
Abstract
The simultaneous liquid chromatographic analysis of water- and fat-soluble vitamins is challenging because of their wide polarity range. Typically, water-soluble vitamins are separated and analyzed by hydrophilic interaction chromatography (HILIC) while fat-soluble vitamins are analyzed by reversed-phase liquid chromatography (RPLC). The combination of these two retention principles in a column coupling or multidimensional liquid chromatography approach seems to be a logical consequence to solve the problem. In this work, a selective comprehensive HILIC × RPLC 2D-LC approach is investigated. In this method, the polar water-soluble vitamins are resolved in the first dimension (1D) on a 2-pyridylurea mixed-mode phase operated by a HILIC gradient and the coeluted fat-soluble vitamins in the early part of the chromatogram are comprehensively transferred in ten 40-µL fractions into a second dimension (2D) separation by RPLC on a C8 core–shell column. This mode of separation is also known as high-resolution sampling. The separations in 1D and 2D were optimized systematically and the retention mechanism on the mixed-mode column interpreted by support of these chromatographic data. The solvent incompatibility of 1D HILIC and 2D RPLC conditions due to sampling of acetonitrile-rich fractions from 1D into 2D RPLC led to severe peak broadening when a direct fraction transfer was carried out. An isocratic refocusing step could partly improve the situation for the stronger retained fat-soluble vitamins. Active solvent modulation with a specifically designed valve which allows a bypass of the weak eluent from the 2D pump to the column head and dilution of the fractionated sample from the sampling loop completely solved the problem and provided perfect peak shapes and chromatographic efficiencies.












Similar content being viewed by others
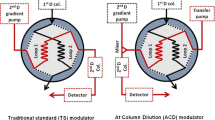
Change history
03 August 2020
In this article, we depicted the structure of the investigated fat-and water-soluble vitamins in Fig.��2.
References
Papadoyannis IN, Tsioni GK, Samanidou VF (1997) Simultaneous determination of nine water- and fat-soluble vitamins after SPE separation and RP-HPLC analysis in pharmaceutical preparations and biological fluids. J Liq Chromatogr Relat Technol 20(19):3203–3231. https://doi.org/10.1080/10826079708000485
Moreno P, Salvado V (2000) Determination of eight water- and fat-soluble vitamins in multi-vitamin pharmaceutical formulations by high-performance liquid chromatography. J Chromatogr A 870(1 + 2):207–215. https://doi.org/10.1016/S0021-9673(99)01021-3
Zbanyszek W, Buszewski B (2002) Determination of different solubility vitamins in pharmaceutical preparations. II. Methods validation. J Liq Chromatogr Relat Technol 25(8):1243–1254. https://doi.org/10.1081/JLC-120004022
Buszewski B, Zbanyszek W (2002) Determination of different solubility vitamins in pharmaceutical preparations. I. HPLC column switching. J Liq Chromatogr Relat Technol 25(8):1229–1241. https://doi.org/10.1081/JLC-120004021
Phinney KW, Rimmer CA, Thomas JB, Sander LC, Sharpless KE, Wise SA (2011) Isotope dilution liquid chromatography-mass spectrometry methods for fat- and water-soluble vitamins in nutritional formulations. Anal Chem (Washington, DC, U S) 83(1):92–98. https://doi.org/10.1021/ac101950r
Li HB, Chen F (2001) Simultaneous determination of twelve water- and fat-soluble vitamins by high-performance liquid chromatography with diode array detection. Chromatographia 54(3):270–273. https://doi.org/10.1007/bf02492256
Klejdus B, Petrlová J, Potěšil D, Adam V, Mikelová R, Vacek J, Kizek R, Kubáň V (2004) Simultaneous determination of water- and fat-soluble vitamins in pharmaceutical preparations by high-performance liquid chromatography coupled with diode array detection. Anal Chim Acta 520(1):57–67. https://doi.org/10.1016/j.aca.2004.02.027
Romain D, Nazanin A, Achim S, Michael L, Wolfgang L (2011) Simultaneous separation and analysis of water- and fat-soluble vitamins on multi-modal reversed-phase weak anion exchange material by HPLC-UV. J Sep Sci 34(7):761–772. https://doi.org/10.1002/jssc.201000793 doi
Gentili A, Caretti F(2013) Analysis of vitamins by liquid chromatography. In Elsevier Inc., pp 477–517. https://doi.org/10.1016/B978-0-12-415806-1.00018-8
Karazniewicz-Lada M, Glowka A (2016) A review of chromatographic methods for the determination of water- and fat-soluble vitamins in biological fluids. J Sep Sci 39(1):132–148. https://doi.org/10.1002/jssc.201501038
Fanali C, D’Orazio G, Fanali S, Gentili A (2017) Advanced analytical techniques for fat-soluble vitamin analysis. TrAC Trends Anal Chem 87:82–97. https://doi.org/10.1016/j.trac.2016.12.001
Santos J, Mendiola JA, Oliveira MBPP, Ibanez E, Herrero M (2012) Sequential determination of fat- and water-soluble vitamins in green leafy vegetables during storage. J Chromatogr A 1261:179–188. https://doi.org/10.1016/j.chroma.2012.04.067
Tayade AB, Dhar P, Kumar J, Sharma M, Chaurasia OP, Srivastava RB (2013) Sequential determination of fat- and water-soluble vitamins in Rhodiola imbricata root from trans-Himalaya with rapid resolution liquid chromatography/tandem mass spectrometry. Anal Chim Acta 789:65–73. https://doi.org/10.1016/j.aca.2013.05.062
Taguchi K, Fukusaki E, Bamba T (2014) Simultaneous analysis for water- and fat-soluble vitamins by a novel single chromatography technique unifying supercritical fluid chromatography and liquid chromatography. J Chromatogr A 1362:270–277. https://doi.org/10.1016/j.chroma.2014.08.003
Ni X, Xing X, Cao Y, Cao G (2014) Rapid analysis of water- and fat-soluble vitamins by electrokinetic chromatography with polymeric micelle as pseudostationary phase. J Chromatogr A 1370:263–269. https://doi.org/10.1016/j.chroma.2014.10.047
Stoll DR, Carr PW (2017) Two-dimensional liquid chromatography: a state of the art tutorial. Anal Chem 89(1):519–531. https://doi.org/10.1021/acs.analchem.6b03506
PB WJ, GA FG, J. SP (2018) Optimizing separations in online comprehensive two-dimensional liquid chromatography. J Sep Sci 41(1):68–98. https://doi.org/10.1002/jssc.201700863 doi
Stoll DR, Shoykhet K, Petersson P, Buckenmaier S (2017) Active solvent modulation: a valve-based approach to improve separation compatibility in two-dimensional liquid chromatography. Anal Chem 89(17):9260–9267. https://doi.org/10.1021/acs.analchem.7b02046
Pursch M, Wegener A, Buckenmaier S (2018) Evaluation of active solvent modulation to enhance two-dimensional liquid chromatography for target analysis in polymeric matrices. J Chromatogr A 1562:78–86. https://doi.org/10.1016/j.chroma.2018.05.059
Stoll DR, Harmes DC, Staples GO, Potter OG, Dammann CT, Guillarme D, Beck A (2018) Development of comprehensive online two-dimensional liquid chromatography/mass spectrometry using hydrophilic interaction and reversed-phase separations for rapid and deep profiling of therapeutic antibodies. Anal Chem 90(9):5923–5929. https://doi.org/10.1021/acs.analchem.8b00776
Lämmerhofer M, Richter M, Wu J, Nogueira R, Bicker W, Lindner W (2008) Mixed-mode ion-exchangers and their comparative chromatographic characterization in reversed-phase and hydrophilic interaction chromatography elution modes. J Sep Sci 31(14):2572–2588. https://doi.org/10.1002/jssc.200800178 doi
Bäurer S, Polnick S, Sánchez-Muñoz OL, Kramer M, Lämmerhofer M (2018) N-Propyl-N′-2-pyridylurea-modified silica as mixed-mode stationary phase with moderate weak anion exchange capacity and pH-dependent surface charge reversal. J Chromatogr A 1560:45–54. https://doi.org/10.1016/j.chroma.2018.05.012
Jandera P, Hájek T, Česla P (2011) Effects of the gradient profile, sample volume and solvent on the separation in very fast gradients, with special attention to the second-dimension gradient in comprehensive two-dimensional liquid chromatography. J Chromatogr A 1218(15):1995–2006. https://doi.org/10.1016/j.chroma.2010.10.095
Stoll DR, O’Neill K, Harmes DC (2015) Effects of pH mismatch between the two dimensions of reversed-phase × reversed-phase two-dimensional separations on second dimension separation quality for ionogenic compounds—I. Carboxylic acids. J Chromatogr A 1383:25–34. https://doi.org/10.1016/j.chroma.2014.12.054
Gargano AFG, Duffin M, Navarro P, Schoenmakers PJ (2016) Reducing dilution and analysis time in online comprehensive two-dimensional liquid chromatography by active modulation. Anal Chem 88(3):1785–1793. https://doi.org/10.1021/acs.analchem.5b04051
Stoll DR, Sajulga RW, Voigt BN, Larson EJ, Jeong LN, Rutan SC (2017) Simulation of elution profiles in liquid chromatography—II: Investigation of injection volume overload under gradient elution conditions applied to second dimension separations in two-dimensional liquid chromatography. J Chromatogr A 1523:162–172. https://doi.org/10.1016/j.chroma.2017.07.041
Acknowledgements
ML is grateful to Agilent Technologies for financial support through an Agilent Research Award. Stephan Buckenmaier (Agilent Technologies, Waldbronn, Germany) is gratefully acknowledged for technical support.
Funding
This study was funded by an Agilent Technologies Research Award (Grant Number Agilent Research Gift #4068).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Author ML has received a research grant from Agilent Technologies. Other authors declare no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Published in Chromatographia’s 50th Anniversary Commemorative Issue.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Bäurer, S., Guo, W., Polnick, S. et al. Simultaneous Separation of Water- and Fat-Soluble Vitamins by Selective Comprehensive HILIC × RPLC (High-Resolution Sampling) and Active Solvent Modulation. Chromatographia 82, 167–180 (2019). https://doi.org/10.1007/s10337-018-3615-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10337-018-3615-0