexamethasone. Based on our results of an attenuated GC-dependent response in samples from patients with BRCA1 mutations, weaker GCs anti-inflammatory activity could be observed in these patients, which may lead to a reduced breast cancer-protective effect, but needs to be further explored. In their recent paper, Conzen and coworkers [14] reported opposite patterns of the levels of GR expression and prognosis of breast cancers according to ER expression. Loss of GR was associated with a better prognosis in ER-negative tumors. This could suggest that among the TN tumors, the tumor subsets which keep GR expression could have a worse prognosis. It was reported that in TN tumors, the lack of immune-responsive phenotype could be associated with a worse prognosis, or the recently identified immunomodulatory subtype among TN molecular signatures with a better prognosis [41, 42]. So far, no correlation with these subtypes and BRCA1 mutations has been released and data on the level of GR expression and the various TN breast cancer subtypes are lacking. The level of expression of GR and possibly the level of GR P-Ser211 expression could also help to stratify therapeutic strategies among tumors with different ER expression.
A relation between GR signalization pathway and BRCA1 was also previously described by Antonova et al. [12], who demonstrated that hydrocortisone was able to decrease BRCA1 expression in non-malignant mouse mammary cells. In our experiments involving MCF-7 cells, downregulation of BRCA1 under dexamethasone treatment was not detected (Figs. 5b, 7a, S3B). However, in conditions of GR inactivation, through p38 phosphorylation inhibition, an increase in BRCA1 level was observed suggesting that a crosstalk between GR and BRCA1 is also occurring in human breast cells (Fig. 7a).
Glucocorticoid receptor signaling is regulated by MAPK cascade, on GR Ser211 and GR Ser203 phosphorylation by p38 and p42/44, respectively [23, 43]. The interaction between BRCA1 and various MAPK has been reported previously [32]. BRCA1 induces the phosphorylation of different kinases including p38 [33]. In this study, we showed that under GR activation, BRCA1 regulates GR-dependent genes and GR-dependent cellular functions (Figs. 2, 3, 4, 5, 6, 7). Interestingly, the regulation was impaired on positively regulated genes, but was not altered on negatively regulated genes in conditions of BRCA1 overexpression. This could be related to the different responsive sequence elements for GR-mediated repression as recently reported [44]. Our results, although preliminary, suggest that the BRCA1 positively interacts with p38 activation. This would be crucial for the stimulation of GR-dependent signaling through GR P-Ser211 activation. The abrogation of Ser211 phosphorylation in the deficient form of BRCA1 should correlate with GCs/GR pathway inhibition.
Under BRCA1 forced expression, and in the absence of dexamethasone, GR total protein content was increased, but remains in the cytoplasm (Fig. 6c), whereas GR Ser-211 phosphorylated form was found enhanced in the nucleus as compared to control. These data suggest that BRCA1 could constitutively maintain a basal level of GR by promoting GR Ser 211 phosphorylation, in the absence of ligand.
In conditions where GR P-Ser211 is drastically decreased, the constitutive synthesis of GR would also be impaired. The decrease in GR total and GR P-Ser211 basal protein level upon the suppression of BRCA1 expression could be the consequence of a lack of GR transcription auto-regulation when GR P-Ser211 is decreased. Transpose to the clinical context, these data would suggest that a deficient BRCA1 will impair the level of GR P-Ser211, as observed in patients with BRCA1 mutation carrier (Fig. 1A, B). In these patients, a deficient BRCA1 protein would also impaired the GC responses, as shown in our cell experiments.
As previously shown for other promoters, BRCA1 may modulate directly the activity of GR promoters by interacting with various coactivators [45, 46]. This additional mechanism cannot be excluded to modulate GR basal transcriptional activity. In patients with BRCA1 mutation carriers, the level of GR total showed only a tendency to be less expressed as compared to normal patients in the TN cancers (Fig. 1B). Basal GR promoter regulation is not a major parameter to evaluate the impact of GC responses between BRCA1 mutant and non-mutant patients.
Accordingly, GR Ser211 phosphorylation staining in cancer patients bearing BRCA1 mutation could constitute a key diagnostic factor. To confirm this, the loss of GR Ser211 phosphorylation in triple-negative tumors in BRCA1 carrier should be addressed in a larger series with correlation of the molecular profiling of the tumors. The extension of this finding on sporadic cancers and the possible detection of lost BRCA1 expression or in BRCAness tumors, using a simpler method such as immunohistochemistry, would quickly facilitate the optimization of potential therapeutic strategies. The treatment using poly (ADP-ribose) polymerase inhibitors which have proved to be beneficial on BRCA1-mutated breast cancer patients could be proposed to a larger set of patients [47].
In conclusion, using human normal and cancerous breast tissues, we established a mechanistic link between the multifunctional protein BRCA1 and GR signaling. Our results suggest that the lack of GR Ser211 phosphorylation in tumors may be a potential diagnostic marker for a BRCA1-mutated carrier.
References
Antoniou A, Pharoah PD, Narod S, Risch HA, Eyfjord JE, Hopper JL, Loman N, Olsson H, Johannsson O, Borg A et al (2003) Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case Series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet 72:1117–1130
Easton DF, Ford D, Bishop DT (1995) Breast and ovarian cancer incidence in BRCA1-mutation carriers. Breast Cancer Linkage Consortium. Am J Hum Genet 56:265–271
Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey SS et al (2001) Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA 98:10869–10874
Sorlie T, Tibshirani R, Parker J, Hastie T, Marron JS, Nobel A, Deng S, Johnsen H, Pesich R, Geisler S et al (2003) Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci USA 100:8418–8423
Fan S, Ma YX, Wang C, Yuan RQ, Meng Q, Wang JA, Erdos M, Goldberg ID, Webb P, Kushner PJ et al (2001) Role of direct interaction in BRCA1 inhibition of estrogen receptor activity. Oncogene 20:77–87
Ma Y, Katiyar P, Jones LP, Fan S, Zhang Y, Furth PA, Rosen EM (2006) The breast cancer susceptibility gene BRCA1 regulates progesterone receptor signaling in mammary epithelial cells. Mol Endocrinol 20:14–34
Park JJ, Irvine RA, Buchanan G, Koh SS, Park JM, Tilley WD, Stallcup MR, Press MF, Coetzee GA (2000) Breast cancer susceptibility gene 1 (BRCAI) is a coactivator of the androgen receptor. Cancer Res 60:5946–5949
Gorski JJ, Kennedy RD, Hosey AM, Harkin DP (2009) The complex relationship between BRCA1 and ERalpha in hereditary breast cancer. Clin Cancer Res 15:1514–1518
Mote PA, Leary JA, Avery KA, Sandelin K, Chenevix-Trench G, Kirk JA, Clarke CL (2004) Germ-line mutations in BRCA1 or BRCA2 in the normal breast are associated with altered expression of estrogen-responsive proteins and the predominance of progesterone receptor A. Genes Chromosomes Cancer 39:236–248
Bramley M, Clarke RB, Howell A, Evans DG, Armer T, Baildam AD, Anderson E (2006) Effects of oestrogens and anti-oestrogens on normal breast tissue from women bearing BRCA1 and BRCA2 mutations. Br J Cancer 94:1021–1028
Ritter HD, Antonova L, Mueller CR (2012) The unliganded glucocorticoid receptor positively regulates the tumor suppressor gene BRCA1 through GABP beta. Mol Cancer Res 10:558–569
Antonova L, Mueller CR (2008) Hydrocortisone down-regulates the tumor suppressor gene BRCA1 in mammary cells: a possible molecular link between stress and breast cancer. Genes Chromosomes Cancer 47:341–352
Vilasco M, Communal L, Mourra N, Courtin A, Forgez P, Gompel A (2011) Glucocorticoid receptor and breast cancer. Breast Cancer Res Treat 130:1–10
Pan D, Kocherginsky M, Conzen SD (2011) Activation of the glucocorticoid receptor is associated with poor prognosis in estrogen receptor-negative breast cancer. Cancer Res 71:6360–6370
Courtin A, Communal L, Vilasco M, Cimino D, Mourra N, de Bortoli M, Taverna D, Faussat AM, Chaouat M, Forgez P et al (2011) Glucocorticoid receptor activity discriminates between progesterone and medroxyprogesterone acetate effects in breast cells. Breast Cancer Res Treat 131(1):49–63
Wu W, Pew T, Zou M, Pang D, Conzen SD (2005) Glucocorticoid receptor-induced MAPK phosphatase-1 (MPK-1) expression inhibits paclitaxel-associated MAPK activation and contributes to breast cancer cell survival. J Biol Chem 280:4117–4124
Moran TJ, Gray S, Mikosz CA, Conzen SD (2000) The glucocorticoid receptor mediates a survival signal in human mammary epithelial cells. Cancer Res 60:867–872
Pang D, Kocherginsky M, Krausz T, Kim SY, Conzen SD (2006) Dexamethasone decreases xenograft response to Paclitaxel through inhibition of tumor cell apoptosis. Cancer Biol Ther 5:933–940
Burnstein KL, Bellingham DL, Jewell CM, Powell-Oliver FE, Cidlowski JA (1991) Autoregulation of glucocorticoid receptor gene expression. Steroids 56:52–58
Govindan MV, Pothier F, Leclerc S, Palaniswami R, Xie B (1991) Human glucocorticoid receptor gene promotor-homologous down regulation. J Steroid Biochem Mol Biol 40:317–323
Galliher-Beckley AJ, Cidlowski JA (2009) Emerging roles of glucocorticoid receptor phosphorylation in modulating glucocorticoid hormone action in health and disease. IUBMB Life 61:979–986
Vandevyver S, Dejager L, Libert C (2012) On the trail of the glucocorticoid receptor: into the nucleus and back. Traffic 13:364–374
Miller AL, Webb MS, Copik AJ, Wang Y, Johnson BH, Kumar R, Thompson EB (2005) p38 Mitogen-activated protein kinase (MAPK) is a key mediator in glucocorticoid-induced apoptosis of lymphoid cells: correlation between p38 MAPK activation and site-specific phosphorylation of the human glucocorticoid receptor at serine 211. Mol Endocrinol 19:1569–1583
Chen W, Dang T, Blind RD, Wang Z, Cavasotto CN, Hittelman AB, Rogatsky I, Logan SK, Garabedian MJ (2008) Glucocorticoid receptor phosphorylation differentially affects target gene expression. Mol Endocrinol 22:1754–1766
Allred DC, Harvey JM, Berardo M, Clark GM (1998) Prognostic and predictive factors in breast cancer by immunohistochemical analysis. Mod Pathol 11:155–168
Malet C, Gompel A, Yaneva H, Cren H, Fidji N, Mowszowicz I, Kuttenn F, Mauvais-Jarvis P (1991) Estradiol and progesterone receptors in cultured normal human breast epithelial cells and fibroblasts: immunocytochemical studies. J Clin Endocrinol Metab 73:8–17
Wan Y, Nordeen SK (2002) Overlapping but distinct gene regulation profiles by glucocorticoids and progestins in human breast cancer cells. Mol Endocrinol 16:1204–1214
Mattern J, Buchler MW, Herr I (2007) Cell cycle arrest by glucocorticoids may protect normal tissue and solid tumors from cancer therapy. Cancer Biol Ther 6:1345–1354
Mikosz CA, Brickley DR, Sharkey MS, Moran TW, Conzen SD (2001) Glucocorticoid receptor-mediated protection from apoptosis is associated with induction of the serine/threonine survival kinase gene, sgk-1. J Biol Chem 276:16649–16654
Yudt MR, Cidlowski JA (2002) The glucocorticoid receptor: coding a diversity of proteins and responses through a single gene. Mol Endocrinol 16:1719–1726
Arnett-Mansfield RL, Graham JD, Hanson AR, Mote PA, Gompel A, Scurr LL, Gava N, de Fazio A, Clarke CL (2007) Focal subnuclear distribution of progesterone receptor is ligand dependent and associated with transcriptional activity. Mol Endocrinol 21:14–29
Yan Y, Haas JP, Kim M, Sgagias MK, Cowan KH (2002) BRCA1-induced apoptosis involves inactivation of ERK1/2 activities. J Biol Chem 277:33422–33430
Gilmore PM, McCabe N, Quinn JE, Kennedy RD, Gorski JJ, Andrews HN, McWilliams S, Carty M, Mullan PB, Duprex WP et al (2004) BRCA1 interacts with and is required for paclitaxel-induced activation of mitogen-activated protein kinase kinase kinase 3. Cancer Res 64:4148–4154
Rennstam K, Ringberg A, Cunliffe HE, Olsson H, Landberg G, Hedenfalk I (2010) Genomic alterations in histopathologically normal breast tissue from BRCA1 mutation carriers may be caused by BRCA1 haploinsufficiency. Genes Chromosomes Cancer 49:78–90
Reiche EM, Nunes SO, Morimoto HK (2004) Stress, depression, the immune system, and cancer. Lancet Oncol 5:617–625
Lillberg K, Verkasalo PK, Kaprio J, Teppo L, Helenius H, Koskenvuo M (2003) Stressful life events and risk of breast cancer in 10,808 women: a cohort study. Am J Epidemiol 157:415–423
Hermes GL, Delgado B, Tretiakova M, Cavigelli SA, Krausz T, Conzen SD, McClintock MK (2009) Social isolation dysregulates endocrine and behavioral stress while increasing malignant burden of spontaneous mammary tumors. Proc Natl Acad Sci USA 106:22393–22398
Michael YL, Carlson NE, Chlebowski RT, Aickin M, Weihs KL, Ockene JK, Bowen DJ, Ritenbaugh C (2009) Influence of stressors on breast cancer incidence in the Women’s Health Initiative. Health Psychol 28:137–146
Kricker A, Price M, Butow P, Goumas C, Armes JE, Armstrong BK (2009) Effects of life event stress and social support on the odds of a > or =2 cm breast cancer. Cancer Causes Control 20:437–447
Communal L, Vilasco M, Hugon-Rodin J, Courtin A, Mourra N, Lahlou N, Dumont S, Chaouat M, Forgez P, Gompel A (2012) Ulipristal acetate does not impact human normal breast tissue. Hum Reprod 27:2785–2798
Fernandez-Ramires R, Sole X, De Cecco L, Llort G, Cazorla A, Bonifaci N, Garcia MJ, Caldes T, Blanco I, Gariboldi M et al (2009) Gene expression profiling integrated into network modelling reveals heterogeneity in the mechanisms of BRCA1 tumorigenesis. Br J Cancer 101:1469–1480
Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, Pietenpol JA (2011) Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Investig 121:2750–2767
Takabe S, Mochizuki K, Goda T (2008) De-phosphorylation of GR at Ser203 in nuclei associates with GR nuclear translocation and GLUT5 gene expression in Caco-2 cells. Arch Biochem Biophys 475:1–6
Surjit M, Ganti KP, Mukherji A, Ye T, Hua G, Metzger D, Li M, Chambon P (2011) Widespread negative response elements mediate direct repression by agonist-liganded glucocorticoid receptor. Cell 145:224–241
Gorski JJ, James CR, Quinn JE, Stewart GE, Staunton KC, Buckley NE, McDyer FA, Kennedy RD, Wilson RH, Mullan PB et al (2010) BRCA1 transcriptionally regulates genes associated with the basal-like phenotype in breast cancer. Breast Cancer Res Treat 122:721–731
Gorski JJ, Savage KI, Mulligan JM, McDade SS, Blayney JK, Ge Z, Harkin DP (2011) Profiling of the BRCA1 transcriptome through microarray and ChIP-chip analysis. Nucleic Acids Res 39:9536–9548
Javle M, Curtin NJ (2011) The potential for poly (ADP-ribose) polymerase inhibitors in cancer therapy. Ther Adv Med Oncol 3:257–267
Acknowledgments
This research was supported by grants from INSERM-UPMC, HRA Pharma, Paris, and the Institut National du Cancer (INCa, France). Laudine Communal was the recipient of a CIFRE grant from HRA Pharma and the government. Myriam Vilasco was the recipient of a post-doc fellowship (INCa). We are very grateful to Sylvie Dumont and Aurélie Scriva for their technical assistance in immunohistochemistry. We thank Steve K. Nordeen (University of Colorado, Denver, USA) for sequences of IEX-1 primers and Wayne V. Vedeckis (University of Louisiana Health Sciences Center, New Orleans, USA) for providing the hGR 1A and 1B promoter constructs. We also thank Dr Alexandre A. Zukiwski, Dr Philippe le Rouzic, and Neil Insdorf for their kind help in editing the manuscript.
Author information
Authors and Affiliations
Consortia
Corresponding authors
Additional information
The members of BRACAPS are listed in the Appendix.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Appendix
Appendix
The members of BRACAPS are as follows: Jean Feunteun, Université Paris XI, Institut Gustave-Roussy; Suzette Delaloge, Institut Gustave-Roussy; Pascal Pujol, INSERM 824, CHU Arnaud de Villeneuve, Montpellier; Marie-Pierre Chauvet, Centre Oscar Lambret, Lille; Murielle Perrault de Jotemps, Clinique Hartmann, Neuilly; and Marc Chaouat, Hopital Saint Louis, Paris.
Rights and permissions
About this article
Cite this article
Vilasco, M., Communal, L., Hugon-Rodin, J. et al. Loss of glucocorticoid receptor activation is a hallmark of BRCA1-mutated breast tissue. Breast Cancer Res Treat 142, 283–296 (2013). https://doi.org/10.1007/s10549-013-2722-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-013-2722-8