
Abstract
Thermal modification of wood is an environment-friendly alternative method for improving several properties of wood without the use of chemicals. This paper deals with the examination of color and chemical changes in spruce (Picea abies L.) and oak wood (Quercus robur F.) that occur due to thermal treatment. The thermal modification was performed at 160, 180, and 210 °C according to thermowood process. The color changes were measured by the spectrophotometer and described in the L*a*b* color system. Chemical changes were examined by wet chemistry methods, infrared spectroscopy and liquid chromatography. During the experiment, oak samples showed smaller color changes than spruce samples at all temperature values. During thermal modification, the content of cellulose, lignin, and extractives increases; however, the hemicellulose content drops by 58.85% (oak) and by 37.40% (spruce). In addition to deacetylation, new carbonyl and carboxyl groups are formed as a result of oxidation. Bonds in lignin (mainly β-O-4) and methoxyl groups are cleaved, and lignin is condensed at higher temperatures.
Similar content being viewed by others
Introduction
Wood is one of the oldest and the most abundant natural composite materials on Earth. In its structure, wood is a natural composite material formed from a chemical complex of cellulose, hemicelluloses, and extractives. These polymeric substances are not uniformly distributed within the wood cell wall and their concentration changes from one morphological region to another [1,2,3,4]. Wood as a universal and renewable material is currently used in many areas, such as construction and tiling materials, furniture production, fuel, and chemical processing (pulp, paper, bioethanol, etc.) [5].
Its color can be an indicator of the quality of thermally modified wood, and thus determine its final use on the market. The color characteristics mostly depend on the specific chemical composition of the wood in interaction with the light [6]. From a chemical viewpoint, wood is composed of cellulose (40–50%), hemicelluloses (20–30%), lignin (20–30%) and extractives (3–10%). These amounts vary in different wood species [7]. High temperatures during thermal modification cause the degradation of hemicelluloses and lignin, resulting in the production of water, carbon dioxide, formic acid, acetic acid, and other substances that can be involved in condensation reactions, and chromophore groups are formed [8]. The color of the wood darkens during thermal modification; however, the degree of color change depends on the conditions of the thermal modification, especially the temperature and duration of the treatment [9,10,11]. Thermal processes also affect some physical and mechanical properties (dimensional stability, hygroscopicity, durability, MOR—modulus of rupture, MOE—modulus of elasticity, etc.) [12,13,14,15,16]. Interest in thermally modified wood increases especially because of its higher durability compared to untreated wood and also because of the legal restrictions relating to use of toxic substances [17].
In practice, different types of wood are used under different technological conditions, depending on the wood species and its final use. Thermal modification temperatures usually range between 160 and 260 °C, but differences between individual modification methods consist of the use of a gaseous environment (nitrogen, steam, etc.), ambient humidity, the use of oil, etc. [18,19,20]. The modified wood has better properties for exterior and interior use, especially preservation against biotic factors, etc. [21, 22].
The aim of this paper was to determine the effect of the thermal modification temperature on color changes and the chemical composition of spruce and oak wood in terms of their exterior and interior use, using the thermowood process.
Experimental
Materials
The experiment consisted of two basic sets of test samples (spruce—Picea abies L., and oak—Quercus robur F.). Each set was divided into subsets according to the thermal modification temperature (20, 160, 180, and 210 °C) and consisted of ten 100 (tangential) × 20 (radial) × 200 (longitudinal) mm samples (Figs. 1, 2). These samples were air-conditioned (RH 65 ± 3% and temperature 20 ± 2 °C) for more than 6 months to achieve an equilibrium moisture content (EMC) of 12%.

Samples of the original and thermally modified spruce wood

Samples of the original and thermally modified oak wood
Methods
Thermal modification
-
1.
Heating and drying: In this phase, the temperature in the oven is increased to approximately 100 °C with steam. The drying medium is hot air. Throughout this stage, the wood is dried to approximately zero moisture content.
-
2.
Thermal modification: In the second phase, the temperature is increased to 185–230 °C for 2–3 h. The temperature and duration of the effects are given by the thermowood product class (Thermo-S and Thermo-D). The letter ‘S’ in ‘Thermo-S’ stands for ‘stability’ and it is classified as relatively durable according to the standard EN 113 standard [23]. The letter ‘D’ in ‘Thermo-D’ stands for ‘durability’ and it is classified as durable according to the EN 113 standard [23].
-
3.
Cooling and climatization: In the third phase, the wood is gradually cooled to 80–90 °C, and the humidity is stabilized so that the final moisture content stabilizes at 4–7%.
These three-phase processes are presented in Fig. 3 for spruce wood and in Fig. 4 for oak wood.

Thermal modification process of spruce wood

Thermal modification process of oak wood
The wood samples were modified in the thermal chamber, type S400/03 (LAC Ltd., Rajhrad, Czech Republic).
Color measurement
After thermal modification, samples were relaxed for 3 h in in a chamber APT Line II (Binder, Tuttlingen, Germany) to an EMC of 8% at a temperature of 20 °C and relative humidity (RH) of 42%.
Color values of wood surface were measurement by CM-700D spectrophotometer (Konica Minolta, Osaka, Japan) (10° standard observer, D65 standard illuminate, color difference format ΔE*ab). To quantify the color, we used the three-dimensional colorimetric system L*a*b*. This color space consists of three mutually perpendicular axes: axis L* determines the lightness from 0 (black) to 100 (white), axis a* determines the ratio of red (positive) to green (negative), and axis b* specifies the ratio of yellow (positive) to blue (negative). To ensure the most accurate results the color was measured on three specific places of each sample before and after thermal modification. To assess the difference between two colors, we used a total color difference ΔE* (expressing the distance between two points in the L*a*b* system). The ΔE* was evaluated according to [24,25,26] and calculated using the formula (Eq. 1):
where ΔL*, Δa*, and Δb* are differences in individual axes (difference between the values measured after thermal modification of the sample and the reference sample). Untreated samples at 20 °C were chosen as references for each treatment. The measured color values were evaluated in Statistica 10 software by a two-factor analysis, and the analysis factors were the wood species and the temperature.
Chemical analyses
Reference (untreated, denoted as 20 °C) and thermally modified samples were mechanically disintegrated to sawdust, and the fraction of the size 0.5–1.0 mm was extracted in the Soxhlet apparatus with a mixture of ethanol and toluene (2:1, v/v) for 8 h, according to the NREL (National Renewable Energy Laboratory) laboratory analytical procedure [27]. The lignin content was determined according to the NREL laboratory analytical procedure based on Klason method [28]. Briefly, the samples were hydrolyzed in a two-stage process. In the first stage, 72% (w/w) H2SO4 at a temperature of 30 °C was used for 2 h, and in the second stage, the samples were refluxed after dilution to 4% (w/w) H2SO4 for 4 h. Acid-insoluble residue was filtered, and dried at 105 ± 3 °C until a constant weight. Holocellulose was determined using the Wise et al. [29], and cellulose by the Seifert method using a mixture of acetylacetone, dioxane, and hydrochloric acid (6:2:1.5) under reflux for 30 min for delignification of wood samples [30]. Hemicelluloses were calculated as the difference between holocellulose and cellulose. All measurements were performed on four replicates per sample. The data were presented as percentages of oven-dry weight (ODW) per unextracted wood.
Analysis of saccharides
The qualitative and quantitative analyses of saccharides were carried out using high-performance liquid chromatography (HPLC) according to the NREL laboratory analytical procedure [28]. The samples were hydrolyzed in a two-stage process. In the first stage, 72% (w/w) H2SO4 at a temperature of 30 °C was used for 1 h, and in the second stage, the formed oligomers were hydrolyzed to monosaccharides after a dilution to 4% (w/w) H2SO4 at a temperature of 121 °C for 1 h. The analyses were performed with Agilent 1200 HPLC chromatograph (Agilent Technologies, Santa Clara, USA), equipped with an Aminex HPX-87P column (Bio-Rad Laboratories, Hercules, USA) at a temperature of 80 °C, and a mobile phase (deionized water) flow rate of 0.6 ml min−1. Two samples were hydrolysed in parallel, and each hydrolysate was analyzed twice.
FT-IR analysis of the wood’s main components
Fourier transform-infrared spectroscopy (FT-IR) spectra of the wood surface were recorded on the Nicolet iS10 FT-IR spectrometer, equipped with Smart iTR using an attenuated total reflectance (ATR) sampling accessory attached to a diamond crystal (Thermo Fisher Scientific). The spectra were from 4000 to 650 cm−1 at a spectral resolution of 4 cm−1, and 64 scans were used. Measurements were performed on four replicates per sample.
Results and discussion
In the color changes, value on axis L* (lightness) showed the most significant change out of all the evaluated parameters (Fig. 5; Table 1). Thermally modified samples of both wood species showed a very significant drop in values due to the increasing temperature of the thermal modification. After thermal modification at 210 °C, all the tested sets of samples had a reduced of L* value by approximately 50% relative to the values obtained from untreated samples. Our values are comparable to those reported for oak, birch, pine, black alder, and beech wood [31, 32].

Graphical representation of the L* values’ dependence on the temperature
The results of Duncan’s test shown in Table 2 show that the difference in all L* values affected by the applied temperatures was statistically very different, with a significance level of p = 0.001.
The value of a* (ratio of red to green) changed more significantly in spruce samples than in oak samples (Table 3). The tested wood species also exhibited different change tendencies with respect to the temperature (Fig. 6).

Graphical representation of the a* values dependence on the temperature
The effect of the temperature applied during thermal modification on the values of a* in spruce wood was very significant in all monitored cases, with a significance level of p = 0.001 (Table 4). In the case of oak wood, the difference in the measured values of coordinate a* at temperatures of 20 and 160 °C was not observed; in this case, the significance level was p = 0.718. At other temperatures, a significant difference in the measured values was confirmed (Table 4).
The value of b* (the ratio of yellow to blue) achieved the same values in all tested samples before thermal modification, but their subsequent changes exhibited different tendencies and intensities (Fig. 7; Table 5). Oak wood exhibited the same decreasing tendency of changes with respect to the thermal modification temperatures. In spruce wood, the value of coordinate b* significantly increased in the case of thermal modification at 160 and 180 °C, by up to about 35%. This increase was followed by a significant decrease of up to 45% during thermal modification at 210 °C.

Graphical representation of the b* values’ dependence on the temperature
The results of Duncan’s test shown in Table 6 show that the difference in all b* coordinate values affected by the applied temperatures was statistically very different, with a significance level of p = 0.001.
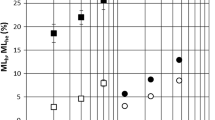
The total color difference ΔE* varies greatly with the rising thermal modification temperature (Fig. 8, Table 7). The ΔE* was also affected by the type of modified wood species. Spruce and oak achieved very similar results up to a temperature of 180 °C, but at a temperature of 210 °C, oak exhibited a smaller total color change.

Graphical representation of the ΔE* values’ dependence on the temperature
The results of Duncan’s test shown in Table 8 show that the difference in all total color change values affected by the applied temperatures was statistically very different, with a significance level of p = 0.001.
The total color difference ΔE* varied considerably in the dependence of the temperature, and it was also affected by the wood species. At 180 °C, spruce and oak samples had very similar results (the woods have the highest color stability at thermal modification temperatures of 180 and 210 °C); however, at 210 °C, the oak sample had a smaller total color change.
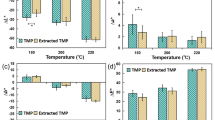
Thermal modification causes decrease of polysaccharide amount in both wood species, mostly by the degradation of hemicelluloses. The amount of thermally more stable polymers (lignin, cellulose) in modified wood slightly increases. When wood is exposed to high temperatures, certain extractives decompose, but new extractives are created due to decomposition of main components, and their total quantity in thermally modified wood increases (Table 9). According to Nuopponen et al. [33] and Hill [34], high temperatures cause the decomposition of ellagitannins in oak wood, resulting in the formation of ellagic acid. In pine wood at temperatures of 100–160 °C, fats and waxes moved to the surface of the wood, and at 180 °C they decomposed. Resin acids decomposed at temperatures above 200 °C. In our experiment, the least stable component of wood during thermal treatment was hemicelluloses. Their amount decreases in modified wood, accompanied by a relative increase of cellulose and lignin (Table 9). Hemicelluloses in spruce wood dropped by 37.40% at 210 °C, and by 58.85% in oak wood, which suggests that hemicelluloses in coniferous wood are thermally more stable than hemicelluloses in deciduous wood. Our results are consistent with the findings that hemicelluloses in deciduous wood species decompose at lower temperatures, and the decomposition temperature also reduces the amount of acetyl groups [35, 36].
The greatest degradation of spruce saccharides under thermal treatment was found in mannose, to a smaller extent in xylose, and the least in galactose [37]. In our experiment, similar results of saccharide degradation in spruce wood were found (mannose > xylose ~ galactose > arabinose). In oak wood, the least stable saccharides were galactose, arabinose, and mannose, almost completely decomposed by thermal modification (Table 10).
Chemical changes in deciduous and coniferous wood species during thermal modification depend on many conditions, especially the temperature, duration, and atmosphere, taking into account that many competitive reactions occur simultaneously, and they particularly depend on the prevailing experimental conditions. For this reason, several authors have found different results during the thermal modification of wood. The C=O linkage exhibits strong absorptions in FT-IR spectra between 1750 and 1700 cm−1, and the precise wavenumber depends on the functional group (carboxylic acid: at about 1725–1700 cm−1; ester, ketone: 1725–1705 cm−1, aldehyde: 1740–1720 cm−1) and of its structural location, with lower wavenumbers for conjugated C=O [38]. In our conditions, the increase and shifting absorbance at around 1730 cm−1 to smaller wavenumbers with increasing treatment severity may be due to an increase of carbonyl or carboxyl groups in lignin or carbohydrates by oxidation (Figs. 9, 10, 11, 12). In thermally treated beech, spruce, and pine wood, the band increased at 1730–1732 cm−1 in deciduous wood species; in coniferous wood species the opposite trend was observed [39]. During the thermal modification of eucalyptus and pine wood, Esteves et al. [38] first observed a decline, followed by a rise in the peak at 1730 cm−1. This decline may be due to the cleavage of acetyl groups, especially hemicelluloses; on the other hand, carbonyl and carboxyl groups can form due to oxidation [38, 40].

FT-IR spectra of spruce wood before and after thermal modification

Differential FT-IR spectra of spruce wood

FT-IR spectra of oak wood before and after thermal modification

Differential FT-IR spectra of oak wood
Absorbance at 1655 cm−1 (conjugated C–O in quinones coupled with C=O stretching in various groups) [41, 42] decreases in spruce wood treated at the temperature of 180 °C, conversely it increases in oak wood at the temperature of 210 °C. This phenomenon can be caused by the cleavage of acetyl groups from hemicelluloses, resulting in the release of acetic acid, which occurs during the degradation of less ordered saccharides, as well as during the degradation and condensation reactions of lignin [42].
Bands at 1505 cm−1 (C=C stretching of the aromatic skeletal vibrations in lignin) have different bands from 1504 to 1512 cm−1, similar to eucalyptus wood [39], which may be caused by the cleavage of methoxyl groups or the loss of syringyl units, as these are less condensed by C–C bonds than guaiacyl and more easily degraded at high temperatures [39, 43, 44]. In spruce wood, the absorbance decreased; a similar trend was observed in spruce and pine wood [38]. Some authors reported the increase of absorbance around 1505 cm−1 in thermally treated beech, teak, and oak wood [38, 43, 45]. On the other hand, in our experiment, the decrease of absorbance at 1505 cm−1 was observed, similar to eucalypt wood [37]. The decrease in bands at 1026–1030 cm−1 (C–O ester stretching vibrations in methoxyl and β-O-4 linkages in lignin) in both wood species suggests the cleavage of β-O-4 bonds and the cleavage of methoxylates from lignin. A similar decrease was found in teak and linden wood [43, 44].
From the obtained results it can be concluded that heat treatment caused significant changes in the chemical composition and structure of both spruce and oak wood, by changing polysaccharides and lignin. Due to bond cleavage and subsequent oxidation and dehydration in polysaccharides new chromophores are formed (particularly, carbonyl and carboxyl groups). Bonds in lignin (mainly β-O-4) and methoxyl groups are cleaved, and lignin is condensed at higher temperatures. These changes in wood’s main components and higher amounts of extractives in thermally treated wood can affect its color.
Conclusions
During thermal modification of spruce and oak wood, the color changes were observed. Surface lightness (L*) decreases with increase of treatment temperature by about 50% in both wood species (53.23% for spruce and 52.96% for oak, respectively). Chromaticity values (a* and b*) have seen bigger changes in spruce than in oak wood. For both wood species, the highest total color difference (ΔE*) at 210 °C was achieved (rated as very significant color changes). During thermal modification of spruce and oak wood, the content of cellulose, lignin, and extractives increases. The least stable component of the wood at high temperatures is hemicelluloses with larger decrease in oak wood (58.86%) than in spruce wood (37.40%). In spruce wood, the thermal stability declines in the following order: mannose > xylose ~ galactose > arabinose. In oak wood, the least stable saccharides were galactose, arabinose, and mannose, which were almost completely decomposed by the thermal modification. In addition to the cleavage of acetyl groups from polysaccharides, new chromophores (particularly, carbonyl and carboxyl groups) are formed in the modified wood due to oxidation. Bonds in lignin (mainly β-O-4) and methoxyl groups are cleaved, and lignin is condensed at higher temperatures. Higher amounts of extractives in thermally treated wood can also affect its color. Thus, understanding the influence of chemical changes on wood color during thermal loading is useful for enhancing the applicability of thermally modified wood, e.g., in furniture manufacturing as an alternative to expensive tropical species.
References
Saka S (1993) Structure and chemical composition of wood as a natural composite material. In Shiraishi N, Kajita H, Norimoto M (eds). Recent research on wood and wood-based materials: current Japanese materials research, vol 11. Elsevier, Barking, pp 1–20. ISBN 0-444-81691-7
Montero C, Clair B, Alméras T, Van Der Lee A, Gril J (2012) Relationship between wood elastic strain under bending and cellulose crystal strain. Compos Sci Technol 72(2):175–181
Lee KY, Aitomäki Y, Berglund LA, Oksman K, Bismarck A (2014) On the use of nanocellulose as reinforcement in polymer matrix composites. Compos Sci Technol 105:15–27
Sorieul M, Dickson A, Hill SJ, Pearson H (2016) Plant fibre: molecular structure and biomechanical properties, of a complex living material, influencing its deconstruction towards a biobased composite. Materials 9(8):618–654
Boonstra M (2008) A two-stage thermal modification of wood. Ph.D. dissertation in cosupervision Ghent University (Ghent Belgium) and Université Henry Poincaré, Nancy, France
Hon DNS, Shiraishi N (2001) Wood and cellulosic chemistry. Marcel Dekker, Inc., New York, pp 385–442. ISBN 0-8247-0024-4
Fengel D, Wegener G (2003) Wood: chemistry, ultrastructure, reactions. Kessel Verlag, Remagen, pp 26–65. ISBN 3-935638-39-6
Hakkou M, Pétrissans M, Zoulalian A, Gérardin P (2005) Investigation of wood wettability changes during heat treatment on the basis of chemical analysis. Polym Degrad Stabil 89(1):1–5
Chen Y, Fan Y, Gao J, Stark NM (2012) The effect of heat treatment on the chemical and color change of black locust (Robinia pseudoacacia) wood flour: a review. Bioresources 7(1):1157–1170
Lovrić A, Zdravković V, Furtula M (2014) Influence of thermal modification on color of poplar (Populus × euramericana) rotary cut veneer. Wood Res Slovak 59(2):661–670
Mitani A, Barboutis I (2013) Heat treatment effect on color changes of beech (Fagus sylvatica) wood. Pro Ligno 9(4):140–145
Bekhta P, Niemz P (2003) Effect of high temperature on the change of color, dimensional stability and mechanical properties of spruce wood. Holzforschung 57(5):539–546
Kamdem PD, Pizzi A, Jermannaud A (2002) Durability of heat-treated wood. Holz als Roh Werkstoff 60(1):1–6
Kačíková D, Kačík F, Čabalová I, Ďurkovič J (2013) Effects of thermal treatment on chemical, mechanical and colour traits in Norway spruce wood. Bioresour Technol 144:669–674
Candelier K, Thevenon MF, Petrissans A, Dumarcay S, Gerardin P, Petrissans M (2016) Control of wood thermal treatment and its effects on decay resistance—a review. Ann For Sci 73(3):1–13
Zawadzki J, Gawron J, Antczak A, Klosinska T, Radomski A (2016) The influence of heat treatment on the physico-chemical properties of pinewood (Pinus sylvestris L.). Drewno 59(196):49–57
Reinprecht L (2016) Wood deterioration, protection and maintenance. Wiley, New York. ISBN 9781119106531
Esteves BM, Pereira HM (2009) Wood modification by heat treatment: a review. Bioresources 4(1):370–404
Tolvaj L, Papp G, Varga D, Lang E (2012) Effect of steaming on the colour change of softwoods. Bioresources 7(3):2799–2808
Militz H (2002) Thermal treatment of wood: European processes and their background. The International Group on Wood Preservation, Document No. IRG/WP 02–40241
Boonstra MJ, Acker VJ, Tjeredsma BF, Kegel EV (2007) Strength properties of thermally modified softwoods and its relation to polymeric structural wood constituents. Ann For Sci 64(7):679–690
Pelit H, Yalc M (2017) Resistance of mechanically densified and thermally post-treated pine sapwood to wood decay fungi. J Wood Sci 63:514–522
EN 113 (1996) Wood preservatives. Test method for determining the protective effectiveness against wood destroying basidiomycetes. Determination of the toxic values, European Committee for Standardization, Brussels
Cividini R, Travan L, Allegretti O (2007) White beech: a tricky problem in the drying process Procs. In: International scientific conference on hardwood processing (ISCHP), Québec City, Canada, 24–26 September, pp 135–140
ISO 11664-4 (2008) Colorimetry—part 4: CIE 1976 L*a*b* colour space. International Organization for Standardization, London
ISO 11664-6 (2013) Colorimetry—part 6: CIEDE2000 colour-difference formula. International Organization for Standardization, London
Sluiter A, Ruiz R, Scarlata C, Sluiter J, Templeton D (2008) Determination of extractives in biomass: laboratory analytical procedure (LAP). NREL/TP-510-42619. National Renewable Energy Laboratory, Golden
Sluiter A, Hames B, Ruiz R, Scarlata C, Sluiter J, Templeton D, Crocker D (2011) Determination of structural carbohydrates and lignin in biomass: laboratory analytical procedure (LAP). NREL/TP-510-42618. National Renewable Energy Laboratory, Golden
Wise LE, Murphy M, D’Addieco AA (1946) Chlorite holocellulose, its fractionation and bearing on summative wood analysis and on studies on the hemicelluloses. Pap Trade J 122(35):35–34
Seifert VK (1956) About a new method for rapid determination of pure cellulose (in German). Das Papier 10(13):301–306
Barcík Š, Gašparík M, Razumov EY (2015) Effect of temperature on the color changes of wood during thermal modification. Cell Chem Technol 49(9–10):789–798
Salca EA, Kobori H, Inagaki T, Kojima Y, Suzuki S (2016) Effect of heat treatment on colour changes of black alder and beech veneers. J Wood Sci 62:297–304
Nuopponen M, Vuorinen T, Jamsa S, Viitaniemi P (2004) Thermal modification in softwood studied by FT-IR and UV resonance Raman spectroscopies. J Wood Chem Technol 24(1):13–26
Hill CA (2006) Wood modification: chemical, thermal and other processes. vol 5. Wiley, New York
Shimizu K, Teratani F, Hashi M, Miyazaki K (1972) Effect of the thermal treatment on wood hemicelluloses. VI. Studies on the thermal analysis of arabinogalactan, and O-acetyl-and deacetylated-galactoglucomannans. Mokuzai Gakkaishi 18(2):79–84
Zaman A, Alén R, Kotilainen R (2000) Thermal behaviour of Scots pine (Pinus sylvestris) and Silver birch (Betula pendula) at 200–230 °C. Wood Fiber Sci 32(2):138–143
Čabalová I, Kačík F, Kačíková D, Oravec M (2014) The influence of cross-section of spruce wood beams on saccharides changes at thermal loading. Acta Facultatis Xylologiae 56(2):81–86
Esteves B, Velez Marques A, Domingos I, Pereira H (2013) Chemical changes of heat treated pine and eucalypt wood monitored by FTIR. Maderas-Cienc Tecnol 15(2):245–258
Özgenç Ö, Durmaz S, Boyaci IH, Eksi-Kocak H (2017) Determination of chemical changes in heat-treated wood using ATR-FTIR and FT Raman spectrometry. Spectrochim Acta A 171:395–400
Solár R, Reinprecht L, Kačík F, Melcer J, Horský D (1987) Comparison of some physico-chemical and chemical properties of carbohydrate and lignin part of contemporary and subfossile oak wood. Cell Chem Technol 21(5):513–524
González-Peña MM, Curling SF, Hale MD (2009) On the effect of heat on the chemical composition and dimensions of thermally-modified wood. Polym Degrad Stabil 94(12):2184–2193
Popescu MC, Froidevaux J, Navi P, Popescu CM (2013) Structural modifications of Tilia cordata wood during heat treatment investigated by FT-IR and 2D IR correlation spectroscopy. J Mol Struct 1033:176–186
Li MY, Cheng SC, Li D, Wang SN, Huang AM, Sun SQ (2015) Structural characterization of steam-heat treated Tectona grandis wood analyzed by FT-IR and 2D-IR correlation spectroscopy. Chinese Chem Lett 26(2):221–225
Kačík F, Kubovský I (2011) Chemical changes of beech wood due to CO2 laser irradiation. J Photoch Photobiol A 222(1):105–110
Čabalová I, Kačík F, Lagaňa R, Výbohová E, Bubeníková T, Čaňová I, Ďurkovič J (2018) Effect of thermal treatment on the chemical, physical, and mechanical properties of Pedunculate oak (Quercus robur L.) wood. Bioresources 13(1):157–170
Acknowledgements
The authors are grateful for the support of “Advanced research supporting the forestry and wood-processing sector´s adaptation to global change and the 4th industrial revolution”, no. CZ.02.1.01/0.0/0.0/16_019/0000803 financed by OP RDE and by the of the University-wide Internal Grant Agency (CIGA) of the Faculty of Forestry and Wood Sciences, project no. 2016-4309 (70%) and by the Slovak Research and Development Agency under the contract no. APVV-16-0326 (30%).
Author information
Authors and Affiliations
Corresponding author
About this article

Cite this article
Sikora, A., Kačík, F., Gaff, M. et al. Impact of thermal modification on color and chemical changes of spruce and oak wood. J Wood Sci 64, 406–416 (2018). https://doi.org/10.1007/s10086-018-1721-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10086-018-1721-0