
Abstract
Human bocavirus (HBoV) is a novel parvovirus, often associated with respiratory tract diseases in children. This study explored the epidemiological characteristics and molecular evolution of HBoV-1 in southeastern China. Nasopharyngeal aspirates were collected from children admitted to hospital with acute respiratory tract infections. HBoV-1 was detected using real-time reverse transcription polymerase chain reaction and further characterized by complete genome sequences analysis. Among the 3,022 recruited children, 386 (12.77 %) were HBoV-1-positive and 300 (77.72 %) had co-detection with other respiratory viruses. Seasonal prevalence peaked in summer. HBoV-1 presence was significantly associated with asthma attack [odds ratio = 1.74; 95 % confidence interval: 1.30, 2.31; p < 0.001]. Similar results were obtained when either single detection or co-detection of HBoV-1 was considered, demonstrating the minor impact of co-detection on the clinical characteristics or epidemic pattern. Phylogenetic analysis based on the complete genome sequences showed that all the HBoV-1 sequences clustered together and no branch was formed that was supported by bootstrap value ≥750. The overall evolutionary rate of the complete genome of HBoV-1 was estimated at 1.08 × 10−4 nucleotide substitutions per site per year (s/s/y) [95 % highest probability density: (0.40–1.86) × 10−4 s/s/y]. Selective pressure analysis showed that all the ω-values were less than 1, suggesting that HBoV-1 was under negative selective pressure. Site-by-site analysis identified the codon site 40 of the VP1 gene under positive selection. In conclusion, our study disclosed the epidemiological and genetic dynamics of HBoV-1 epidemics in southeastern China in the most recent 3 years, the information of which might help to further improve our understanding of HBoV-1 infection and guide better surveillance and control strategies in the future.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Acute respiratory tract infections (ARTI) are a major cause of significant morbidity worldwide, especially in infants and children. Viruses are leading pathogens of ARTI. Although some viruses, such as influenza virus, respiratory syncytial virus (RSV), and human adenovirus (HAdV), are responsible for most ARTI, etiological causes remain unknown in a proportion of ARTI [1].
Human bocavirus (HBoV) is a novel parvovirus first described by Allander et al. in 2005 [2]. It has been classified in the family Parvoviridae, subfamily Parvovirinae, and genus Bocavirus by genetic organization and sequence homology. Other parvoviruses infecting humans included the adeno-associated viruses, the well-known pathogen parvovirus B19, and the recently discovered human parvovirus 4 [3]. Based on the phylogenetic analysis of viral genomes, four species of HBoVs (HBoV1–4) have been identified [4], among which HBoV-1 was mainly identified in respiratory specimens from children with acute respiratory disease [5–7]. According to previous studies, HBoV-1 has a global distribution, with prevalences varying considerably from 1.5 % to 45.7 % in ARTI patients [5, 6, 8–14]. However, its role as a causative agent of respiratory tract diseases is frequently questioned due to its concurrent detection with other potential pathogens. Possible interactions between HBoV-1 and other viruses remained rarely investigated.
In China, HBoV-1 was detected in children with positive rates ranging from 4.6 % to 31.0 % [6, 14–18]. However, HBoV-1 was co-detected frequently with other viruses in patients with respiratory or enteric infections; therefore, the impact of HBoV-1 on ARTI occurrence remains unclear. The seasonal patterns of HBoV-1 infection in China, especially the surveillance data from the most recent 4 years, are scarcely reported, probably owing to the lack of a systematic and prolonged surveillance.
This study was aimed to explore the epidemiology pattern and clinical characteristics of HBoV-1 infection in Chinese children, as well as the molecular evolutionary pattern, for HBoV-1, by performing a 4-year laboratory surveillance of ARTI cases.
Patients and methods
The laboratory surveillance was performed to detect the viral infection in children with ARTI between June 2009 and May 2013 at the Children’s Hospital of Chongqing Medical University, China. The ARTI was determined based on syndromes of cough, rhinorrhea and/or dyspnea, and/or fever of >37.5 °C. Fever caused by known chronic medical conditions was excluded. The pneumonia was defined by the presence of patchy alveolar opacities on chest radiographs, in addition to presenting symptoms of cough, dyspnea (lower chest wall indrawing), or tachypnea (in infants, >50–60 breaths/min; in older children, >40 breaths/min). Severe pneumonia was defined as pneumonia plus hypoxemia (maintained SaO2 < 92 % in air) or rising respiratory and pulse rates with clinical evidence of respiratory distress and exhaustion with or without raised PaCO2. The definition of an attack was used as an episode of respiratory symptoms which prompts an urgent consultation with a doctor, is of sufficient severity to prevent the patient working/attending school/performing domestic duties/playing, and results in increased use of anti-asthma medication.
The recruited patients had nasopharyngeal aspirates (NPA) collected the day they were hospitalized. A standardized form was applied to extract the patients’ information regarding demographic characteristics, underlying medical conditions, selected laboratory tests, radiographic findings, clinical manifestation, treatment course, and outcome. Written informed consent was obtained from their parents.
Viral RNA and DNA were extracted from 200 μl of the NPA by using QIAamp® MinElute Virus Spin Kits (Qiagen, Hilden, Germany). The cDNA was synthesized by using the SuperScript®III First-Strand Synthesis System for reverse transcription polymerase chain reaction (RT-PCR) (Invitrogen, Carlsbad, CA, USA). All samples were routinely screened for HBoV-1 with the real-time PCR method [19]. Partial HBoV-1-positive samples were whole-genome sequenced following the method previously described [20].
Genomic sequences were assembled using Lasergene’s DNA SeqMan software (version 7.1.0, DNA Star Inc., Madison, WI, USA). Previously published GenBank sequences of HBoV-1 were used for comparison. All alignments and phylogenetic tree construction were performed by the neighbor-joining (NJ) method with 1,000 bootstrap replicates using CLC Genomic Workbench 5.5. Similarities between strains were calculated by using BioEdit version 7.13 (North Carolina State University, Raleigh, NC, USA; http://www.mbio.ncsu.edu/bioedit/bioedit.html). The substitution rates, the divergence over time, and the time to the most recent common ancestor (TMRCA) of the current HBoVs were estimated using a Bayesian Markov chain Monte Carlo (MCMC) approach, as implemented in the Bayesian Evolutionary Analysis Sampling Trees (BEAST) package version 1.8.0.
The value of ω and the individual site-specific selection pressure were measured by using the single likelihood ancestor counting (SLAC), fixed effects likelihood (FEL), internal fixed effects likelihood (IFEL), and fast unconstrained Bayesian approximation (FUBAR) methods implemented in the hypothesis testing using phylogenies (HyPhy) package performed at Datamonkey online (http://www.datamonkey.org/). The sites were confirmed when they were selected by more than two methods. A single breakpoint recombination (SBP) test with both Akaike information criterion (AIC) and Bayesian information criterion (BIC) calculation was conducted. The overall ω-value and 95 % confidence interval (CI) were estimated based on NJ trees. Selective pressure was defined as neutral evolution with ω = 1, purifying or negative selection with ω < 1, and positive selection with ω > 1. A p-value of less than 0.10 was considered as the threshold for strong evidence of selection in SLAC, FEL, and IFEL, and posterior probability ≥0.9 in FUBAR, respectively.
Statistical analysis was done by the Chi-square test for categorical variables. Logistic regression models were used to explore the factors associated with higher HBoV-1 detection and to determine the association between clinical characteristics and HBoV-1 infection, with clinical disease as the dependent variable and HBoV-1 infection as the independent variable. Odds ratios (ORs) and their 95 % CIs were estimated using maximum likelihood methods. Statistical significance was set at a p-value of less than 0.05. All the tests were performed by using SAS 9.13 (SAS Institute Inc., Cary, NC, USA).
Results
From June 2009 to May 2013, 3,022 NPA samples of hospitalized infants and children were collected. The median age was 8 months (range 1 months to 16 years), and 2,016 (66.71 %) were boys. The median delay before hospital entrance was 9 days (range 1–60 days). The hospitalization duration [mean ± standard deviation (SD)] was 7 ± 4 days. Among the 3,022 children, 450 (14.90 %) displayed severe pneumonia and 25 (0.83 %) had severe asthma. In total, 386 (12.77 %) children were positive for HBoV-1. The children aged between 7 and 24 months had higher detection rates of HBoV-1 than children of the other age groups (Table 1). When compared with the >25 months group, the ORs for HBoV-1-positive detection were 1.92 (95 % CI: 1.32, 2.80) for the 7–12 months group and 2.57 (95 % CI: 1.75, 3.76) for the 13–24 months group after adjustment for the confounding factors. The HBoV-1 detection rate was significantly higher in children having asthma attack (OR = 1.74; 95 % CI: 1.30, 2.31).
Among the 386 children with positive results, 86 (22.28 %) had single HBoV-1 detection and the remaining 300 (77.72 %) were co-detected with other viruses, including human rhinovirus, enterovirus, human adenovirus, influenza virus, metapneumovirus, RSV, parainfluenza virus, and coronaviruses. The most frequently presented co-detection occurred between HBoV-1 with parainfluenza virus (30.05 %), followed by with RSV (28.50 %) and influenza virus (26.17 %). The patients with single detection had a median age of 11 months (range 1 months to 13 years) and 265 (68.65 %) were boys, which were comparable with the subjects with HBoV-1 detection as a whole. The patients with HBoV-1 detection presented clinical manifestations of cough (291, 97.00 %), sputum (226, 75.33 %), and diarrhea (91, 30.33 %). These syndromes were presented with highly comparable frequencies among patients with HBoV-1 single detection [cough (83, 96.51 %), sputum (63, 73.26 %), and diarrhea (26, 30.23 %)]. The occurrence of severe pneumonia and severe asthma were identified in 53 (13.73 %) and 4 (1.04 %) HBoV-1-detected patients, among whom 12 and four had single detection, respectively, which is comparable with the patients with co-detection.
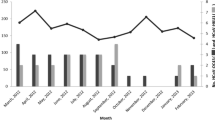
The monthly positive rates of HBoV-1 displayed specific seasonal distribution, as plotted in Fig. 1. One seasonal peak was observed in summer, demonstrated by remarkably higher infection rates during May–August. An exception was noticed in 2010, when another low peak was also observed in the winter season from November to December. When the HBoV-1 single infection was plotted, a similar seasonal pattern was derived, with a clearly increased activity of HBoV demonstrated in summer.

Distribution of monthly positive rates of HBoV-1 infection during 2009–2012
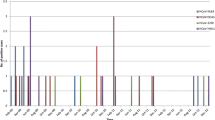
The phylogenetic tree based on 5,157 bp of the whole genome sequences showed that all the HBoV-1 sequences, regardless of whether they were single detection or co-detection, clustered together and no other branch was formed that was supported by bootstrap value ≥750 (Fig. 2). The identities for the HBoV-1 complete genome were 99.4–100 %. The phylogenetic trees were similarly established for the NP1, NS1, VP1, and VP2 genes (Supplemental Figure 1). The identities of HBoV-1 were 97.8–100 % for the NP1 gene, 99.6–100 % for NS1, 98.9–100 % for VP1, and 98.8–100 % for VP2. All the HBoV-1 viruses in the investigated localities were dominated by a single viral lineage, which had been dominant in most parts of China in recent years. Neither a spatial nor a temporal specific branch was formed based on the whole-genome analysis.

Phylogenetic tree constructed based on 5,157 bp of the complete genome sequences of HBoV-1 with the neighbor-joining method using CLC Main Workbench 5.5. The strain name in this study began with CQ. HBoV-1 is represented by the black line, HBoV-2 by the blue line, HBoV-3 by the green line, and HBoV-4 by the red line
The overall evolutionary rate was estimated at 1.08 × 10−4 nucleotide substitutions per site per year (s/s/y) [95 % highest probability density (HPD): (0.40–1.86) × 10−4 s/s/y] based on the complete genome analysis. The highest evolutionary rate was derived from the NP gene, while the lowest was from the NS gene sequences (Table 2).
The SBP test showed no evidence of recombination for any of the four genes. Selective pressure analysis showed that all the ω-values were less than 1 (Table 3), indicating that these HBoV-1 genes were under negative selective pressure and the evolutionary process is shaped mainly by purifying selection. The NP gene had the largest global rate (0.23, 95 % CI: 0.15–0.33) among the four genes (range 0.08–0.23), suggesting that it is subject to greater selective pressure than the other three genes.
Site-by-site analysis of the full data set consisting of 206 HBoV-1 coding sequences identified only one site on the VP1 gene (codon site 40) undergoing significant positive selection and a number of negatively selected sites. A codon-based maximum likelihood reconstruction of the evolutionary history of codon site 40 under positive selection was carried out (Supplemental Figure 2). The result disclosed that the substitution has occurred mostly in the strains from Asia, especially in China, suggesting that HBoV-1 strains from China undertake more selective pressure than those from other regions.
Discussion
In the current study, we have identified the clinical characteristics and temporal patterns of HBoV-1 in southeastern China through a 4-year surveillance. No role of HBoV-1 in severe pneumonia occurrence was observed, regardless of whether they were single detection or co-detection. In contrast, a significant association between HBoV and asthma was determined, suggesting a possible causal relationship. Based on the genetic analysis, HBoV-1 in southeastern China have similar mean evolutionary rates as other species of the family Parvoviridae and one positive selection site was also disclosed which differs from HBoV of other origins.
According to our results, the prevalence of HBoV-1 in pediatric ARTI patients is higher than those from other hospital-based studies [2, 6, 21, 22], whereas it was lower than that previously detected in persistently wheezing children [16]. HBoV-1 was commonly co-detected with a median rate of 42.5 %, and the most frequent co-detected viruses included rhinovirus, enteroviruses, and influenza [23]. We demonstrated a co-detection rate of 77.72 %, which is similar to the high proportion of co-detection as previously studied [24], yet significantly higher than those from other studies [9, 12]. These discrepancies could be explained by the inherent differences in the population subsets, detection methods, as well as the epidemiological variability relating to geographic origin and climatic factors.
Seasonal circulations of HBoV-1 vary, with most previous studies reporting HBoV-1 peak attained during the winter season [9, 22, 23]. Choi et al. found a higher frequency of HBoV-1 between May and July over a 5-year period among children with ARTI in Korea [25], which, interestingly, is consistent with our results displaying a summer peak. Although this temporal pattern coincided with the epidemic peaks of influenza circulation in southern China (http://www.cnic.org.cn/chn/), the single detection of HBoV also demonstrated a similar temporal pattern after excluding the effect from other respiratory pathogens, therefore indicating the actual seasonal pattern predominantly occurring in summer. An unexpected low peak in the winter of 2010 was demonstrated, probably caused by the extremely cold weather in Chongqing during the winter of that year (http://www.cqmb.gov.cn/ecms/stny/nyqx/2011-03-11/3683.html).
The relative importance of HBoV-1 as a causative pathogen for viral respiratory illnesses has not been determined, but it has been associated with respiratory diseases ranging from upper respiratory tract disease to bronchiolitis, and lower respiratory tract diseases, including pneumonia [11, 15, 26]. Vallet et al. found a high concordance between HBoV-1 detection and exacerbation of asthma requiring eventual hospitalization [27]. In our study, HBoV-1 detection was not associated with pneumonia. Instead, HBoV-1 tended to cause more upper respiratory tract infections, and its infection was also related with asthma exacerbation. Although a causal relationship between HBoV-1 infection and asthma cannot be concluded based on the statistical results, their significant association suggest the need for further investigation. Prospective longitudinal studies will hopefully increase our understanding of what, if any, role HBoV plays in asthma.
The epidemiology of the HBoV-1 circulation in China has been previously studied; however, the viral gene sequence data acquired from these studies were rare and insufficient to reveal any temporal or spatial characteristics or any evolutionary dynamics. Analysis of the genomic and epidemiological data of HBoV-1 from the current study, as well as those available from different localities, revealed limited heterogeneity, and high conservation was demonstrated from both spatial and temporal perspectives. Zehender et al. reported that the estimated mean evolutionary rate of the HBoV-1 VP2 gene was 8.6 × 10−4 s/s/y using 48 non-recombinant isolates [28], demonstrating that HBoV-1 was characterized by a rapid evolution. The rate identified in the current study is slower, which might be partially caused by the sample size of analyzed HBoV-1 sequences. A few of the previous studies explored the estimated evolutionary rates of parvoviruses in carnivore parvoviruses (7.9 × 10−5–1.7 × 10−4 s/s/y) [29] and human parvovirus B19 (1.1 × 10−4–2.6 × 10−4 s/s/y) [30]. The estimated mean evolutionary rates of the complete genome, VP1, VP2, and NS genes of HBoV-1 were similar to those of carnivore parvoviruses and human parvovirus B19. The NS gene was highly conservative among the four genes. Surprisingly, the NP gene showed a slightly higher substitution rate than the other genes.
The icosahedral capsid consists of two structural proteins, VP1 and VP2, which are identical except for a certain number of amino acids at the amino-terminal end of the VP1 protein, the so-called VP1 unique region (VP1U). A conserved phospholipase A2 (PLA2) motif, which resembles the catalytic motif of secreted PLA2 (sPLA2), has been identified in the VP1U of most parvoviruses [31]. For HBoV-1, VP1 and VP2 have the same sequence of 542 aa at the C termini, while VP1 has an additional 129 aa peptide at the N-terminus. One study indicated that the VP1U of HBoV-1 also had sPLA2-like enzymatic activity, and these residues are crucial for its sPLA2-like activity [32]. Mutation of one of the amino acids (21Pro, 41His, 42Asp, or 63Asp) in the VP1U almost eliminated the sPLA2 activity of the HBoV-1 VP1U. Interestingly enough, one positive selection site (codon 40) was identified near codon 41 and 42 in the current study, which indicated a high probability of VP1U undergoing selective pressure. In spite of this finding, the associations between the positive selection site and sPLA2 activity or amplification of DNA need further investigation.
In conclusion, our study, for the first time, disclosed the epidemiological characteristics and genetic dynamics of HBoV-1 in southeastern China in recent years. The disclosure of the temporal pattern of HBoV infection provided the season when HBoV infection could be highly suspected and surveillance should be strengthened. The current data also demonstrated that HBoV was co-detected with a high prevalence; therefore, the sole presence of HBoV in patients should be interpreted carefully as to its causal role in causing disease. This information might help to further improve our understanding of HBoV-1 infection and guide better surveillance and control strategies in the future, especially among children.
References
Pavia AT (2011) Viral infections of the lower respiratory tract: old viruses, new viruses, and the role of diagnosis. Clin Infect Dis 52(Suppl 4):S284–S289
Allander T, Tammi MT, Eriksson M, Bjerkner A, Tiveljung-Lindell A, Andersson B (2005) Cloning of a human parvovirus by molecular screening of respiratory tract samples. Proc Natl Acad Sci U S A 102(36):12891–12896
Allander T (2008) Human bocavirus. J Clin Virol 41(1):29–33
Peltola V, Söderlund-Venermo M, Jartti T (2013) Human bocavirus infections. Pediatr Infect Dis J 32(2):178–179
Guido M, Quattrocchi M, Campa A, Zizza A, Grima P, Romano A, De Donno A (2011) Human metapneumovirus and human bocavirus associated with respiratory infection in Apulian population. Virology 417(1):64–70
Imamura T, Fuji N, Suzuki A, Tamaki R, Saito M, Aniceto R, Galang H, Sombrero L, Lupisan S, Oshitani H (2011) Enterovirus 68 among children with severe acute respiratory infection, the Philippines. Emerg Infect Dis 17(8):1430–1435
Christensen A, Nordbø SA, Krokstad S, Rognlien AG, Døllner H (2010) Human bocavirus in children: mono-detection, high viral load and viraemia are associated with respiratory tract infection. J Clin Virol 49(3):158–162
Allander T, Jartti T, Gupta S, Niesters HG, Lehtinen P, Osterback R, Vuorinen T, Waris M, Bjerkner A, Tiveljung-Lindell A, van den Hoogen BG, Hyypiä T, Ruuskanen O (2007) Human bocavirus and acute wheezing in children. Clin Infect Dis 44(7):904–910
Bastien N, Chui N, Robinson JL, Lee BE, Dust K, Hart L, Li Y (2007) Detection of human bocavirus in Canadian children in a 1-year study. J Clin Microbiol 45(2):610–613
Jacques J, Moret H, Renois F, Lévêque N, Motte J, Andréoletti L (2008) Human Bocavirus quantitative DNA detection in French children hospitalized for acute bronchiolitis. J Clin Virol 43(2):142–147
Ma X, Endo R, Ishiguro N, Ebihara T, Ishiko H, Ariga T, Kikuta H (2006) Detection of human bocavirus in Japanese children with lower respiratory tract infections. J Clin Microbiol 44(3):1132–1134
Smuts H, Hardie D (2006) Human bocavirus in hospitalized children, South Africa. Emerg Infect Dis 12(9):1457–1458
Lindner J, Modrow S (2008) Human bocavirus—a novel parvovirus to infect humans. Intervirology 51(2):116–122
Zheng LS, Yuan XH, Xie ZP, Jin Y, Gao HC, Song JR, Zhang RF, Xu ZQ, Hou YD, Duan ZJ (2010) Human bocavirus infection in young children with acute respiratory tract infection in Lanzhou, China. J Med Virol 82(2):282–288
Chuang CY, Kao CL, Huang LM, Lu CY, Shao PL, Lee PI, Chang LY (2011) Human bocavirus as an important cause of respiratory tract infection in Taiwanese children. J Microbiol Immunol Infect 44(5):323–327
Deng Y, Liu E-M, Zhao X-D, Ding Y, Li Q-B, Luo Z-X, Wang L-J, Huang Y, Yang X-Q (2007) Clinical characteristics of 12 persistently wheezing children with human bocavirus infection. Chin J Pediatr 45(10):732–735
Qu XW, Duan ZJ, Qi ZY, Xie ZP, Gao HC, Liu WP, Huang CP, Peng FW, Zheng LS, Hou YD (2007) Human bocavirus infection, People’s Republic of China. Emerg Infect Dis 13(1):165–168
Wang W, Cavailler P, Ren P, Zhang J, Dong W, Yan H, Mardy S, Cailhol J, Buchy P, Sheng J, Fontanet A, Deubel V (2010) Molecular monitoring of causative viruses in child acute respiratory infection in endemo-epidemic situations in Shanghai. J Clin Virol 49(3):211–218
Koskenvuo M, Möttönen M, Waris M, Allander T, Salmi TT, Ruuskanen O (2008) Human bocavirus in children with acute lymphoblastic leukemia. Eur J Pediatr 167(9):1011–1015
Chieochansin T, Chutinimitkul S, Payungporn S, Hiranras T, Samransamruajkit R, Theamboolers A, Poovorawan Y (2007) Complete coding sequences and phylogenetic analysis of human bocavirus (HBoV). Virus Res 129(1):54–57
Arnold JC, Singh KK, Spector SA, Sawyer MH (2006) Human bocavirus: prevalence and clinical spectrum at a children’s hospital. Clin Infect Dis 43(3):283–288
Cao CQ, Li YN, Jin Y, Xie ZP, Gao HC, Zhou QH, Gao XQ, Zhang YT, Zhang J, Duan ZJ (2011) Detection and clinical characteristics analysis of human bocavirus 1–3 in children for acute respiratory infection in Lanzhou area. Chin J Exp Clin Virol 25(1):5–7
Chow BD, Esper FP (2009) The human bocaviruses: a review and discussion of their role in infection. Clin Lab Med 29(4):695–713
Fry AM, Lu X, Chittaganpitch M, Peret T, Fischer J, Dowell SF, Anderson LJ, Erdman D, Olsen SJ (2007) Human bocavirus: a novel parvovirus epidemiologically associated with pneumonia requiring hospitalization in Thailand. J Infect Dis 195(7):1038–1045
Choi EH, Lee HJ, Kim SJ, Eun BW, Kim NH, Lee JA, Lee JH, Song EK, Kim SH, Park JY, Sung JY (2006) The association of newly identified respiratory viruses with lower respiratory tract infections in Korean children, 2000–2005. Clin Infect Dis 43(5):585–592
Kahn JS, Kesebir D, Cotmore SF, D’Abramo A Jr, Cosby C, Weibel C, Tattersall P (2008) Seroepidemiology of human bocavirus defined using recombinant virus-like particles. J Infect Dis 198(1):41–50
Vallet C, Pons-Catalano C, Mandelcwajg A, Wang A, Raymond J, Lebon P, Gendrel D (2009) Human bocavirus: a cause of severe asthma exacerbation in children. J Pediatr 155(2):286–288
Zehender G, De Maddalena C, Canuti M, Zappa A, Amendola A, Lai A, Galli M, Tanzi E (2010) Rapid molecular evolution of human bocavirus revealed by Bayesian coalescent inference. Infect Genet Evol 10(2):215–220
Shackelton LA, Parrish CR, Truyen U, Holmes EC (2005) High rate of viral evolution associated with the emergence of carnivore parvovirus. Proc Natl Acad Sci U S A 102(2):379–384
Shackelton LA, Holmes EC (2006) Phylogenetic evidence for the rapid evolution of human B19 erythrovirus. J Virol 80(7):3666–3669
Lupescu A, Bock CT, Lang PA, Aberle S, Kaiser H, Kandolf R, Lang F (2006) Phospholipase A2 activity-dependent stimulation of Ca2+ entry by human parvovirus B19 capsid protein VP1. J Virol 80(22):11370–11380
Qu XW, Liu WP, Qi ZY, Duan ZJ, Zheng LS, Kuang ZZ, Zhang WJ, Hou YD (2008) Phospholipase A2-like activity of human bocavirus VP1 unique region. Biochem Biophys Res Commun 365(1):158–163
Acknowledgments
This study was supported by the China Mega-Project for Infectious Diseases grant (2013ZX10004-202) and the Natural Science Foundation of China (81222037).
Conflict of interest
The authors do not have any commercial or other association that might pose a conflict of interest. All authors read and approved the final manuscript.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Article note
Qing-Bin Lu and Ying Wo contributed equally to this work.
Electronic supplementary material
Below are the links to the electronic supplementary material.
Supplemental Figure 1
Phylogenetic tree were constructed based on the four genes sequences of HBoV-1 with the Neighbor-Joining method using CLC Main Workbench 5.5. The strain name in this study began with CQ. HBoV-1 was represented by the black line; HBoV-2 by the blue line; HBoV-3 by the green line; HBoV-4 by the red line. (GIF 6.32 mb)
Supplemental Figure 2
Inferred evolutionary history at sites under positive selection. The trees are rooted to AB480170_JAPAN2009 and ancestral states are inferred by ASR implemented in Datamonkey. Tree branches are highlighted in red under positive pressure. (GIF 7.22 mb)
Rights and permissions
About this article

Cite this article
Lu, QB., Wo, Y., Wang, HY. et al. Epidemic and molecular evolution of human bocavirus in hospitalized children with acute respiratory tract infection. Eur J Clin Microbiol Infect Dis 34, 75–81 (2015). https://doi.org/10.1007/s10096-014-2215-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10096-014-2215-7