
Abstract
Wilson’s disease (WD) is a severe disorder of copper misbalance, which manifests with a wide spectrum of liver pathology and/or neurologic and psychiatric symptoms. WD is caused by mutations in a gene encoding a copper-transporting ATPase ATP7B and is accompanied by accumulation of copper in tissues, especially in the liver. Copper-chelation therapy is available for treatment of WD symptoms and is often successful, however, significant challenges remain with respect to timely diagnostics and treatment of the disease. The lack of genotype-phenotype correlation remains unexplained, the causes of fulminant liver failure are not known, and the treatment of neurologic symptoms is only partially successful, underscoring the need for better understanding of WD mechanisms and factors that influence disease manifestations. Recent gene and protein profiling studies in animal models of WD began to uncover cellular processes that are primarily affected by copper accumulation in the liver. The results of such studies, summarized in this review, revealed new molecular players and pathways (cell cycle and cholesterol metabolism, mRNA splicing and nuclear receptor signaling) linked to copper misbalance. A systems biology approach promises to generate a comprehensive view of WD onset and progression, thus helping with a more fine-tune treatment and monitoring of the disorder.
Similar content being viewed by others


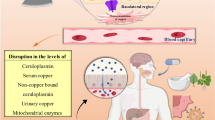
Wilson’s disease (WD) is caused by mutations in the copper-transporting ATPase ATP7B
WD is an autosomal recessive disorder caused by mutations in the gene ATP7B that encodes an ATP-driven copper transporter of the same name (Das and Ray 2006). In a healthy organism, ATP7B is expressed in various tissues, including liver, central nervous system, kidney, mammary gland, and others (Lutsenko et al. 2007). The major physiologic role of ATP7B is in the liver, a key homeostatic organ for copper metabolism. In hepatocytes, ATP7B transports copper from the cytosol into the lumen of the trans-Golgi network (TGN), where copper is post-translationally incorporated into secreted copper-dependent enzymes, such as ceruloplasmin. In addition, ATP7B maintains copper levels in the cytosol. When cytosolic copper exceeds a certain threshold, ATP7B traffics towards the canalicular membrane and sequesters excess copper into vesicles, which subsequently fuse with the canalicular membrane excreting excess copper into the bile (Bartee and Lutsenko 2007).
In WD, ATP7B expression, function, and/or intracellular targeting are disrupted by mutations. As a result, copper delivery to the TGN and copper excretion are both impaired, and copper accumulates to very high levels. It is thought that a tissue’s inability to adequately protect itself from excess of redox active copper causes the development of WD pathology (Ferenci 2005). Recent systematic analysis of genomic and proteomic changes also show that in addition to well-described damaging effects of high copper on DNA, proteins, and lipids, copper misbalance triggers a series of more specific metabolic responses that are likely to significantly influence pathology development. The results of these studies are summarized in this review.
Phenotypic variability and a lack of genotype-phenotype correlations in WD
Since the initial discovery of ATP7B (Bull et al. 1993; Tanzi et al. 1993) and first characterization of its gene structure (Petrukhin et al. 1994), over 380 mutations in this gene have been identified in WD patients world-wide (see WD mutation database http://www.wilsondisease.med.ualberta.ca/database.asp). Most of the WD mutations result in a single base-pair substitution replacing an amino-acid (missense mutations). However, nonsense, insertion, deletion and splice site mutations, which more drastically affect transcript and protein structure, have also been reported (Okada et al. 2010; Wilson et al. 2009; Kusuda et al. 2000). Although none of the WD mutations are predominant, several mutations show higher frequency in different world populations (Ferenci 2006). The best characterized H1069Q mutation is a prevalent mutation in Caucasian population of European origin (Vrabelova et al. 2005), including Lithuanian (Kucinskas et al. 2008), Czech (Vrabelova et al. 2005) Hungarian (Folhoffer et al. 2007), German (Huster et al. 2004) and other groups. In contrast, the R778L mutation is predominant among Korean (Kusuda et al. 2000; Park et al. 2009), Chinese (Mak et al. 2008), and Japanese (Kusuda et al. 2000) patients. Neither H1069Q nor R778L are common in India, where mutation resulting in the replacement of Cys271 with a Stop codon appears to be among the most frequent (Gupta et al. 2005). The carriers’ rate was estimated to be highest among Asians (1:51) followed by African Americans and Caucasians (both groups at 1:87).
Significant effort in examining genotype-phenotype correlations revealed some trends; for example, patients with protein truncating mutations have in general earlier onset of the disease (Merle et al. 2010), and the early onset and predominantly hepatic course was reported for R778L mutation (Wu et al. 2001). In contrast, the H1069Q substitution was suggested to be associated with a late on-set and neurological presentation (Stapelbroek et al. 2004). However, other studies comparing various mutations showed no correlations with a phenotype (Nicastro et al. 2009; Hermann et al. 2002). Furthermore, markedly different disease manifestations (fulminant liver failure versus a pre-symptomatic liver disease) have recently been reported for genetically identical twins homozygous for H1069Q mutation (Kegley et al. 2010; Czlonkowska et al. 2009). Overall, there has been a growing appreciation that epigenetic/environmental and/or dietary factors may play a very important role in modulating the WD phenotype. Little is known about such factors. As we illustrate in this review, a systems biology approach promises to facilitate their discovery.
Molecular mechanism of WD is not well understood
Currently, the generally cited mechanism of pathology development in Wilson’s disease involves oxidative damage due to copper overload (Ferenci 2005). Generation of reactive oxygen species (ROS) as well as lipid oxidation and DNA damage has been detected in the WD liver, particularly at the advanced stages of the disease (Yasuda et al. 2006; Nair et al. 1998; Hayashi et al. 2006). At the same time, radical-mediated damage alone does not explain the lack of genotype-phenotype correlations, the variability of symptoms even among siblings, and worsening neurologic symptoms upon treatment with copper chelators. Studies in bacteria suggest that that Cu(I) can also disrupt metabolic processes by binding to the coordinating sulfur atoms of Fe/S clusters, displacing iron and thus inhibiting the cluster containing enzymes (Macomber and Imlay 2009). Recent studies using animal models of WD uncovered additional players and new pathways that can contribute to pathology development. Although such studies are still very limited, the emerging data focus our attention on the cellular processes, the importance of which for WD has not been previously appreciated (see below).
The age of WD onset varies significantly; however, the disease typically presents in children and young adults. Although copper is always elevated in the WD liver, major disease manifestations vary, and either neurologic or hepatic phenotype may dominate. In the liver, copper overload triggers development of hepatitis, cirrhosis, which may progress to liver failure (Huster 2010). In the brain, globus pallidus, putamen, thalamus, mesencephalon, pons, and corpus collosum are most affected (Trocello et al. 2010). The development of neuropsychiatric symptoms (Involuntary movements, ataxia, depression) usually occurs in older patients and is thought to be secondary to liver malfunction. This view is probably an over-simplification, because ATP7B is expressed in various regions of the brain (Choi and Zheng 2009; Platonova 2005; Ahmed et al. 2005; Barnes et al. 2005) and its inactivation is likely to directly alter the CNS copper metabolism.
The molecular mechanisms behind WD phenotypic variability are not known. Recent proteomics studies in the Atp7b −/− liver identified molecules altered in response to copper accumulation, which have well-established roles in CNS physiology (see below). Thus, it is possible that the molecular machinery responding to copper levels in the brain is similar to one in the liver, and the extent of disease manifestation in different organs depends on a tissue-specific modulation of this machinery in response to overall metabolic state of the organism.
Animal models of Wilson’s disease
Several rodent models of Wilson’s disease have been described (Table 1). Toxic milk mice (tx mice) is an inbred strain with a Met1386Val mutation within the copper translocation pathway of ATP7B (Theophilos et al. 1996). This mutation does not disrupt the ATP7B biosynthesis, but markedly decreases its transport activity (Voskoboinik et al. 2001) and prevents copper-dependent trafficking of the transporter from Golgi to vesicles (La Fontaine et al. 2001). In the tx animals, copper concentration is increased in the liver, spleen, kidney and the brain (Allen et al. 2006). Copper accumulation is associated with morphologic changes in hepatic mitochondria, endoplasmic reticulum, and nuclei as well as accumulation of microvesicular lipid droplets (Biempica et al. 1988). An alteration of mitochondrial enzyme function in animals older than 3 months also occurs (Roberts et al. 2008). The detailed protein or mRNA profiling has not been done for this animal model.
A different variant of the tx mice originates from the C3H/HeJ strain bred at the Jackson Laboratory (hence the name, txJ). The txJ mice contain the Gly712Asp substitution at the cytosolic end of the second trans-membrane segment of ATP7B (Coronado et al. 2001). In addition, the Atp7b gene of txJ mice has nucleotide changes within the exon 2 that produce the Lys107Ser and His108Ala substitutions, which are thought to be polymorphisms. Adult homozygous txJ mice accumulate copper and develop cirrhosis, whereas infant mice are copper deficient and display increased mortality. This animal model is relatively recent and remains to be fully characterized. Interestingly, crossing the txJ mice with the transgenic CRND8 amyloid precursor protein mice decreases the number of plagues and increases survival compared to the latter (Phinney et al. 2003).
Most of the studies dissecting the mechanism of WD pathology have been done using Long-Evans Cinnamon (LEC) rats and, more recently, Atp7b −/− mice. The LEC rats are an inbred strain, which has a large deletion in the 3′ region of the Atp7b gene that abrogates protein production (Levy et al. 2007; Muramatsu et al. 1995). The Atp7b −/− mice are genetically engineered whole-body knockouts maintained on a hybrid C57BLx129S6/SvEv background (Buiakova et al. 1999). In these mice, the insertion of multiple stop codons in exon 2 completely abolishes protein synthesis (Buiakova et al. 1999), similarly to LEC rats. As expected for human WD, the LEC rats and Atp7b −/− mice accumulate copper in the liver, fail to incorporate copper into a copper-dependent enzyme ceruloplasmin (produced by the liver), and develop marked hepatic pathology. At the later stages of the disease, LEC rats develop hepatocellular carcinoma (Marquez et al. 2007), whereas old Atp7b −/− animals do not have hepatocellular carcinoma, but show marked proliferation and tumor development in bile ducts (Huster et al. 2006).
Pathology development and corresponding changes in liver transcriptome
Although some aspects of pathology differ between rodent strains as well as between human and murine WD, the general course of liver disease is similar for these species. Specifically, copper accumulation occurs rapidly and precedes visible pathology development. In the 6 weeks old LEC rats and Atp7b −/− mice hepatic copper is already high, whereas liver histology is fairly normal (Huster et al. 2006; Kasai et al. 1990). Subsequently, at around 8–9 weeks the increase in the size of hepatocytes and nuclei becomes apparent (Huster et al. 2006) and, in rats, there is an onset of jaundice (Kasai et al. 1990). In a period from 12 to 20 weeks, the disease is pronounced with a significant inflammation, necrosis and diminished liver function. 30–40% LEC rats die at the age of 4 months, whereas surviving rats develop tumors along with regenerative nodules. Atp7b −/− mice do not show increased mortality even at the height of the disease; at 30–35 weeks their liver begins to regenerate along with a marked proliferation and tumor development in bile ducts. It should be noted that the precise age at which different symptoms manifest may differ between animals bred in different laboratories/countries, which may reflect local conditions in animal housing, water supply, mode of breeding. Defining the stage of the liver disease by physiologic/biochemical parameters (rather than age) is likely to provide a more consistent picture when the results from different laboratories are compared.
Several groups have investigated molecular mechanisms behind copper-induced pathology by characterizing changes in hepatic mRNA profiles (Ralle et al. 2010; Klein et al. 2003; Tsubota et al. 2010; Huster et al. 2007; Marquez-Quinones et al. 2007). The precise gene-to-gene comparison of the results is difficult, because studies used different array platforms and the age of animals and the functional state of their liver did not match exactly. Nevertheless, overall trends (i.e., the major biological processes affected by copper overload) can be identified and compared. In order to make such comparisons with a minimal bias, we analyzed with the same GeneSifter software the mRNA array data that were relatively similar (Klein et al. 2003; Tsubota et al. 2010; Marquez-Quinones et al. 2007). We specifically focused on the earlier stages of pathology development (i.e., when pathology is not yet apparent or mild) to identify initial, and presumably more specific, effects of copper and to minimize differences between strains that become more apparent with age. The results are summarized in Tables 2 and 3. The data from Tsubota et al. represent changes in the 26 weeks LEC rats compared to 4 weeks old LEC rats (i.e., diseased state versus non-diseased state, whereas the data from Huster et al. show changes in the 6 week-old Atp7b −/− compared to the wt mice. These two sets of data are easier to compare because in both studies the entire transcriptome was probed, whereas Klein et al. (2003) used a more limited set of probes on a toxicology specialized Affymetrix RTU34 oligonucleotide array covering 1031 genes (In Tables 2 and 3, we show comparisons of data for 9 weeks to 6 weeks old LEC rats from this latter study).
It is apparent that common processes that accompany disease development in mice and the early-mid stage of disease in rats involve up-regulation of cell cycle and cell division (Table 2), whereas the most significantly down-regulated process is lipid metabolism (steroid metabolism and cholesterol metabolism, Table 3). In fact, in Atp7b −/− mice the transcripts of genes involved in lipid metabolism remain down-regulated throughout the entire course of the disease (Ralle et al. 2010). These changes in the transcriptome are associated with metabolic changes, as demonstrated by the decreased levels of cholesterol in the liver and of VLDL cholesterol in a serum of Atp7b −/− mice (Ralle et al. 2010; Huster et al. 2007). Recent studies in LEC rats also revealed significant lipid misbalance: the decrease in serum cholesterol and abnormalities in both circulating lipoprotein composition and size along with a significantly increased content of triglycerides, free cholesterol, and cholesteryl ester in the liver (Li et al. 2009). It is interesting that the down-regulation of genes associated with cholesterol biosynthesis was also observed in the liver of fish exposed to high copper (Santos et al. 2010), suggesting conservation of hepatic response to copper overload in low and high vertebrates.
Nuclear receptor signaling is significantly dysregulated in response to copper overload
Previous analysis of transcripts down-regulated in Atp7b −/− mice identified SREBP-1, as a factor at the center of down-regulated network (Huster et al. 2006). However, the mRNA levels of SREBP-1 are also decreased (Huster et al. 2006), indicating that factors upstream of SREBP-1 may play important role in altering lipid metabolism. Analysis of down-regulated signaling pathways reveals significant involvement of LXR/RXR/FXR nuclear receptors in hepatocytes response to copper overload (Fig. 1). This observation is particularly interesting because of the dual role of LXR/RXR in lipid metabolism and inflammation (Zhao and Dahlman-Wright 2010; Morello et al. 2009). The LXR and RXR agonists, oxysterols and retinoic acid respectively, promote transcription of lipid metabolism related genes and inhibit the NF-κB dependent activation of inflammatory mediators (Bensinger et al. 2008). Thus, down-regulation of the LXR/RXR signaling may account for the observed changes in lipid metabolism and liver inflammation in WD. Reciprocally, metabolic changes in Atp7b−/− hepatocytes may trigger inflammatory response, which in turn would repress lipid metabolism through the LPS/IL-1 mediated Inhibition of RXR Function (LMIRF). For example, during inflammation RXRs may leave the nucleus, thus suppressing transcription of RXR controlled genes (Ghose et al. 2004).

The top 10 canonical signaling pathways down-regulated in LEC rats (top) and Atp7b−/− mice (bottom). Differentially expressed genes (greater than 1.5 fold) were derived from comparing 6-week old Atp7b−/− to wild-type littermates; or 26 week old LEC rats—to 4 week old LEC rats. Ingenuity Pathway Analysis (IPA) defined 218 genes from Atp7b−/− and 1208 from LEC rats eligible for pathway analysis. These genes were queried against the IPA pathway library to identify over-represented signaling pathways that were significantly down-regulated. The significance [−log(P-value)] of a given pathway is measured by the likelihood that the pathway is associated with the dataset by random chance using the Fischer’s exact test. Striped bars Pathways common to Atp7b−/− mice and LEC rats. Black bars Pathways unique to Atp7b−/− mice or LEC rats. *Pathways involved in lipid or steroid metabolism
Comparison of available datasets also show some differences. Inflammatory and oxidative stress signaling via IL-17 and NRF2, respectively, are among the down-regulated pathways in the 6 weeks old Atp7b −/− mouse but not in the LEC rats (Fig. 1). This difference is likely due to a more advanced age of LEC animals, whose liver at 26 weeks does undergo inflammation. It would be important to compare younger LEC rats to determine if changes in lipid metabolism precede oxidative stress/inflammatory response as seen in the 6 week Atp7b −/− mouse.
Additional information was revealed in the mRNA profiling studies where time-dependent progression of hepatitis in the LEC rat was monitored using animals treated with a copper-chelating drug d-penicillamine as a control (Marquez-Quinones et al. 2007). This study also included useful evaluation of liver morphology and function. Comparison to the chelator-treated rats (rather than LEA rats) is an interesting experimental design, because genetic background of animals in this experiment is the same (an important consideration) and specific effects of copper are likely to be more clearly dissected. At the same time, if transcriptional changes occur in the control d-penicillamine-treated animals, such changes could be missed. Overall, the study has pointed to the upregulation of glutathione-S-transferase P (GSTP) as being characteristic for the stage of hepatitis associated with jaundice development. Among the annotated transcripts the majority (38 out of 59) belong to the “metabolism” category with the most affected metabolic processes being lipid biosynthesis and catabolism, alcohol metabolism and amino-acid biosynthesis (in addition to other less altered pathways) (Marquez-Quinones et al. 2007). Altogether, lipid metabolism is emerging as an important pathway to consider in WD pathology.
Studies in cultured cell lines identify stress-response genes
Another trend reported in the above study of LEC rats was upregulation of protein degradiation machinery along with pathology progression (Marquez-Quinones et al. 2007). This observation resonates with the results of Muller and colleagues, who investigated response of cultured hepatocytes HepG2 to high copper load (Muller et al. 2007). HepG2 cells contain active ATP7B and therefore represent a model of acute copper toxicity rather than WD; nevertheless, some parallels in cell response to high copper may exist. The initial response of HepG2 cells to high copper involves upregulation of metallothionein (also seen in WD patients and in all animal models of WD), whereas longer exposure to copper induces transcripts for proteins involved in proteasomal degradation and oxidative stress response (Muller et al. 2007). In Atp7b −/− mice, changes in the abundance of transcripts for proteosomal components and ubiquitinating enzymes are also seen (for example, ubiquitin specific protease Usp2 is down-regulated early in the disease, whereas the ubiquitin-conjugating enzyme E2 variant 2 is up at the later stage of the disease, our data) along with the upregulation of other soluble and membrane-bound proteases. It appears that the enhanced protein degradation may represent a general protective response of hepatocytes to a prolonged exposure to a very high copper.
Another response to a prolonged injury may involve activation of the NF-κB signaling pathway. The upregulation of this pathway was observed in HepG2 cells in response to a 4 h treatment with 400 or 600 μM copper (McElwee et al. 2009). How closely these conditions resemble WD remains to be determined. In WD liver, copper can be extremely high (reaching mM levels in the cytosol and nuclei (Ralle et al. 2010)), however such accumulation occurs gradually. Analysis of the Atp7b −/− mRNA dataset at the early stage of the disease did not show the NF-κB pathway as the most significantly upregulated (Fig. 2). Instead, CDKN1A, cyclin-dependent kinase inhibitor 1A, appears to play a prominent role. It should also be noted that the mRNA profiling of the entire liver does not discriminate between different cellular populations, and the contribution of cells other than hepatocytes (for example bile ducts’ cells that eventually proliferate) to the network of upregulated genes cannot be excluded. At the same time, upregulated genes linked to NF-κB are present in the early CDKN1A network (Fig. 2); thus more significant involvement of the NF-κB signaling may take place at the later stage of the disease when copper levels exceed liver buffering capacity.

The most populated network of genes up-regulated in 6 week old Atp7b −/− mice as defined by Ingenuity Network analysis. Shaded symbols represent transcripts for which the change in abundance was experimentally demonstrated
RNA processing machinery: a link between hepatic and neurologic symptoms in WD?
Analysis of the transcriptome offers a relatively quick way to generate useful insights into the physiological state of cells and tissue. The mRNA abundance suggests the amounts of corresponding proteins, however this information is indirect as the transcripts are one or more steps removed from key physiologic players: proteins and their metabolites. Consequently, proteomic studies, although technically more challenging, are gaining momentum as the way to generate mechanistic insights into pathology development. Focus on the proteome of specific subcellular compartments can be particularly informative. Within the Atp7b −/− hepatocytes, copper is distributed non-uniformly, and this distribution changes as disease progresses. At the early stage of pathology development the highest copper is preferentially elevated in the cytosol and nuclei (Huster et al. 2006; Ralle et al. 2010). Studies of copper-induced changes in the nuclear proteome of Atp7b −/− mice revealed a remodeling of RNA processing machinery as a possible mechanistic link between copper accumulation in nuclei and changes in the liver transcriptome (Burkhead et al. 2010). It was also found that copper, although markedly elevated in Atp7b −/− nuclei, did not induce large-scale displacement of other metals from the nucleus, suggesting a high buffering capacity of nuclei for metals. Similarly, global protein oxidation was not apparent as no change was seen in the levels of reduced thiols, protein nitration and protein glutathionylation. At the same time, the nuclear-localized Selenoprotein H (SelH) showed an increase in protein abundance (Burkhead et al. 2010). This increase in SelH, which has a glutathione reductase activity, may help to maintain the proper ratio of reduced and oxidized glutathione in the nucleus and may be part of the reason why the large-scale oxidation is not observed despite high copper levels.
The 2D-gel based proteomic analysis of isolated Atp7b −/− nuclei yielded two key observations. First, it became apparent that changes in the abundance and modification are limited to a small set of nuclear proteins, only a few of which were abundant. Second, the most significant group of changed nuclear proteins was one associated with mRNA processing (Burkhead et al. 2010). Upon further analysis by semi-quantitative mass spectrometry, it was found that some of the alteration in the levels of RNA processing proteins could be due to their nucleo-cytoplasmic redistribution. Specifically, the Atp7b −/− nuclei showed an increase in apparent retention of hnRNPs A2/B1, A3 and U, which form a multiprotein complex (Raju et al. 2008). HnRNP A2/B1 is a component of the spliceosome, a marker in hepatocellular carcinoma and other cancer (Raju et al. 2008; Cui et al. 2010; Wu et al. 2003; Peebles et al. 2007), and it functions in nucleocytoplasmic shuttling of mRNA (Munro et al. 1999). In Atp7b −/− liver, distinct alternative splicing-derived variant of hnRNP A2/B1 is upregulated due to increased retention of exon 2 (Burkhead et al. 2010).
Alteration of RNA processing machinery is likely one of the mechanisms that drives the observed changes in the transcriptome. For example, the LDL receptor mRNA is down-regulated in Atp7b −/− livers (Huster et al. 2007) as well as in LEC rats. The LDL receptor transcript can be destabilized by KH-type splicing regulatory protein (KSRP) and hnRNP I through the AU-rich elements in the 3′UTR (Li et al. 2009). Coincidently, KSRP is altered in Atp7b −/− livers (Huster et al. 2007; Burkhead et al. 2010), a finding that offers a potential link to the observed down-regulation of the LDL receptor mRNA. Similarly TDP-43 and hnRNP H1 (both changed in Atp7b −/− nuclei compared to control) have roles in alternative splicing of apolipoprotein-AII (Mercado et al. 2005), another transcript altered in Atp7b −/− livers.
The involvement of RNA processing machinery, particularly TDP-43 and hnRNP A2/B1, in cells’ response to elevated may provide a molecular link between hepatic and neurologic symptoms in WD. Recent data suggest that TDP-43 can be found in the mRNA processing sites in neurons and may have an important role in neurodegenerative disorders (Casafont et al. 2009; King et al. 2010). In turn, hnRNP A2/B1 is well characterized in nucleo-cytoplasmic shuttling of myelin-basic protein (MBP), the most abundant protein component of the myelin sheath in the CNS (Hoek et al. 1998; Shan et al. 2000). It is tempting to speculate that copper-induced alteration of RNA processing machinery may be a contributing factor to pathways leading to neuronal degeneration in WD.
Conclusions
WD is a severe disorder of copper metabolism with a varied age of onset and diverse phenotypic manifestations. Implementation of new data-driven approaches promises to greatly facilitate understanding of WD pathology. The diversity of WD phenotype and complexity of symptoms in humans indicates that the inactivation of ATP7B in WD has far-reaching physiological consequences. The “omics” studies have identified new metabolic and signaling pathways associated with disease progression, such as lipid metabolism, cell cycle regulation, LXR/RXR and CDKN1A signaling and, mRNA splicing. The cellular pathways and physiologic processes that are particularly sensitive to copper overload are likely to contribute to pathology development in WD. The “omics” studies also raise new mechanistic questions about previously unknown roles for copper in normal human physiology. In a future, systems information may allow for more fine-tuned therapies of WD, particularly with respect to symptoms unresponsive to copper chelation.
References
Ahmed S, Deng J, Borjigin J (2005) A new strain of rat for functional analysis of PINA. Brain Res Mol Brain Res 137(1–2):63–69
Allen KJ et al (2006) Chronological changes in tissue copper, zinc and iron in the toxic milk mouse and effects of copper loading. Biometals 19(5):555–564
Barnes N, Tsivkovskii R, Tsivkovskaia N, Lutsenko S (2005) The copper-transporting ATPases, Menkes and Wilson disease proteins, have distinct roles in adult and developing cerebellum. J Biol Chem 280(10):9640–9645
Bartee MY, Lutsenko S (2007) Hepatic copper-transporting ATPase ATP7B: function and inactivation at the molecular and cellular level. Biometals 20(3–4):627–637
Bensinger SJ et al (2008) LXR signaling couples sterol metabolism to proliferation in the acquired immune response. Cell 134(1):97–111
Biempica L, Rauch H, Quintana N, Sternlieb I (1988) Morphologic and chemical studies on a murine mutation (toxic milk mice) resulting in hepatic copper toxicosis. Lab Invest 59(4):500–508
Buiakova OI et al (1999) Null mutation of the murine ATP7B (Wilson disease) gene results in intracellular copper accumulation and late-onset hepatic nodular transformation. Hum Mol Genet 8(9):1665–1671
Bull PC, Thomas GR, Rommens JM, Forbes JR, Cox DW (1993) The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene. Nat Genet 5(4):327–337
Burkhead JL, Ralle M, Wilmarth P, David L, Lutsenko S (2010) Elevated copper remodels hepatic RNA processing machinery in the mouse model of Wilson’s disease. J Mol Biol 406(1):44–58
Casafont I, Bengoechea R, Tapia O, Berciano MT, Lafarga M (2009) TDP-43 localizes in mRNA transcription and processing sites in mammalian neurons. J Struct Biol 167:235–241
Choi BS, Zheng W (2009) Copper transport to the brain by the blood-brain barrier and blood-CSF barrier. Brain Res 1248:14–21
Coronado V, Nanji M, Cox DW (2001) The Jackson toxic milk mouse as a model for copper loading. Mamm Genome 12(10):793–795
Cui H, Wu F, Sun Y, Fan G, Wang Q (2010) Up-regulation and subcellular localization of hnRNP A2/B1 in the development of hepatocellular carcinoma. BMC Cancer 10:356
Czlonkowska A, Gromadzka G, Chabik G (2009) Monozygotic female twins discordant for phenotype of Wilson’s disease. Mov Disord 24(7):1066–1069
Das SK, Ray K (2006) Wilson’s disease: an update. Nat Clin Pract Neurol 2:482–493
Ferenci P (2005) Wilson’s disease. Clin Gastroenterol Hepatol 3(8):726–733
Ferenci P (2006) Regional distribution of mutations of the ATP7B gene in patients with Wilson disease: impact on genetic testing. Hum Genet 120(2):151–159
Folhoffer A et al (2007) Novel mutations of the ATP7B gene among 109 Hungarian patients with Wilson’s disease. Eur J Gastroenterol Hepatol 19(2):105–111
Ghose R, Zimmerman TL, Thevananther S, Karpen SJ (2004) Endotoxin leads to rapid subcellular re-localization of hepatic RXRalpha: a novel mechanism for reduced hepatic gene expression in inflammation. Nucl Recept 2(1):4
Gupta A et al (2005) Molecular pathogenesis of Wilson disease: haplotype analysis, detection of prevalent mutations and genotype-phenotype correlation in Indian patients. Hum Genet 118(1):49–57
Hayashi M et al (2006) Accumulation of copper induces DNA strand breaks in brain cells of Long-Evans Cinnamon (LEC) rats, an animal model for human Wilson Disease. Exp Anim 55(5):419–426
Hermann W et al (2002) Genotype correlation with fine motor symptoms in patients with Wilson’s disease. Eur Neurol 48(2):97–101
Hoek KS, Kidd GJ, Carson JH, Smith R (1998) hnRNP A2 selectively binds the cytoplasmic transport sequence of myelin basic protein mRNA. Biochemistry 37:7021–7029
Huster D (2010) Wilson disease. Best Pract Res Clin Gastroenterol 24(5):531–539
Huster D, Weizenegger M, Kress S, Mossner J, Caca K (2004) Rapid detection of mutations in Wilson disease gene ATP7B by DNA strip technology. Clin Chem Lab Med 42(5):507–510
Huster D et al (2006) Consequences of copper accumulation in the livers of the Atp7b−/− (Wilson disease gene) knockout mice. Am J Pathol 168(2):423–434
Huster D et al (2007) High copper selectively alters lipid metabolism and cell cycle machinery in the mouse model of Wilson disease. J Biol Chem 282(11):8343–8355
Kasai N et al (1990) Clinico-pathological studies of LEC rats with hereditary hepatitis and hepatoma in the acute phase of hepatitis. Lab Anim Sci 40(5):502–505
Kegley KM et al (2010) Fulminant Wilson’s disease requiring liver transplantation in one monozygotic twin despite identical genetic mutation. Am J Transplant 10(5):1325–1329
King A et al (2010) Abnormal TDP-43 expression is identified in the neocortex in cases of dementia pugilistica, but is mainly confined to the limbic system when identified in high and moderate stages of Alzheimer’s disease. Neuropathology 30(4):408–419
Klein D, Lichtmannegger J, Finckh M, Summer KH (2003) Gene expression in the liver of Long-Evans cinnamon rats during the development of hepatitis. Arch Toxicol 77(10):568–575
Kucinskas L et al (2008) High frequency of the c.3207C>A (p.H1069Q) mutation in ATP7B gene of Lithuanian patients with hepatic presentation of Wilson’s disease. World J Gastroenterol 14(38):5876–5879
Kusuda Y et al (2000) Novel mutations of the ATP7B gene in Japanese patients with Wilson disease. J Hum Genet 45(2):86–91
La Fontaine S et al (2001) Effect of the toxic milk mutation (tx) on the function and intracellular localization of Wnd, the murine homologue of the Wilson copper ATPase. Hum Mol Genet 10(4):361–370
Levy E et al (2007) Abnormal hepatobiliary and circulating lipid metabolism in the Long-Evans Cinnamon rat model of Wilson’s disease. Life Sci 80(16):1472–1483
Li H et al (2009) Identification of mRNA binding proteins that regulate the stability of LDL receptor mRNA through AU-rich elements. J Lipid Res 50(5):820–831
Lutsenko S, Barnes NL, Bartee MY, Dmitriev OY (2007) Function and regulation of human copper-transporting ATPases. Physiol Rev 87(3):1011–1046
Macomber L, Imlay JA (2009) The iron-sulfur clusters of dehydratases are primary intracellular targets of copper toxicity. Proc Natl Acad Sci USA 106(20):8344–8349
Mak CM et al (2008) Mutational analysis of 65 Wilson disease patients in Hong Kong Chinese: identification of 17 novel mutations and its genetic heterogeneity. J Hum Genet 53(1):55–63
Marquez A, Villa-Trevino S, Gueraud F (2007) The LEC rat: a useful model for studying liver carcinogenesis related to oxidative stress and inflammation. Redox Rep 12(1):35–39
Marquez-Quinones A et al (2007) Proteasome activity deregulation in LEC rat hepatitis: following the insights of transcriptomic analysis. OMICS 11(4):367–384
McElwee MK, Song MO, Freedman JH (2009) Copper activation of NF-kappaB signaling in HepG2 cells. J Mol Biol 393(5):1013–1021
Mercado PA, Ayala YM, Romano M, Buratti E, Baralle FE (2005) Depletion of TDP 43 overrides the need for exonic and intronic splicing enhancers in the human apoA-II gene. Nucleic Acids Res 33(18):6000–6010
Merle U et al (2010) Truncating mutations in the Wilson disease gene ATP7B are associated with very low serum ceruloplasmin oxidase activity and an early onset of Wilson disease. BMC Gastroenterol 10:8
Morello F et al (2009) LXR-activating oxysterols induce the expression of inflammatory markers in endothelial cells through LXR-independent mechanisms. Atherosclerosis 207:38–44
Muller P et al (2007) Gene expression profiling of liver cells after copper overload in vivo and in vitro reveals new copper-regulated genes. J Biol Inorg Chem 12(4):495–507
Munro TP et al (1999) Mutational analysis of a heterogeneous nuclear ribonucleoprotein A2 response element for RNA trafficking. J Biol Chem 274:34389–34395
Muramatsu Y et al (1995) The rat homologue of the Wilson’s disease gene was partially deleted at the 3′ end of its protein-coding region in Long-Evans Cinnamon mutant rats. Res Commun Mol Pathol Pharmacol 89(3):421–424
Nair J et al (1998) Lipid peroxidation-induced etheno-DNA adducts in the liver of patients with the genetic metal storage disorders Wilson’s disease and primary hemochromatosis. Cancer Epidemiol Biomarkers Prev 7(5):435–440
Nicastro E et al (2009) Genotype-phenotype correlation in Italian children with Wilson’s disease. J Hepatol 50(3):555–561
Okada T et al (2010) High prevalence of fulminant hepatic failure among patients with mutant alleles for truncation of ATP7B in Wilson’s disease. Scand J Gastroenterol 45(10):1232–1237
Park HD, Ki CS, Lee SY, Kim JW (2009) Carrier frequency of the R778L, A874V, and N1270S mutations in the ATP7B gene in a Korean population. Clin Genet 75(4):405–407
Peebles KA, Dwyer-Nield LD, Malkinson AM (2007) Altered expression of splicing factor, heterogeneous nuclear ribonucleoprotein A2/B1, in mouse lung neoplasia. Mol Carcinog 46(11):887–900
Petrukhin K et al (1994) Characterization of the Wilson disease gene encoding a P-type copper transporting ATPase: genomic organization, alternative splicing, and structure/function predictions. Hum Mol Genet 3(9):1647–1656
Phinney AL et al (2003) In vivo reduction of amyloid-beta by a mutant copper transporter. Proc Natl Acad Sci USA 100(24):14193–14198
Platonova NA et al (2005) In vivo expression of copper transporting proteins in rat brain sections. (Trans from Rus) Izv Akad Nauk Ser Biol 2:141–154 (in Russian)
Raju CS et al (2008) In cultured oligodendrocytes the A/B-type hnRNP CBF-A accompanies MBP mRNA bound to mRNA trafficking sequences. Mol Biol Cell 19:3008–3019
Ralle M et al (2010) Wilson disease at a single cell level: intracellular copper trafficking activates compartment-specific responses in hepatocytes. J Biol Chem 285(40):30875–30883
Roberts EA, Robinson BH, Yang S (2008) Mitochondrial structure and function in the untreated Jackson toxic milk (tx-j) mouse, a model for Wilson disease. Mol Genet Metab 93(1):54–65
Santos EM et al (2010) Identifying health impacts of exposure to copper using transcriptomics and metabolomics in a fish model. Environ Sci Technol 44(2):820–826
Shan J et al (2000) Binding of an RNA trafficking response element to heterogeneous nuclear ribonucleoproteins A1 and A2. J Biol Chem 275(49):38286–38295
Stapelbroek JM et al (2004) The H1069Q mutation in ATP7B is associated with late and neurologic presentation in Wilson disease: results of a meta-analysis. J Hepatol 41:758–763
Tanzi RE et al (1993) The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene. Nat Genet 5(4):344–350
Theophilos MB, Cox DW, Mercer JF (1996) The toxic milk mouse is a murine model of Wilson disease. Hum Mol Genet 5(10):1619–1624
Trocello JM et al (2010) Corpus callosum abnormalities in Wilson’s disease. J Neurol Neurosurg Psychiatry
Tsubota A et al (2010) IQGAP1 and vimentin are key regulator genes in naturally occurring hepatotumorigenesis induced by oxidative stress. Carcinogenesis 31(3):504–511
Voskoboinik I, Greenough M, La Fontaine S, Mercer JF, Camakaris J (2001) Functional studies on the Wilson copper P-type ATPase and toxic milk mouse mutant. Biochem Biophys Res Commun 281(4):966–970
Vrabelova S, Letocha O, Borsky M, Kozak L (2005) Mutation analysis of the ATP7B gene and genotype/phenotype correlation in 227 patients with Wilson disease. Mol Genet Metab 86(1–2):277–285
Wilson AM, Schlade-Bartusiak K, Tison JL, Macintyre G, Cox DW (2009) A minigene approach for analysis of ATP7B splice variants in patients with Wilson disease. Biochimie 91(10):1342–1345
Wu ZY et al (2001) Mutation analysis and the correlation between genotype and phenotype of Arg778Leu mutation in Chinese patients with Wilson disease. Arch Neurol 58(6):971–976
Wu S et al (2003) hnRNP B1 protein may be a possible prognostic factor in squamous cell carcinoma of the lung. Lung Cancer 41(2):179–186
Yasuda J et al (2006) Reactive oxygen species modify oligosaccharides of glycoproteins in vivo: a study of a spontaneous acute hepatitis model rat (LEC rat). Biochem Biophys Res Commun 342(1):127–134
Zhao C, Dahlman-Wright K (2010) Liver X receptor in cholesterol metabolism. J Endocrinol 204(3):233–240
Acknowledgments
This work was supported by National Institute of Health grants R21 DK075659 and P01 GM067166 to SL. JLB is supported the sub-award to P20RR016466-10; LG is a recipient of the NRSA fellowship F31DK084730-02.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Burkhead, J.L., Gray, L.W. & Lutsenko, S. Systems biology approach to Wilson’s disease. Biometals 24, 455–466 (2011). https://doi.org/10.1007/s10534-011-9430-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10534-011-9430-9