
Abstract
The natural product triostin A is known as an antibiotic based on specific DNA recognition. Structurally, a bicyclic depsipeptide backbone provides a well-defined scaffold preorganizing the recognition motifs for bisintercalation. Replacing the intercalating quinoxaline moieties of triostin A by nucleobases results in a potential major groove binder. The functionalization of this DNA binding triostin A analog with a metal binding ligand system is reported, thereby generating a hybrid molecule with DNA binding and metal coordinating capability. Transition metal ions can be placed in close proximity to dsDNA by means of non-covalent interactions. The synthesis of the nucleobase-modified triostin A analog is described containing a propargylglycine for later attachment of the ligand by click-chemistry. As ligand, two [1,4,7]triazacyclononane rings were bridged by a phenol. Formation of the proposed binuclear zinc complex was confirmed for the ligand and the triostin A analog/ligand construct by high-resolution mass spectrometry. The complex as well as the respective hybrid led to stabilization of dsDNA, thus implying that metal complexation and DNA binding are independent processes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
DNA binding peptides have received considerable attention as promising drug candidates (Vázquez et al. 2003). Valuable lead structures in drug design are derived from peptidic natural products. Numerous methods exist for peptide modification with respect to sequence, backbone or side chains, also incorporating non-proteinogenic amino acids (Gante 1994). Next to actinomycin D as one of the most prominent examples of a DNA binding peptidic natural product used in anti-cancer therapy (Farber 1966; Lewis 1972), the quinoxaline antibiotics echinomycin, thiocoraline, and triostin A (1) are well established (Lee and Waring 1978; Waring and Wakelin 1974). Transcription and replication are efficiently blocked by these sequence-specific double strand DNA bisintercalators, thus possessing antibiotic and cytotoxic activity (Katagiri et al. 1975). The disulfide-bridged depsipeptide backbone of triostin A provides a rigid scaffold preorganizing two quinoxaline intercalators in a distance of 10.5 Å. Therefore, bisintercalation of dsDNA spanning a dinucleotide is entropically favored (Addess and Feigon 1994). In earlier studies, the peptide scaffolds of triostin A, des-N-tetramethyltriostin A (2, TANDEM) (Ciardelli and Olsen 1977), and the peptide analog aza-TANDEM (3) (Dietrich and Diederichsen 2005) were used as rigid templates for functional modification in order to generate a new class of DNA binders (Fig. 1). As part of our studies to change the DNA binding mode of triostin A and its derivatives from bisintercalation to specific major groove recognition, the rigid templating depsipeptide scaffold is used to organize nucleobases or other recognition moieties instead of the native quinoxaline moieties (Lorenz and Diederichsen 2004).

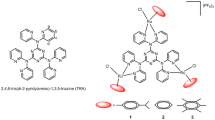
Structures of triostin A (1), TANDEM (2), aza-TANDEM (3), and hybrid of the nucleobase substituted aza-TANDEM 4 with a ligand capable of forming binuclear transition metal complexes
With the intention to use the DNA recognition unit for guiding a functional metal complex in proximity to the oligonucleotide backbone, synthesis of a hybrid molecule was intended. Comparable examples for the use of the hybrid approach in drug design are the combination of a DNA binding motif with a cell penetrating peptide (Langer et al. 2001) or a cytotoxic warhead (Xie et al. 1996; Jia et al. 1999; Baraldi et al. 1998). We synthesized a conjugate of a triostin A derivative as DNA binder and a ligand capable of forming binuclear transition metal complexes. Such a hybrid molecule 4 was expected to retain its DNA binding properties, and additionally, being able to recruit loosely bound copper and zinc ions from cellular pools. Positioning a binuclear metal center in close proximity of DNA strands can be considered as an opportunity to generate, e.g., reactive oxygen species (ROS), which ultimately cause cytotoxic DNA damage (Sensi et al. 1999; Manzl et al. 2004).
A common strategy for the attachment of ligands to peptides is based on the incorporation of artificial amino acid building blocks by solid phase peptide synthesis (Dirscherl and König 2008; Levadala et al. 2004; Cheng et al. 2005; Damian et al. 2010). Bipyridine- and phenanthroline-substituted amino acids or functionalized lysine and tyrosine derivatives are reported (Jiang et al. 2005; Iranzo et al. 2007). In addition, site-specific coupling of ligand systems with amino acid side chains can be achieved by disulfide formation (Su et al. 2006), C–C coupling reactions (Albrecht et al. 2003) or copper-mediated [3 + 2] cycloaddition of an azide to an alkyne (click-chemistry) (Best 2009; Nadler et al. 2009). Click-chemistry can be applied especially on sterically hindered or conformational rigid systems. Therefore, it was used for the ligation of our triostin A analog with the ligand providing construct 4 as a chimera. A propargylglycine containing the aza-TANDEM derivative was ligated with an azide-functionalized ligand composed of two [1,4,7]triazacyclononane (TACN) moieties and a bridging phenol. Similar ligands have been shown to form binuclear zinc(II) and copper(II) complexes (Rossi et al. 1998) and to catalyze hydrolysis (O’Donoghue et al. 2006). Two metal ions are likely to be arranged in a distance of 3.4 Å which is well suitable for cooperative processes.
Materials and methods
General remarks
The solvents used were of the highest grade available. Dichloromethane and triethylamine were distilled from calcium hydride prior to use. DMF was purchased dry and stored over molecular sieves (4 Å). All reagents were of analytical grade and used without further purification. NMR spectra were recorded with a Varian Unity 300 or Varian INOVA-600 instrument. Chemical shifts are referenced to the residual solvent peaks of [D6]DMSO (1H: δ = 2.49 ppm; 13C: δ = 39.5 ppm) or CDCl3 (1H: δ = 7.24 ppm; 13C: δ = 77.0 ppm). ESI-MS data were measured with a LCQ Finnigan spectrometer. HRMS data were determined with a Bruker APEX-Q IV 7T spectrometer. Chromatographic separations were performed using silica gel 60 (0.040–0.063 mm, Merck). HPLC separations were carried out with a Pharmacia Äkta Basic using J’sphere ODS-H80 RP-C18 columns for analytical samples (250 × 4.6 mm, S-4 μm, 8 nm, 1 mL min−1) and for preparative samples (250 × 20 mm, S-4 μm, 8 nm, 10 mL min−1) with a linear gradient of A (0.1% TFA in H2O) to B (MeCN/H2O, 9:1 + 0.1% TFA). Analytical thin-layer chromatography was performed using Merck silica gel 60 F254 precoated aluminum plates. Visualization was achieved with UV light (254 nm) or by dyeing with ninhydrin (3% in ethanol).
Di-tertbutoxycarbonyl-[1,4,7]triazacyclononane (BBT, 12)
Dry Et3N (5.18 mL, 37.1 mmol) was added to a cooled solution (−5°C) of TACN (2.40 g, 18.6 mmol) in dry DCM (60 mL). A solution of Boc2O (tert-butyloxycarbonyl anhydride, 8.10 g, 37.1 mmol) in dry DCM (20 mL) was added over a period of 1 h and stirred for 30 min at −5°C. All volatile components were removed in vacuo and the residue was dissolved in MeOH/EtOAc = 1:8 and purified by flash chromatography (4 × 18 cm, MeOH/EtOAc = 1:8) to yield a yellow oil (3.28 g, 54%). 1H-NMR (300 MHz, CDCl3): δ = 1.45 (s, 18 H, Boc-CH3), 2.87–2.97 (m, 4 H, 2, 9-H2), 3.17–3.32 (m, 4 H, 3, 8-H2), 3.39–3.52 (m, 4 H, 5, 6-H2) ppm. MS (ESI, MeOH): m/z (%) = 330 (100) [M + H]+, 352 (10) [M + Na]+.
4-Bromo-2,6-di-(bromomethyl)-phenol (14)
HBr in acetic acid (33 wt%, 37.0 mL, 0.21 mol) was added to 4-bromo-2,6-dihydroxy-phenol (13, 10.0 g, 42.9 mmol) at 0°C and stirred for 20 min at 0°C. The solution was allowed to warm to rt and was stirred for another 24 h at rt. Water (50 mL) was added and the precipitate was filtered off, washed with water (2× 40 mL) and dried in vacuo to yield 92% (14.2 g, 39.6 mmol) of compound 14 which was used for the synthesis of 15 without further purification.
4-Bromo-2,6-bis-(di-tertbutoxycarbonyl-[1,4,7]triazacyclononane-1-yl-methyl)-phenol (15)
Potassium carbonate (1.01 g, 7.30 mmol) was added to a solution of 4-bromo-2,6-bis-bromomethyl-phenol (1.25 g, 3.48 mmol), BBT 12 (2.40 g, 7.30 mmol) in dry MeCN (25 mL) and stirred for 16 h at 60°C. The remaining solid was separated by filtration and the filtrate evaporated to dryness. The residue was purified by flash chromatography (4 × 20 cm, EtOAc/pentane = 1:1) to yield a yellow solid (2.42 g, 81%). 1H-NMR (300 MHz, [D6]DMSO, 100°C): δ = 1.44 (s, 36 H, Boc-CH3), 2.67–2.74 (m, 8 H, TACN-6,6′,8,8′-H2), 3.25–3.30 (m, 8 H, TACN-5,5′,9,9′-H2), 3.40 (s, 8 H, TACN-2,2′,3,3′-H2), 3.75 (s, 4 H, CH2), 7.22 (s, 2 H, Ar-H) ppm. 13C-NMR (150 MHz, [D6]DMSO, 100°C): δ = 27.7 (Boc-CH3), 48.5 (TACN-CH2), 52.3 (TACN-CH2), 55.4 (CH2), 78.4 (Boc-C), 109.3 (Ar-CBr), 126.0 (Ar-CCH2), 130 (Ar-CH), 154.2 (Boc-CO), 154.5 (Ar-COH) ppm. MS (ESI, MeOH): m/z (%) = 855 (100) [M + H]+. HRMS (ESI): calcd. for C40H68BrN6O9 ([M + H]+): 855.42257, found: 855.42230.
4-Bromo-2,6-bis-([1,4,7]triazacyclononane-1-yl-methyl)-phenol (16)
A solution of ligand 15 (48 mg, 56 μmol) in TFA/DCM = 1:1 (2 mL) was stirred at rt for 1 min. The mixture was evaporated to dryness in vacuo and purified by HPLC (preparative, 0–60% MeOH + 0.1% TFA in 30 min, R t = 21.61 min) to yield a yellowish solid (21.7 mg, 85%). 1H-NMR (300 MHz, [D6]DMSO): δ = 2.80–2.87 (m, 8 H, TACN-2,2′,9,9′-H2), 3.06–3.15 (m, 8 H, TACN-3,3′,8,8′-H2), 3.43–3.51 (m, 8 H, TACN-5,5′,6,6′-H2), 3.80 (s, 4 H, CH2), 7.43 (s, 2 H, Ar-H), 9.13 (s br, 4 H, NH) ppm. 13C-NMR (150 MHz, [D6]DMSO): δ = 42.8 (TACN-2,2′,9,9′-CH2), 43.9 (TACN-3,3′,8,8′-CH2), 48.4 (TACN-5,5′,6,6′-CH2), 52.9 (CH2), 110.5 (Ar-CBr), 126.9 (Ar-CCH2), 133.4 (Ar-CH), 153.9 (Ar-COH) ppm. HRMS (ESI): calcd. for C20H35BrN6O ([M + H]+): 455.2128, found: 455.2120.
4-Azido-2,6-bis-(di-tertbutoxycarbonyl-[1,4,7]triazacyclononane-1-yl-methyl)-phenol (6)
An argon-saturated solution of ligand 15 (0.52 g, 0.60 mmol) and dimethylethylenediamine (64.9 μL, 0.60 mmol) in EtOH/H2O = 7:3 (50 mL) was added to sodium azide (0.12 g, 1.80 mmol), sodium ascorbate (0.36 g, 1.80 mmol) and copper iodide (0.35 g, 1.80 mmol) and refluxed for 24 h. The mixture was filtered over silica, the filtrate evaporated to dryness and dried in vacuo to yield a brown-yellow solid (0.34 g, 70%). 1H-NMR (300 MHz, [D6]DMSO, 100°C): δ = 1.44 (s, 36 H, Boc-CH3), 2.66–2.77 (m, 8 H, TACN-6,6′,8,8′-H2), 3.24–3.31 (m, 8 H, TACN-5,5′,9,9′-H2), 3.38–3.45 (m, 8 H, TACN-2,2′,3,3′-H2), 3.75 (s, 4 H, CH2), 6.83 (s, 2 H, Ar-H) ppm. MS (ESI, MeOH): m/z (%) = 818 (100) [M + H]+, 840 (25) [M + Na]+. HRMS (ESI): calcd. for C40H68N9O9 ([M + H]+): 818.51345, found: 818.51336.
Cyclo[β-d-Dap(adenine-9-yl-acetyl)-l-Ala-l-Cys-l-[1-(4-hydroxy-3,5-bis-[1,4,7]triazacyclonane-1-yl-methyl-phenyl)-1H-[1,2,3]triazole-4-yl]-Gly-β-d-Dap(thymine-1-yl-acetyl)-l-Ala-l-Cys-l-Val]-disulfide (4)
A solution of 2,6-lutidine in dry DMF (5 mL, 8 mM) was saturated with argon; cyclopeptide 5 (0.5 mg, 0.4 μmol), ligand 6 (0.9 mg, 1.1 μmol), [CuIP(OEt)3] (catalytic) and tris-(1-benzyl-1H-[1,2,3]triazole-4-yl-methyl)amine (TBTA) (catalytic) were placed in a 0.5 mL Eppendorf cap equipped with a magnetic stirrer. 2,6-Lutidine in dry DMF (100 μL, 0.8 mmol) was added under argon. The mixture was stirred for 5 days at rt. The solution was evaporated to dryness in vacuo and the residue was dialyzed for 7 h (ZelluTrans/Roth V-Series, MWCO 1000, H2O/MeCN = 1:1) and purified by HPLC (analytical, 50–100% B in 30 min, R t = 12.2 min) to yield a yellowish solid of the Boc- and Z-protected intermediate. HRMS (ESI): calcd. for C90H130N26O23S2 ([M + 2H]2+): 1003.46163, found: 1003.46029; calcd. for C90H131N26O23S2 ([M + 3H]3+): 669.31018, found: 669.31026. For Boc and Z deprotection, the intermediate was dissolved in TFA (2 mL) and ethanedithiol (0.1 mL) was added. The solution was stirred for 48 h at rt, and afterward, evaporated to dryness in vacuo. The residue was purified by HPLC (analytical, 12–28% B, R t = 13.6 min) to yield target compound 4 as yellowish solid (0.1 mg, 16%). MS (ESI, MeOH): m/z (%) = 368 (49) [M + 4H]4+, 491 (100) [M + 3H]3+, 736 (73) [M + 2H]2+. HRMS (ESI): calcd. for C62H91N26O13S2 ([M + H]+): 1471.6700, found: 1471.6689.
[Zn2(4-Bromo-2,6-bis-(di-tertbutoxycarbonyl-[1,4,7]triazacyclononane-1-yl-methyl)-phenol)] (17)
To a solution of 16 in H2O (5 mM, 10 μL), a solution of ZnSO4 in H2O (4.8 mM, 20.8 μL) was added and the reaction mixture agitated for 10 min at rt. HRMS (ESI): calcd. for C20H34BrN6O5SZn2 ([M + SO4]+): 677.00722, found: 677.00732.
[Cu2(4-Bromo-2,6-bis-(di-tertbutoxycarbonyl-[1,4,7]triazacyclononane-1-yl-methyl)-phenol)] (18)
To a solution of 16 in H2O (5 mM, 10 μL), a solution of CuSO4 in H2O (4.8 mM, 20.8 μL) was added and the reaction mixture stirred for 10 min at rt. HRMS (ESI): calcd. for C20H34BrN6O5SCu2 ([M + SO4]+): 675.00813, found: 675.00919.
[Zn2(Cyclo[β-d-Dap(adenine-9-yl-acetyl)-l-Ala-l-Cys-l-[1-(4-hydroxy-3,5-bis-[1,4,7]triazacyclonane-1-yl-methyl-phenyl)-1H-[1,2,3]triazole-4-yl]-Gly-β-d-Dap(thymine-1-yl-acetyl)-l-Ala-l-Cys-l-Val]-disulfide)] (19)
To a solution of 4 in H2O (0.68 mM, 10 μL), a solution of ZnSO4 in H2O (1.36 mM, 10 μL) was added and the reaction mixture agitated for 10 min at rt. HRMS (ESI): calcd. for C64H92N26O17S2Zn2 ([M + 2HCOO]2+): 844.2576, found: 844.2569.
Results and discussion
To enable click-chemistry as the ligation step generating the hybrid compound 4, the propargylglycine containing aza-TANDEM derivative 5 and the azide-functionalized ligand system 6 were synthesized (Scheme 1). The phenol-bridged bis-TACN ligand was substituted with an azide in the para position with respect to the phenolic hydroxyl group in order to achieve a spatial separation of the metal binding site and the triazole moiety formed during the click-reaction. Thus, involvement of the triazole ring as nitrogen donor in metal coordination should be avoided.

Ligation of the aza-TANDEM scaffold 5 with ligand 6 by means of copper-mediated [3 + 2] cycloaddition
The aza-TANDEM scaffold required alkyne functionalization at a position not being essential for DNA recognition. Therefore, in peptide 5, a valine from the original triostin A sequence that is well suited for side chain modifications (Addess and Feigon 1994) was replaced by propargylglycine. Side chain functionalization to hybrid 4 was now feasible using click-chemistry with no need for further protecting groups (Tornøe et al. 2002).
Synthesis of the nucleobase-functionalized bicyclic aza-TANDEM scaffold 5
The octapeptide 7 was synthesized by manual Fmoc-SPPS according to an already reported protocol (Dietrich and Diederichsen 2005) with modifications concerning coupling conditions and amino acid sequence (Scheme 2). The coupling of Fmoc-protected propargylglycine was achieved by coupling with 1-hydroxy-7-aza-benzotriazole (HOAt) and O-(7-azabenzotriazolyl)-tetramethyluronium hexafluorophosphate (HATU) using three equivalents of amino acid. Resin loading before and after coupling was determined by UV spectroscopy to confirm quantitative conversion (Gude et al. 2002).

Synthesis of the propargylglycine-functionalized aza-TANDEM analog 5
Orthogonal protection of the two d-diaminopropionic acid (d-Dap) α-amino functionalities was required as different nucleobases were attached after macrocyclization. Boc and benzyloxycarbonyl (Z) protecting groups were chosen both being orthogonal to the Fmoc-SPPS protocol. The linear peptide was obtained in an overall yield of 87%. Formation of the disulfide bridge (26%) and macrocyclization yielded the triostin analog scaffold 8 (30%) according to a reported protocol (Dietrich and Diederichsen 2005).
For nucleobase functionalization with thymine and adenine derivatives by amide formation, substituted acetic acids, (thymine-1-yl)acetic acid (9) and (N6-Z-adenine-1-yl)acetic acid (10) were prepared as reported by Nielsen and co-workers (Dueholm et al. 1994). The Boc-protecting group of d-Dap in peptide 8 was cleaved under mild conditions in order to preserve Z protection. Subsequent coupling of (thymine-1-yl)acetic acid was achieved with HOAt, diisopropylcarbodiimide (DIC) and N-methylmorpholine (NMM) providing nucleopeptide 11 in 47% yield. Cleavage of the Z group and subsequent coupling of (N6-Z-adenine-9-yl)acetic acid with bromo-tris-pyrrolidino-phosphonium hexafluorophosphate (PyBroP) yielded 37% of the click precursor 5. Reaction details and analytical data concerning the peptide synthesis can be found in the supporting information.
Synthesis of the ligand system 6 and assembly of the hybrid compound 4
The design of the ligand system attached to the triostin A scaffold allows for binuclear metal complexation employing two TACN moieties bridged by a phenol. The phenol derivative 13 was prepared by hydroxymethylation with formaldehyde and KOH starting from 4-bromophenol. Subsequently, it was converted into the corresponding 2,6-dibromomethyl compound 14 (92%) by treatment with HBr. Attachment of two BBT moieties (12) obtained from TACN by Boc protection of two amino functions (Kimura et al. 2001) was achieved in 81% yield to give compound 15. The required azide for the click-reaction was obtained by mild halogen azide exchange employing dimethyldiethyldiamine and copper iodide (Andersen et al. 2005). In order to provide an additional ligand for metal binding studies, compound 15 was Boc deprotected to yield the TACN derivative 16 (Scheme 3).

Synthesis of the metal chelating building block 6 and unprotected ligand 16
Copper-mediated [3 + 2] cycloaddition was applied in order to generate the desired hybrid molecule 4 containing the triostin A analog recognition unit and the binuclear metal binding site. For ligation of the azide containing ligand 6 to the alkynyl side chain of cyclopeptide 5, 2,6-lutidine was employed as base, the copper(I) complex [CuIP(OEt3)] as catalytic precursor and TBTA as ligand for the in situ generated active copper complex (Chan et al. 2004). The crude product was dialyzed. Subsequent deprotection of the TACN and adenine moieties was achieved with TFA and ethanedithiol (EDT) as scavenger (Scheme 1). After HPLC purification target compound 4 was obtained in 16% yield (over two steps) and characterized by high-resolution mass spectrometry.
Metal binding and DNA recognition
First evidence for metal coordination was obtained with ligand 16 by generation of zinc and copper complexes. The binuclear zinc complex 17 and the respective copper complex 18 were obtained from a 2:1 mixture of metal sulfate and ligand 16 as indicated by high-resolution mass spectrometry. The hybrid molecule 4 was also shown to form a binuclear zinc complex 19. The incorporation of two zinc ions can be deduced from the isotope pattern observed in the high-resolution mass spectrum, which is in accordance with the calculated spectrum for the binuclear zinc complex (Fig. 2). No higher aggregates or complexes containing one or three zinc ions were observed, thereby suggesting that transition metal ions are solely coordinated in the binding pockets formed by the TACN nitrogen atoms and the phenolic hydroxyl group.

High-resolution MS of complex 19 (left) in comparison with the respective isotopic distribution obtained by calculation (right)
Temperature-dependent UV spectroscopy was used to probe for interaction between the hybrid 4 and dsDNA (Pörschke 1971). The melting temperature of the DNA stem loop 5′CGCGATACGCAAAAAGCGTATCGCG3′ (20, T m = 71°C) was found to be significantly increased (∆T = 4°C) by addition of a tenfold excess of hybrid 4 (Fig. 3). The degree of stabilization is comparable to triostin analogs with adenine/thymine functionalization lacking the metal coordination site. A comparable stabilization (T m = 74°C) was observed for the binuclear zinc complex of the hybrid molecule 19. Thus, it is likely that neither the TACN moieties nor the metal complex interferes with the DNA binding properties of the triostin recognition unit. Furthermore, simultaneous DNA and transition metal binding of hybrid 4 is indicated.

UV-melting curves of DNA 20 with the hybrid molecule 4 and the respective binuclear zinc complex 19 (2 μM DNA 20, 2 mM HEPES, pH 7.0, 10 mM NaCl)
Conclusion
The synthesis of a hybrid molecule composed of a DNA binder and a ligand for binuclear transition metal complexes is described. On the one hand, DNA recognition is provided by a rigid bicyclic peptide scaffold derived from triostin A that contains nucleobases to allow for binding in the major groove of dsDNA. On the other hand, a ligand for a binuclear transition metal complex provides a functional unit that was ligated to the triostin A analog by click-chemistry. The ligation of these sterically demanding molecules was enabled by introducing propargylglycine in the cyclopeptide and azide functionalization of the ligand. Binuclear transition metal complexes were obtained for the hybrid as well as for the isolated ligand system. DNA recognition by the hybrid was found to be not affected by metal complexation, thus suggesting that recognition and metal ligation are not interfering. The new metal containing triostin A analogs promise sequence-specific DNA recognition bringing functional metal complexes in proximity to the backbone. Potential pharmaceutical relevance with respect to directed DNA damaging and the decoration of the helical dsDNA with metal centers for catalytic purposes are applications that are currently under investigation.
References
Addess AJ, Feigon J (1994) Solution structure of a complex between [N-MeCys3,N-MeCys7]TANDEM and [d(GATATC)]2. Biochemistry 33:12386–12396
Albrecht M, Stortz P, Nolting R (2003) Peptide/metal–ligand hybrids for the metal-assisted stabilization of peptide-microstructures. Synthesis 9:1307–1320
Andersen J, Madsen U, Björkling F, Liang X (2005) Rapid synthesis of aryl azides from aryl halides under mild conditions. Synlett 14:2209–2213
Baraldi PG, Cacciari B, Guiotto A, Leoni A, Romagnoli R, Spalluto G, Mongelli N, Howard PW, Thurston DE, Bianchi N, Gambari R (1998) Design, synthesis and biological activity of a pyrrolo [2,1-c][1,4]benzodiazepine (PBD)-distamycin hybrid. Bioorg Med Chem Lett 8:3019–3024
Best MD (2009) Click chemistry and bioorthogonal reactions: unprecedented selectivity in the labeling of biological molecules. Biochemistry 48:6571–6584
Chan TR, Hilgraf R, Sharpless KB, Fokin VV (2004) Polytriazoles as copper(I)-stabilizing ligands in catalysis. Org Lett 6:2853–2855
Cheng SY, Huang Y, Zhang GL, Cheng H, Xia CQ, Ma LJ, Yu XQ (2005) Synthesis of a cyclen functionalized α-amino acid and its incorporation into peptide sequence. Synthesis 12:888–892
Ciardelli TL, Olsen RK (1977) Synthesis of des-N-tetramethyltriostin A, a bicyclic octadepsipeptide related to the quinoxaline antibiotics. J Am Chem Soc 99:2806–2807
Damian MS, Hedman HK, Elmroth SKC, Diederichsen U (2010) Synthesis and DNA interaction of platinum complex/peptide chimera as potential drug candidates. Eur J Org Chem. doi:10.1002/ejoc.201000677
Dietrich B, Diederichsen U (2005) Synthesis of cyclopeptidic analogues of triostin A with quinoxalines or nucleobases as chromophores. Eur J Org Chem 2005:147–153
Dirscherl G, König B (2008) The use of solid-phase synthesis techniques for the preparation of peptide–metal complex conjugates. Eur J Org Chem 2008:597–634
Dueholm KL, Egholm M, Behrens C, Christensen L, Hansen HF, Vulpins T, Petersen KH, Berg RH, Nielsen PE, Buchardt O (1994) Synthesis of peptide nucleic acid monomers containing the four natural nucleobases: thymine, cytosine, adenine, and guanine and their oligomerization. J Org Chem 59:5767–5773
Farber SJ (1966) Chemotherapy in the treatment of leukemia and Wilms’ tumor. J Am Med Assoc 198:826–836
Gante J (1994) Peptidomimetics-tailored enzyme inhibitors. Angew Chem Int Ed Engl 33:1699–1720
Gude M, Ryf J, White PD (2002) An accurate method for the quantitation of Fmoc-derivatized solid phase supports. Lett Pept Sci 9:203–206
Iranzo O, Cabello C, Pecoraro VL (2007) Heterochromia in designed metallopeptides: geometry-selective binding of CdII in a de novo peptide. Angew Chem Int Ed 46:6688–6691
Jia G, Iida H, Lown JW (1999) Synthesis of an unsymmetrical bis-lexitropsin-1,2,9,9a-tetrahydrocyclopropa[c]benzo[e]indol-4-one (CBI) conjugate. Chem Commun 2:119–120
Jiang H, O’Neil EJ, DiVittorio KM, Smith BD (2005) Anion mediated transfer of zinc(II)-coordinated tyrosine derivatives. Org Lett 7:3013–3016
Katagiri K, Yoshida T, Sato K (1975) Quinoxaline antibiotics. In: Corcoran JW et al (eds) Antibiotics. III, pp 234–251
Kimura S, Bill E, Bothe E, Weyermüller T, Wieghardt K (2001) Phenylthiyl radical complexes of gallium(III), iron(III) and cobalt(III) and comparison with their phenoxyl analogues. J Am Chem Soc 123:6025–6039
Langer M, Kratz F, Rothen-Rutishauser B, Wunderli-Allenspach H, Beck-Sickinger AG (2001) Novel peptide conjugates for tumor-specific chemotherapy. J Med Chem 44:1341–1348
Lee JS, Waring MJ (1978) Bifunctional intercalation and sequence specificity in the binding of quinomycin and triostin antibiotics to deoxyribonucleic acid. Biochem J 173:115–128
Levadala MK, Banerjee SR, Maresca KP, Babich JW, Zubieta J (2004) direct reductive alkylation of amino acids: synthesis of bifunctional chelates for nuclear imaging. Synthesis 11:1759–1766
Lewis JL (1972) Chemotherapy of gestational choriocarcinoma. Cancer 30:1517–1521
Lorenz KB, Diederichsen U (2004) Solution-phase synthesis of nucleobase-substituted analogues of triostin A. J Org Chem 69:3917–3927
Manzl C, Enrich J, Ebner H, Dallinger R, Krumschnabel G (2004) Copper-induced formation of reactive oxygen species causes cell death and disruption of calcium homeostasis in trout hepatocytes. Toxicology 196:57–64
Nadler A, Hain C, Diederichsen U (2009) Histidine analog amino acids providing metal binding sites derived from bioinorganic model systems. Eur J Org Chem 2009:4593–4599
O’Donoghue A, Pyun SY, Yang M-Y, Morrow JR, Richard JP (2006) Substrate specificity of an active dinuclear Zn(II) catalyst for cleavage of rna analogues and a dinucleoside. J Am Chem Soc 128:1615–1621
Pörschke D (1971) Cooperative nonenzymic base recognition. II. Thermodynamics of the helix-coil transition of oligoadenylic + oligouridylic acids. Biopolymers 10:1989–2013
Rossi P, Felluga F, Scrimin P (1998) A new ligand α-amino acid: (S)-2-amino-3-[1-(1,4,7-triazacyclononane)]propanoic acid. Tet. Lett 39:7159–7162
Sensi SL, Yin HZ, Carriedo SG, Rao SS, Weiss JH (1999) Preferential Zn2+ influx through Ca2+-permeable AMPA/kainite channels triggers prolonged mitochondrial superoxide production. Proc Natl Acad Sci USA 96:2414–2419
Su X-C, Huber T, Dixon NE, Otting G (2006) Site-specific labelling of proteins with a rigid lanthanide-binding tag. ChemBioChem 7:1599–1604
Tornøe CW, Christensen C, Meldal M (2002) Peptidotriazoles on solid phase: [1,2,3]-Triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J Org Chem 67:3057–3064
Vázquez ME, Caamano AM, Mascarenas JL (2003) From transcription factors to designed sequence-specific DNA-binding peptides. Chem Soc Rev 32:338–349
Waring MJ, Wakelin LP (1974) Echinomycin: a bifunctional intercalating antibiotic. Nature 252:653–657
Xie G, Gupta R, Atchison K, Lown JW (1996) Bisindolylmaleimides linked to DNA minor groove binding lexitropsins: synthesis, inhibitory activity against topoisomerase I, and biological evaluation. J Med Chem 39:1049–1055
Acknowledgments
Financial support from the Deutsche Forschungsgemeinschaft (Di 542/7-1 and the IRTG 1422 ‘Metal Sites in Biomolecules’) is gratefully acknowledged.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Sachs, EF., Nadler, A. & Diederichsen, U. Triostin A derived hybrid for simultaneous DNA binding and metal coordination. Amino Acids 41, 449–456 (2011). https://doi.org/10.1007/s00726-010-0764-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-010-0764-3