
Abstract
Receptor tyrosine kinase c-Kit and its ligand stem cell factor (SCF) regulate resident vascular wall cells and recruit circulating progenitors. We tested whether SCF may be induced by oxidized palmitoyl-arachidonoyl-phosphatidylcholine (OxPAPC) known to accumulate in atherosclerotic vessels. Gene expression analysis demonstrated OxPAPC-induced upregulation of SCF mRNA and protein in different types of endothelial cells (ECs). Elevated levels of SCF mRNA were observed in aortas of ApoE−/− knockout mice. ECs produced biologically active SCF because conditioned medium from OxPAPC-treated cells stimulated activation (phosphorylation) of c-Kit in naïve ECs. Induction of SCF by OxPAPC was inhibited by knocking down transcription factor NRF2. Inhibition or stimulation of NRF2 by pharmacological or molecular tools induced corresponding changes in SCF expression. Finally, we observed decreased levels of SCF mRNA in aortas of NRF2 knockout mice. We characterize OxPLs as a novel pathology-associated stimulus inducing expression of SCF in endothelial cells. Furthermore, our data point to transcription factor NRF2 as a major mediator of OxPL-induced upregulation of SCF. This mechanism may represent one of the facets of pleiotropic action of NRF2 in vascular wall.
Similar content being viewed by others


Introduction
Stem cell factor (SCF, KIT ligand, steel factor) is a growth factor activating receptor tyrosine kinase c-Kit, which is structurally related to the platelet-derived growth factor (PDGF) receptor and is widely recognized for its role in stem cell biology [1]. SCF produced in bone marrow by endothelial and perivascular stromal cells is a major player in forming a niche for c-Kit-positive hematopoietic stem cells [2]. Furthermore, SCF and c-Kit are important for differentiation and function of mast cells [3] and demonstrate multiple other biological activities. In particular, rapidly accumulating data point to the role of SCF/c-Kit in regulation of vascular wall homeostasis, which is however less investigated as compared to other effects of SCF and c-Kit.
Major vascular wall cells such as endothelial cells (EC) and vascular smooth muscle cells (VSMC) are both producers and targets of SCF [4,5,6,7,8]. Treatment of ECs with SCF stimulates pro-angiogenic reactions including survival and migration of ECs, as well as formation of endothelial capillary tubes [9]. In vivo data show that c-Kit deficiency inhibits proliferation of ECs and suppresses angiogenesis [10]. Furthermore, SCF and c-Kit protect VSMCs from apoptosis [11] and regulate contractile phenotype of these cells [5]. SCF and c-Kit play a role in vascular disease and repair as suggested by enhanced expression of c-Kit by resident cells, as well as recruitment of circulating c-Kit-positive cells in various vascular pathologies including atherosclerosis [12, 13], in-stent restenosis [14], restenosis of artificial vessel grafts [15], aortic aneurysms [16], idiopathic pulmonary arterial hypertension [17] and aging [18]. Altogether, available data support the notion that SCF and c-Kit play important and multifaceted roles in vascular biology and pathology thus justifying research into the factors and mechanisms regulating expression and function of these proteins.
Accumulation of oxidized low-density lipoprotein (LDL) within arterial wall is a hallmark of atherosclerosis. Phospholipids within LDL particles are prone to oxidation due to the high contents of polyunsaturated fatty acids. Oxidized phospholipids (OxPLs) are generated by enzymatic or non-enzymatic oxidation of esterified fatty acids and demonstrate a variety of activities in vitro and in vivo [19, 20]. The relevance of OxPLs to vascular pathology is illustrated by their ability to stimulate monocyte–endothelial interactions, inhibit endothelium-dependent relaxation of vessels, promote formation of foam cells, induce phenotypic modulation and migration of VSMCs, enhance thrombogenic activity of ECs and platelets and stimulate angiogenesis [21]. These effects are mediated by multiple signaling pathways including cellular stress reactions called unfolded protein response (UPR) and electrophilic stress response (ESR). The major transcriptional factors mediating UPR and ESR in OxPL-treated ECs are ATF4 and NRF2, respectively [22, 23]. OxPLs are known to accumulate in human and animal atherosclerotic vessels at high concentrations comparable to those inducing biological effects in vitro [24]. In this work, we asked a question whether OxPLs can stimulate production of SCF. Our data show that phospholipid oxidation products increase expression of SCF by endothelial and monocytic cells and that transcription factor NRF2 plays an important role in OxPL-induced upregulation of SCF.
Results
OxPAPC upregulates SCF mRNA and protein in endothelial cells and a monocytic cell line
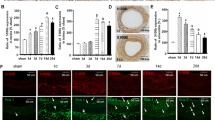
Treatment of ECs with OxPAPC is known to regulate expression of hundreds of genes in HAECs [25]. We applied microarray hybridization in order to analyze effects of OxPAPC on gene expression in another type of arterial ECs, i.e., HCAEC. One of physiologically important genes that were upregulated by OxPAPC in HCAECs was KIT ligand (KITL), more often referred to as stem cell factor (SCF). Elevation of SCF mRNA levels in HCAECs was observed in three independent experiments (Fig. 1a). Real-time PCR analysis confirmed upregulation of SCF mRNA in another type of aortic ECs (HAEC), as well as in venous ECs (HUVEC, Fig. 1a). Furthermore, OxPAPC upregulated SCF mRNA in the human monocytic cell line THP-1 (Suppl. Fig. 1). In addition to OxPAPC, SCF was upregulated in HUVECs by OxLDL and lipid peroxidation product such as electrophilic isoprostaglandin A2 (Suppl. Fig. 2).

SCF mRNA is induced in endothelial and monocytic cells by OxPLs and PGA2 in a time- and concentration-dependent manner a Endothelial cells were stimulated with OxPAPC (100 µg/ml, 6 h). Total RNA was extracted using Trizol reagent and analyzed by microarray hybridization (HCAEC; three independent experiments and hybridizations) or qRT-PCR (HAEC, HUVEC; quadruplicate samples from one representative experiment out of three). Levels of SCF mRNA were normalized to β2-microglobulin mRNA. b HUVECs were stimulated with OxPAPC (100 µg/ml) for indicated time periods, followed by analysis of SCF mRNA. c HUVECs were treated with indicated concentrations of OxPAPC for 6 h. qRT-PCR was used for SCF mRNA quantification. d, e Levels of SCF mRNA were analyzed in aortas of aged (12 months old) wild type and ApoE−/− mice (d) or in aortas of wild type and ApoE−/− young animals fed for 8 weeks either with chow (6.5% fat) or high-fat diet (15% fat) (e). Total RNA was prepared from homogenized aortas using Trizol reagent. SCF mRNA expression was analyzed by qRT-PCR and normalized to β2-microglobulin mRNA levels
Upregulation of SCF mRNA was time-dependent (Fig. 1b) and was observed within the range of OxPAPC concentrations known to be present in atheroma [24] (Fig. 1c). Furthermore, we observed statistically significant elevation of SCF mRNA in aortas from aged ApoE knockout mice (Fig. 1d), as well as in younger animals fed high-fat diet (Fig. 1e).
The analysis of SCF mRNA splice variants demonstrated the presence of two forms. One of them is known to encode both membrane and soluble variants of SCF, while another mainly produces the membrane form [26] (Fig. 2a). In a good agreement with this finding, Western blotting detected two anti-SCF positive bands corresponding to expected sizes of the two isoforms (Suppl. Fig. 3). Furthermore, SCF protein was detected both in conditioned medium from OxPAPC-treated cells and in cell lysates (Fig. 2b). Thus, we hypothesize that OxPAPC upregulated both soluble and membrane isoforms of SCF.

OxPAPC induces expression of cell-associated and secreted SCF protein and stimulates c-Kit phosphorylation. a Two forms of SCF mRNA were quantified in OxPAPC-stimulated HUVECs (100 µg/ml, 6 h) by qRT-PCR using primers selective for individual splice variants. b SCF protein was analyzed by ELISA in extracts of HUVECs or in conditioned media after 12 h of stimulation with 100 µg/ml OxPAPC. c Conditioned media produced by cells stimulated with indicated concentrations of OxPAPC stimulate phosphorylation of c-Kit receptor in HUVECs. d Panel presents ELISA data on the levels of SCF protein in conditioned media. e Pretreatment of cells with c-Kit tyrosine kinase inhibitor imatinib (30 min, 20 µM) attenuated c-Kit phosphorylation in HUVECs incubated with conditioned media from OxPAPC-stimulated cells
We further tested whether SCF produced by OxPAPC-treated cells was biologically active. To this end, HUVECs were stimulated by conditioned medium from OxPAPC-treated cells, followed by analysis of activation (autophosphorylation) of the SCF receptor, c-Kit. Conditioned medium induced phosphorylation of c-Kit (Fig. 2c). The phosphorylation developed within the same range of OxPAPC concentrations where it stimulated SCF secretion (Fig. 2d) and was inhibited by c-Kit inhibitor imatinib (Fig. 2e). These data suggest that OxPAPC stimulated HUVECs to produce biologically active SCF.
Oxidized phospholipids upregulate SCF via the NRF2 pathway
OxPLs are known to activate electrophilic and unfolded protein stress responses (ESR and UPR, respectively), which play important role in regulation of gene expression by these lipids [22, 23]. The knockdown of two major components of UPR, i.e., PERK and ATF4, did not significantly influence induction of SCF by OxPAPC (Fig. 3a, b), although expression of VEGF, known as a target of PERK and ATF4 [23], was suppressed (Suppl. Fig. 4a, b). Furthermore, chemically different inducers of UPR such as tunicamycin, brefeldin and homocysteine upregulated VEGF but did not change SCF mRNA levels (Suppl. Fig. 5a, b). In contrast to the UPR pathway, transfection of cells with siRNA against the key transcriptional mediator of ESR, i.e., NRF2, resulted in significant inhibition of the OxPAPC-induced upregulation of SCF (Fig. 3c) and a reference NRF2 target gene OKL38 [27] (Suppl. Fig. 6a). The knockdown of the NRF2 inhibitor KEAP-1 did not enhance SCF or OKL38 induction by OxPAPC but reproducibly elevated basal mRNA levels of these genes (Fig. 3d and Suppl. Fig. 6b). These data allow hypothesizing that NRF2 is a signaling mediator of OxPAPC-induced upregulation of SCF.

Induction of SCF by OxPAPC depends on the transcription factor NRF2. a, b, c and d HUVECs were transfected with siRNAs targeting ATF4 (a), PERK (b), NRF2 (c), or KEAP (d). Twenty-four hours after transfection cells were stimulated with OxPAPC (100 µg/ml, 6 h). Levels of SCF mRNA were analyzed by qRT-PCR in total RNA prepared using Trizol reagent and normalized to β2-microglobulin mRNA. e Protein kinase CK2 inhibitor TBB attenuates induction of SCF by OxPAPC. Cells were pretreated with TBB (20 µM, 30 min) and thereafter stimulated with OxPAPC (100 µg/ml, 6 h); SCF mRNA was quantified as described above. f miR-155 potentiates induction of SCF by OxPAPC. HUVECs were transfected with the RNA oligonucleotide mimicking miR-155 for 24 h and stimulated by OxPAPC (100 µg/ml, 6 h). Levels of SCF mRNA were quantified by qRT-PCR. g Steady-state levels of SCF mRNA are decreased in aortas of NRF2−/− mice. Total RNA was prepared from aortas of 6 months old NRF2−/− or wild-type mice and analyzed by qRT-PCR method. The levels of NRF2 mRNA were normalized to β2-microglobulin mRNA
Protein kinase CK2 plays an important role in control of ESR in HUVECs treated with OxPLs [28]. In agreement with these data, pretreatment of HUVECs with a specific CK2 inhibitor, TBB, significantly inhibited OxPAPC-induced upregulation of SCF (Fig. 3e) and a reference NRF2 target gene OKL38 (Suppl. Fig. 6c).
Levonen and colleagues have shown that microRNA miR-155 inactivated transcriptional regulator BACH1 [29]. In contrast to NRF2, which enhances transcription by binding at the antioxidant response element (ARE) of target genes, BACH1 acts as a repressor of ARE-dependent transcription [30]. We used oligonucleotides mimicking miR-155 as tools to check the importance of ARE-driven transcription in induction of SCF mRNA by OxPAPC. In agreement with the proposed mechanism, the oligonucleotide mimicking miR-155 elevated basal levels and enhanced OxPAPC-induced upregulation of SCF (Fig. 3f) and a reference NRF2 target gene OKL38 (Suppl. Fig. 6d).
Finally, we found that the levels of SCF mRNA in aortas of the NRF2−/− mice were lower than in the wild-type animals (Fig. 3g), which further support our hypothesis about the involvement of NRF2 in upregulation of SCF under the conditions of oxidative stress and lipid oxidation.
Discussion
Production of SCF by ECs is stimulated by inflammatory agonists and bacterial products [7, 12, 31]. In this work, we describe for the first time additional stimuli promoting expression of SCF, namely OxPLs and electrophilic prostanoids. These compounds accumulate in atherosclerotic vessels and are relevant to the initiation and progression of the disease [21, 32]. We observed upregulation of SCF in genetically hypercholesterolemic mice, which supports the notion that oxidized lipids upregulate SCF in atherosclerotic vessels. Induction of SCF by OxPLs potentially can have broad impact on atherogenesis due to ability of this growth factor to regulate viability, migration and differentiation of ECs and VSMCs, as well as recruit local and circulating progenitor and stem cells [5, 6, 9]. Furthermore, SCF is a key factor regulating differentiation of mast cells, as well as their recruitment into the arterial wall where mast cells play a role both in initiation and advanced stages of atherogenesis [33]. The importance of mast cells for atherogenesis was confirmed by decreased atherosclerosis in hyperlipidemic mice deficient in mast cells [34,35,36]. Altogether, available data suggest that upregulation of SCF can modulate several key mechanisms promoting development and progression of lesions.
OxPLs do not induce the major inflammatory pathway NFκB that is activated by inflammatory cytokines and bacterial products [19, 21], suggesting the involvement of additional mechanisms of SCF induction. In support of this possibility, we characterize NRF2-dependent transcription as a potential mechanism of SCF regulation by OxPLs. The involvement of NRF2 was shown using several approaches targeting different steps both upstream and downstream of NRF2 activation, including inhibition of protein kinase CK2 that is important for activation of electrophilic stress response by OxPLs [28], knockdown of key players in ESR such as KEAP-1 and NRF2, silencing of transcriptional repressor BACH1 by miR-155 and analysis of SCF expression in vivo in arteries of NRF2 knockout mice. All these data consistently support our hypothesis about the importance of the NRF2 pathway in upregulation of SCF by lipid oxidation products.
Previously, we have shown that the ESR pathway plays an important role in upregulation of VEGF in ECs treated with OxPLs [37]. Both VEGF and SCF protect endothelium from apoptosis and stimulate its regeneration by local mechanisms (migration, proliferation) as well as by mobilizing and attracting circulating progenitor cells. Thus, we hypothesize that the expression of VEGF and SCF in response to cellular stress induced by oxidized lipids represents a compensatory reaction aiming at the endothelial protection and repair. Our data show that these protective effects are mediated via the NRF2 pathway. However, prolonged secretion of VEGF and SCF potentially can attract monocytes, smooth muscle and mast cells and also stimulate plaque neovascularization thus leading to disease progression. Further in vivo studies are required in order to dissect specific role of OxPL-induced SCF in early and advanced stages of atherosclerosis.
Materials and methods
Materials, cell culture and lipid preparation
Tunicamycin, brefeldin, homocysteine, 4,5,6,7-tetrabromo-2-azabenzimidazole (TBB), imatinib and polyethylenimine (PEI) were obtained from Sigma-Aldrich. Human umbilical vein ECs (HUVECs), human carotid artery ECs (HCAECs) and human aortic ECS (HAECs) (all from Lonza) were grown at 37 °C in 5% CO2 in medium M199 containing 20% FCS, 1 U/ml heparin, ECGS growth supplement (Promocell), 2 mmol/l glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin and used up to passage 5. 1-Palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine (PAPC) (Avanti Polar Lipids) was oxidized by an exposure to air. Formation of OxPAPC was controlled by electrospray ionization-mass spectrometry and thin-layer chromatography [38]. For cell stimulation, the lipids were resuspended in medium M199 containing 2% fetal calf serum (FCS).
cDNA synthesis and qRT-PCR analysis of RNA
ECs were treated either with indicated concentration of OxPAPC in medium M199 containing 2% serum or with control medium for 4 h. Trizol reagent (Invitrogen) was used for RNA isolation. Microarray (Affymetrix Gene Profiling Array) profiling of mRNAs and data analysis were done at the Medical University of Vienna Genomics Core Facility. GeneAmp RNA-PCR kit and Fast SYBR Green Master Mix were used for analysis of mRNA by qRT-PCR. All these reagents were purchased from Applied Biosystems. Sequences of primers are available upon request. StepOnePlus real-time PCR cycler (Applied Biosystems) was used for quantitative real-time PCR.
Transfection with siRNA and miRNA mimic
ECs were transfected with 50 nM of either siRNA, miRNA mimic, or control oligonucleotide (all from Qiagen) in plain M199 medium for 4 h using polyethylenimine (PEI) reagent [39]. All stimulations were performed 24 h after transfection.
ELISA and Western blotting
R&D Systems ELISA kit was used for measurements of the SCF level in cell lysates and conditioned medium. The degree of c-Kit autophosphorylation was analyzed by PathScan® Phospho-c-Kit (Tyr719) sandwich ELISA kit. Signal detection was performed spectrophotometrically at 450 nm. For SCF analysis, protein samples were denatured in Laemmli buffer and separated in SDS-polyacrylamide gels with following electroblotting to PVDF membrane (Millipore). Protein blots were probed with anti-SCF antibodies (Cell Signaling Technology). Horseradish peroxidase conjugated IgG (GE Healtcare) and SuperSignal West Femto Substrate (Pierce) were used for detection of bound primary antibodies. Chemiluminescense was detected by using FluorChem HD2 imager (Alpha Innotech).
Mice
Sixteen weeks old male C57BL/6 and ApoE−/− mice (six animals per group) were fed either a standard laboratory chow diet (6.5% fat) or a proatherogenic diet containing 15% fat and 1.25% cholesterol (Sniff) for 8 weeks. Twelve months old C57BL/6 and ApoE−/− mice hold on chow diet were used for comparison of mRNA levels in aged animals. Six months old C57BL/6 and NRF2−/− mice hold on chow diet were used for comparison of the SCF mRNA levels. Total RNA was prepared from homogenized aortas using Trizol reagent (Invitrogen) and analyzed as described above. Experiments were approved by the Medical University of Vienna animal experimentation committee and the Austrian Ministry of Science.
Statistical analysis
Two-tailed Student’s t test was used for analysis; p value less than 0.05 was considered significant. All data are represented as means ± standard deviations.
Abbreviations
- OxPAPC:
-
Oxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine
- SCF:
-
Stem cell factor
- ESR:
-
Electrophilic stress response
- UPR:
-
Unfolded protein response
- ECs:
-
Endothelial cells
- OxPLs:
-
Oxidized phospholipids
References
Lennartsson J, Rönnstrand L (2012) Stem cell factor receptor/c-Kit: from basic science to clinical implications. Physiol Rev 92:1619–1649. https://doi.org/10.1152/physrev.00046.2011
Ding L, Saunders TL, Enikolopov G, Morrison SJ (2012) Endothelial and perivascular cells maintain haematopoietic stem cells. Nature 481:457–462. https://doi.org/10.1038/nature10783
El-Agamy DS (2012) Targeting c-kit in the therapy of mast cell disorders: current update. Eur J Pharmacol 690:1–3. https://doi.org/10.1016/j.ejphar.2012.06.030
Broudy VC, Kovach NL, Bennett LG et al (1994) Human umbilical vein endothelial cells display high-affinity c-kit receptors and produce a soluble form of the c-kit receptor. Blood 83:2145–2152
Davis BN, Hilyard AC, Nguyen PH et al (2009) Induction of microRNA-221 by platelet-derived growth factor signaling is critical for modulation of vascular smooth muscle phenotype. J Biol Chem 284:3728–3738. https://doi.org/10.1074/jbc.M808788200
Hollenbeck ST, Sakakibara K, Faries PL et al (2004) Stem cell factor and c-kit are expressed by and may affect vascular SMCs through an autocrine pathway. J Surg Res 120:288–294. https://doi.org/10.1016/j.jss.2004.01.005
Koenig A, Yakisan E, Reuter M et al (1994) Differential regulation of stem cell factor mRNA expression in human endothelial cells by bacterial pathogens: an in vitro model of inflammation. Blood 83:2836–2843
Miyamoto T, Sasaguri Y, Sasaguri T et al (1997) Expression of stem cell factor in human aortic endothelial and smooth muscle cells. Atherosclerosis 129:207–213
Matsui J, Wakabayashi T, Asada M et al (2004) Stem cell factor/c-kit signaling promotes the survival, migration, and capillary tube formation of human umbilical vein endothelial cells. J Biol Chem 279:18600–18607. https://doi.org/10.1074/jbc.M311643200
Fang S, Wei J, Pentinmikko N et al (2012) Generation of functional blood vessels from a single c-kit+ adult vascular endothelial stem cell. PLoS Biol 10:e1001407. https://doi.org/10.1371/journal.pbio.1001407
Wang C-H, Verma S, Hsieh I-C et al (2007) Stem cell factor attenuates vascular smooth muscle apoptosis and increases intimal hyperplasia after vascular injury. Arterioscler Thromb Vasc Biol 27:540–547. https://doi.org/10.1161/01.ATV.0000257148.01384.7d
Dentelli P, Rosso A, Balsamo A et al (2007) C-KIT, by interacting with the membrane-bound ligand, recruits endothelial progenitor cells to inflamed endothelium. Blood 109:4264–4271. https://doi.org/10.1182/blood-2006-06-029603
Torsney E, Mandal K, Halliday A et al (2007) Characterisation of progenitor cells in human atherosclerotic vessels. Atherosclerosis 191:259–264. https://doi.org/10.1016/j.atherosclerosis.2006.05.033
Hibbert B, Chen Y-X, O’Brien ER (2004) c-kit-immunopositive vascular progenitor cells populate human coronary in-stent restenosis but not primary atherosclerotic lesions. Am J Physiol Heart Circ Physiol 287:H518–H524. https://doi.org/10.1152/ajpheart.00002.2004
Tsai T-N, Kirton JP, Campagnolo P et al (2012) Contribution of stem cells to neointimal formation of decellularized vessel grafts in a novel mouse model. Am J Pathol 181:362–373. https://doi.org/10.1016/j.ajpath.2012.03.021
Shen YH, Hu X, Zou S et al (2012) Stem cells in thoracic aortic aneurysms and dissections: potential contributors to aortic repair. Ann Thorac Surg 93:1524–1533. https://doi.org/10.1016/j.athoracsur.2012.01.063
Montani D, Perros F, Gambaryan N et al (2011) C-kit-positive cells accumulate in remodeled vessels of idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med 184:116–123. https://doi.org/10.1164/rccm.201006-0905OC
Ferlosio A, Arcuri G, Doldo E et al (2012) Age-related increase of stem marker expression influences vascular smooth muscle cell properties. Atherosclerosis 224:51–57. https://doi.org/10.1016/j.atherosclerosis.2012.07.016
Bochkov V, Gesslbauer B, Mauerhofer C et al (2016) Pleiotropic effects of oxidized phospholipids. Free Radic Biol Med. https://doi.org/10.1016/j.freeradbiomed.2016.12.034
Mauerhofer C, Philippova M, Oskolkova OV, Bochkov VN (2016) Hormetic and anti-inflammatory properties of oxidized phospholipids. Mol Aspects Med 49:78–90. https://doi.org/10.1016/j.mam.2016.02.003
Bochkov VN, Oskolkova OV, Birukov KG et al (2010) Generation and biological activities of oxidized phospholipids. Antioxid Redox Signal 12:1009–1059. https://doi.org/10.1089/ars.2009.2597
Jyrkkanen H-K, Kansanen E, Inkala M et al (2008) Nrf2 regulates antioxidant gene expression evoked by oxidized phospholipids in endothelial cells and murine arteries in vivo. Circ Res 103:e1–e9. https://doi.org/10.1161/CIRCRESAHA.108.176883
Oskolkova OV, Afonyushkin T, Leitner A et al (2008) ATF4-dependent transcription is a key mechanism in VEGF up-regulation by oxidized phospholipids: critical role of oxidized sn-2 residues in activation of unfolded protein response. Blood 112:330–339. https://doi.org/10.1182/blood-2007-09-112870
Oskolkova OV, Afonyushkin T, Preinerstorfer B et al (2010) Oxidized phospholipids are more potent antagonists of lipopolysaccharide than inducers of inflammation. J Immunol 185:7706–7712. https://doi.org/10.4049/jimmunol.0903594
Gargalovic PS, Imura M, Zhang B et al (2006) Identification of inflammatory gene modules based on variations of human endothelial cell responses to oxidized lipids. Proc Natl Acad Sci U S A 103:12741–12746. https://doi.org/10.1073/pnas.0605457103
Flanagan JG, Chan DC, Leder P (1991) Transmembrane form of the kit ligand growth factor is determined by alternative splicing and is missing in the Sld mutant. Cell 64:1025–1035
Li R, Chen W, Yanes R et al (2007) OKL38 is an oxidative stress response gene stimulated by oxidized phospholipids. J Lipid Res 48:709–715. https://doi.org/10.1194/jlr.M600501-JLR200
Afonyushkin T, Oskolkova OV, Binder BR, Bochkov VN (2011) Involvement of CK2 in activation of electrophilic genes in endothelial cells by oxidized phospholipids. J Lipid Res 52:98–103. https://doi.org/10.1194/jlr.M009480
Pulkkinen KH, Ylä-Herttuala S, Levonen A-L (2011) Heme oxygenase 1 is induced by miR-155 via reduced BACH1 translation in endothelial cells. Free Radic Biol Med 51:2124–2131. https://doi.org/10.1016/j.freeradbiomed.2011.09.014
Dhakshinamoorthy S, Jain AK, Bloom DA, Jaiswal AK (2005) Bach1 competes with Nrf2 leading to negative regulation of the antioxidant response element (ARE)-mediated NAD(P)H:quinone oxidoreductase 1 gene expression and induction in response to antioxidants. J Biol Chem 280:16891–16900. https://doi.org/10.1074/jbc.M500166200
Buzby JS, Knoppel EM, Cairo MS (1994) Coordinate regulation of Steel factor, its receptor (Kit), and cytoadhesion molecule (ICAM-1 and ELAM-1) mRNA expression in human vascular endothelial cells of differing origins. Exp Hematol 22:122–129
Lee S, Birukov KG, Romanoski CE et al (2012) Role of phospholipid oxidation products in atherosclerosis. Circ Res 111:778–799. https://doi.org/10.1161/CIRCRESAHA.111.256859
Kovanen PT (2007) Mast cells: multipotent local effector cells in atherothrombosis. Immunol Rev 217:105–122. https://doi.org/10.1111/j.1600-065X.2007.00515.x
Heikkilä HM, Trosien J, Metso J et al (2010) Mast cells promote atherosclerosis by inducing both an atherogenic lipid profile and vascular inflammation. J Cell Biochem 109:615–623. https://doi.org/10.1002/jcb.22443
Smith DD, Tan X, Raveendran VV et al (2012) Mast cell deficiency attenuates progression of atherosclerosis and hepatic steatosis in apolipoprotein E-null mice. Am J Physiol Heart Circ Physiol 302:H2612–H2621. https://doi.org/10.1152/ajpheart.00879.2011
Sun J, Sukhova GK, Wolters PJ et al (2007) Mast cells promote atherosclerosis by releasing proinflammatory cytokines. Nat Med 13:719–724. https://doi.org/10.1038/nm1601
Afonyushkin T, Oskolkova OV, Philippova M et al (2010) Oxidized phospholipids regulate expression of ATF4 and VEGF in endothelial cells via NRF2-dependent mechanism: novel point of convergence between electrophilic and unfolded protein stress pathways. Arterioscler Thromb Vasc Biol 30:1007–1013. https://doi.org/10.1161/ATVBAHA.110.204354
Watson AD, Leitinger N, Navab M et al (1997) Structural identification by mass spectrometry of oxidized phospholipids in minimally oxidized low density lipoprotein that induce monocyte/endothelial interactions and evidence for their presence in vivo. J Biol Chem 272:13597–13607
Baker A, Saltik M, Lehrmann H et al (1997) Polyethylenimine (PEI) is a simple, inexpensive and effective reagent for condensing and linking plasmid DNA to adenovirus for gene delivery. Gene Ther 4:773–782. https://doi.org/10.1038/sj.gt.3300471
Acknowledgements
Open access funding provided by Austrian Science Fund (FWF). Authors thank Dr. Florian Gruber (Medical University of Vienna, Austria) for providing NRF2−/− mice.
Funding
The work was supported by the grants from Austrian Science Fund (P23016-B11 to T.A. and P27682-B30 to V.N.B.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All applicable national and institutional guidelines for the care and use of animals were used.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Afonyushkin, T., Oskolkova, O.V. & Bochkov, V.N. Oxidized phospholipids stimulate production of stem cell factor via NRF2-dependent mechanisms. Angiogenesis 21, 229–236 (2018). https://doi.org/10.1007/s10456-017-9590-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10456-017-9590-5