
Abstract
Viruses, despite being relatively simple in structure and composition, have evolved to exploit complex cellular processes for their replication in the host cell. After binding to their specific receptor on the cell surface, viruses (or viral genomes) have to enter cells to initiate a productive infection. Though the entry processes of many enveloped viruses is well understood, that of most non-enveloped viruses still remains unresolved. Recent studies have shown that compared to direct fusion at the plasma membrane, endocytosis is more often the preferred means of entry into the target cell. Receptor-mediated endocytic pathways such as the dynamin-dependent clathrin and caveolar pathways are well characterized as viral entry portals. However, many viruses are able to utilize multiple uptake pathways. Fluid phase uptake, though relatively non-specific in terms of its cargo, potentially aids viral infection by its ability to intersect with the endocytic pathway. In fact, many viruses despite using specialized pathways for entry are still able to generate productive infection via fluid phase uptake. Macropinocytosis, a major fluid uptake pathway found in epithelial cells and fibroblasts, is stimulated by growth factor receptors. Many viruses can induce these signaling cascades in cells leading to macropinocytosis. Though endocytic trafficking is utilized by both enveloped and non-enveloped viruses, key differences lie in the way membranes are traversed to deposit the viral genome at its site of replication. This review will discuss recent developments in the rapidly evolving field of viral entry.
Similar content being viewed by others


Introduction
Viruses, like all obligate intracellular pathogens, have to find the means to cross cellular membranes. This is the key to initiating their infectious cycle, and involves a number of discrete steps like receptor binding and entry, capsid destabilization and genome uncoating, culminating in the release of viral nucleic acids at their site of replication. Many of these changes result from conformational alterations in metastable viral structures. Virus binding to and/or cross-linking their specific receptors can also lead to activation of downstream signaling events (Greber 2002). These signals often induce changes that promote entry, prepare the cell for invasion and neutralize host defences. In animal cells, enveloped viruses achieve entry in two principal ways: (1) by direct fusion with the plasma membrane, or (2) by an internalization process into endosomes. For viruses using the first strategy, fusion between the viral and cellular membranes occurs after receptor docking and before the virus core penetrates the cell. Recent developments in membrane trafficking have demonstrated the existence of multiple endocytic pathways at the plasma membrane (Marsh and Helenius 2006). Indeed, most viruses prefer to enter cells via endocytosis since the endocytic network confers an additional advantage of specific localization within the cell for a successful infection. In the case of endocytic entry, the virus must penetrate or fuse with the endosomal membrane to be released into the cytoplasm. Studies indicate that endocytosis serves as an entry portal for both enveloped and non-enveloped viruses. While there have been many advances in understanding how enveloped viruses achieve membrane fusion, the penetration mechanisms of most non-enveloped viruses still lack clarity. This review will give an update on the viral entry pathways and highlight the common themes and key differences between the strategies deployed by enveloped and non-enveloped viruses to achieve productive cell entry.
Role of attachment factors
In a typical animal virus, the nucleic acid is condensed in an icosahedral or helical nucleoprotein complex called capsid. Enveloped viruses bud through cellular membranes and as a result the nucleocapsid is surrounded by an additional lipid bilayer. The cellular membrane is modified by the virus and contains viral glycoproteins that appear as spikes on the surface of the virus particle. Many viruses also carry accessory protein molecules that aid in infection, like reverse transcriptase, RNA polymerase, kinases, etc. In addition, viruses also incorporate host cellular proteins into or onto newly formed particles. This could be due to specific interactions between viral and cellular proteins during virus assembly, or even the presence of cellular membrane proteins in the immediate vicinity of virus budding from cells. Host cell proteins commonly incorporated into virions are Tsg101 and other proteins of the multi-vesicular body pathway that are involved in viral egress, APOBEC3G, a protein of the RNA editing machinery, cyclophilins, etc. Cyclophilin A is involved in T-cell activation and is thought to provide a chaperone function, is incorporated in many viruses like HIV-1, vaccinia virus and vesicular stomatitis virus (VSV) (Cantin et al. 2005). Studies have shown that HIV infectivity is finely tuned by the expression levels of host Cyclophilin A. There also exist multiple host membrane proteins that are inserted in mature virions. The HIV particle contains proteins like MHC-I, MHC-II, ICAM-1, cytoskeleton-associated proteins like actin, ezrin, moesin and cofilin, and glycosylphosphatidylinositol (GPI)-linked proteins Thy-1 and CD59 (Cantin et al. 2005). Many of these proteins enhance receptor binding efficacy and kinetics and thus play a crucial role in determining the efficiency of infection.
For the majority of viruses, the initial stage of the entry process is the binding of a viral attachment protein to a generalized receptor, followed by interaction with a more specific host cell receptor. These generalized receptors, more commonly known as attachment factors, concentrate virus particles on the host cell and create conditions favourable for receptor binding. The molecules to which viruses bind constitute a diverse collection of cellular proteins, carbohydrates and lipids, and play an important role in viral entry (Lonberg-Holm and Philipson 1974). Heparan sulfate proteoglycans (HSPGs) serve as attachment factors for a large number of viruses (Bernfield et al. 1999) that include include the herpes simplex virus (WuDunn and Spear 1989), vaccinia virus (Chung et al. 1998), flaviviruses such as the dengue virus (Chen et al. 1997) and hepatitis C virus (HCV) (Barth et al. 2003), retroviruses such as the human immunodeficiency virus (HIV) (Mondor et al. 1998) and T-cell leukemia virus (Pinon et al. 2003), as well as non-enveloped viruses like adeno-associated virus (Dechecchi et al. 2000), papillomavirus (Giroglou et al. 2001) and norovirus (Tamura et al. 2004). Recent studies from our own laboratory have also shown the non-enveloped hepatitis E virus to attach to hepatic cells through HSPGs (Kalia et al. 2009).
The HSPGs are present almost ubiquitously on cell surfaces but are extensively heterogenous with respect to their composition and quantity among different species, cell types, tissues and developmental stages. These variations include modifications in the length, degree of sulfation, and positions of the sulphate groups in the disaccharide repeats (Lindahl et al. 1998). In many cases, the binding of a particular ligand to heparan sulfate (HS) depends on its specific sulfation pattern (Lyon and Gallagher 1998). It has been proposed that the tissue tropism of some viruses may be determined by the heparan sulphate fine structure (Shukla et al. 1999). Many viruses also bind to sialic acid containing groups (Olofsson and Bergstrom 2005), for which the key examples include the influenza virus (Skehel and Wiley 2000), coronaviruses (Schwegmann-Wessels and Herrler 2006) and non-enveloped viruses like orthoreoviruses (Guglielmi et al. 2006) and rotavirus (Isa et al. 2006). Gangliosides (GM1, GD1a and GT1b) are well established attachment factors for non-enveloped polyoma viruses like SV40 (Tsai et al. 2003). Viruses like HIV, dengue virus, HCV and Ebola virus have high mannose N-linked glycans in their envelope glycoproteins, which can bind cell surface lectins such as DC-SIGN and L-SIGN (Feinberg et al. 2001; Pohlmann et al. 2003; Simmons et al. 2003). DC-SIGN is highly expressed on dendritic cells present in mucosal tissues and binds to the HIV-1 envelope glycoprotein gp120 to efficiently capture the virus in the periphery. This facilitates HIV-1 transport to secondary lymphoid organs that are rich in T cells, to enhance infection in trans of these target cells (Geijtenbeek et al. 2000).
The individual interaction between a virus and a single attachment factor or receptor can be weak and relatively non-specific (Skehel and Wiley 2000), but serves the purpose of concentrating viruses on the specific host cells. Multiple binding leads to increased avidity, and this makes virus binding virtually irreversible.
Receptor binding
Following attachment, receptor binding is often the second and more critical step in viral infection. Cellular receptors vary from one virus to the next and are in many cases cell specific. Viruses from the same family can have selectivity for different receptors, while viruses from different families can use the same protein as their cellular receptor. An example of this is the coxsackie-adenovirus receptor (CAR) protein, which is an immunoglobulin superfamily cell surface protein that is used by coxsackie virus, a RNA virus belonging to family Picornaviridae and adenovirus, a DNA virus of the family Adenoviridae. The presence of receptors determines to a large extent the tissue and species tropism of the virus. Viruses with a high mutation rate can switch receptors or adapt to use alternate receptors when the primary receptor is absent (Vlasak et al. 2005).
Integrins are prime examples of physiologically important receptors that have been exploited by enveloped and non-enveloped viruses alike for attachment and/or cell entry. Many, but not all integrins recognize RGD sequences displayed on the exposed loops of viral capsid proteins or extracellular matrix proteins. A striking feature of the interaction of non-enveloped viruses with integrins is that this often involves very similar geometry or spacing of receptor engagement around the five-fold axis of the virion (Stewart and Nemerow 2007). Virus binding can induce clustering and/or conformational changes in integrin quaternary structure and elicit cell-signaling events that increase ligand affinity as well as promote cytoskeletal rearrangement and virus internalization. Viruses that bind integrins include the human cytomegalovirus (Feire et al. 2004), hantavirus (Gavrilovskaya et al. 1998), adenovirus (Wickham et al. 1994), rotavirus (Graham et al., 2005), echovirus-1 (EV-1) (Evans and Almond 1998) and several other picornaviruses like coxsackie virus, foot and mouth-disease virus, etc. The HIV-1 envelope glycoprotein gp120 was recently shown to bind to activated integrin α4β7 that is preferentially expressed on gut CD4+ T cells. This results in activation of LFA-1, the key integrin involved in establishment of virological synapses, and enables efficient spread of HIV-1 leading to massive depletion of gut CD4+ cells (Arthos et al. 2008).
Apart from integrins, there are numerous other cell surface molecules that act as viral receptors. Receptor binding leads to uptake of virus, most often into endosomes and also triggers signaling events in the cell. It also serves as a cue that induces conformational changes necessary for membrane fusion and penetration. Some viruses use multiple attachment factors and receptors in parallel or in succession. In enveloped viruses, the spike glycoproteins play a role as membrane fusion factors and/or receptor destroying enzymes. Specific examples of this are the hemagglutinin of influenza virus with bound sialic acid (Skehel and Wiley 2000) and the gp120 of HIV-1 bound to CD4 (Kwong et al. 1998). After gp120 binding CD4 and the coreceptor (CXCR4 or CCR5), the ectodomain of the fusogenic gp41 peptide undergoes a conformational change leading to its activation. This initiates membrane fusion by exposing a hydrophobic N-terminal core of the trimeric gp41 (Furuta et al. 1998). The activity of the gp41/gp120 complex is precisely regulated. At the level of its amino acid sequence, gp41 shares many features with other viral envelope glycoproteins involved in membrane fusion. In non-enveloped viruses, projections or indentations on the capsid surface are the receptor binding domains. Adenoviruses have prominent homotrimeric fibres with globular knobs that project from each of the 12 vertices. The X-ray crystal structure of the adenovirus knob, together with the N-terminal domain of the coxsackie and adenovirus receptor (CAR), shows a large contact area on the lateral side of each subunit in the knob (Bewley et al. 1999). The penton base of many adenovirus subfamilies contains an exposed RGD sequence that associates with integrins (Stewart et al. 2003). In rhinoviruses and entroviruses, including the poliovirus, the receptors bind in a cleft in the capsid surface called the “canyon” (Rossmann et al. 2002). For some viruses, binding may cause destabilization of the particle, which is the first step towards uncoating.
Endocytic routes for virus internalization
Although some viruses release their genomes into the cell by direct fusion with the plasma membrane, most viruses enter cells via endocytosis (Pelkmans and Helenius 2003). Several viruses like murine leukaemia virus, avian leucosis virus, HIV and VSV use the cortical actin cytoskeleton, together with myosin II, for directed movement along microvilli or filopodia towards endocytic ‘hot spots’ on the cell.
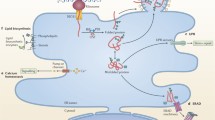
Multiple endocytic pathways simultaneously operate at the cell surface and differ in their mechanism of formation, molecular machinery and cargo destination (Fig. 1). Endocytic compartments are dynamic structures that undergo complex trafficking and sorting events, which are regulated by inherent signals on the internalized receptor, membrane composition and signaling events. After internalization, virus particles are sequestered in endocytic organelles until the proper conditions are met for release of the viral genome. Endosomes offer a convenient and often rapid transit system across the plasma membrane and through a crowded cytoplasm via the cellular microtubular network. It also provides protection to the virus from detection by the host’s innate immune defences. For viruses that replicate in the nucleus, the endosome can deliver its viral cargo to the vicinity of the nuclear pore, ready for translocation into the nucleoplasm (Whittaker and Helenius 1998).

Multiple endocytic pathways operate at the plasma membrane and are utilized in viral entry. The clathrin coated pit pathway is the best characterized endocytic pathway and is used for entry by many viruses like the influenza virus, the herpes simplex virus (HSV), HIV-1, adenovirus and poliovirus. The induced caveolar pathway is the main entry portal for polyomaviruses like SV40 and echovirus. Many viruses are also internalized by clathrin and caveolin independent pathways, which are still not well understood in terms of cargo specificity and molecular players. Though the cartoon shows influenza virus, HSV, poliovirus and SV40 virus internalization via a clathrin and caveolin independent pathway, the independent pathway utilized by one virus may differ from that used by another virus. Macropinocytosis is triggered by virus binding and utilized by vaccinia virus and adenovirus
For many enveloped viruses a low pH in the sorting/late endosome is the trigger for conformational changes necessary to initiate membrane fusion events leading to release of the viral genome at its replication site. This is well established for influenza virus, VSV and the Semliki Forest virus (Gaudin et al. 1995). Among non-enveloped viruses, several families, including picornaviruses and polyomaviruses do not require a low pH for penetration (Hogle 2002).
Viruses in turn have also served as important tools to understand the functioning of endocytic pathways. Together with rapid advances in high-resolution and multicolour live cell imaging techniques, our understanding of viral entry pathways and interactions between viral and cellular structures has substantially improved. We present here a brief overview of the major endocytic pathways operating in mammalian cells and their exploitation by viruses to gain access into cells.
Clathrin coated pit pathway
Clathrin-mediated endocytosis is considered to be the major pathway for receptor internalization in metazoans, and a large array of protein and lipid interactions have been deciphered (Sorkin 2004). This pathway can mediate the constitutive uptake of ligands such as transferrin and low-density lipoprotein (LDL) as well as ligand-triggered uptake of receptor proteins such as the epidermal growth factor receptor. New imaging technologies have provided a real-time, detailed view that allows a deeper understanding of clathrin-mediated endocytosis (Merrifield et al. 2002). Clathrin and associated proteins assemble on the intracellular face of the plasma membrane to form invaginations that pinch off through the action of the large GTPase dynamin. The clathrin-coated pit (CCP) endocytic pathway (Fig. 2) is an equally important gate for entry of many viruses and transports viruses together with their receptors into early endosomes (Brodsky et al. 2001). As they mature into early endosomes, clathrin-coated vesicles shed their protein coat and become acidified (Fig. 2). Early endosomes are major sorting stations for internalized cargo, which can be recycled to the plasma membrane or progress to late endosomes and subsequently to lysosomes. Depending upon the pH threshold, the site of penetration can be at the early or late endosomes. Several viruses, including influenza virus and VSV have been identified in clathrin-coated vesicles by electron microscopy (Marsh and Helenius 2006). Many non-enveloped viruses also use the CCP pathway for entry and infection. Well-studied examples include the reovirus, which in common with influenza virus enters by de novo formation of CCPs, poliovirus, which was observed in coated vesicles by electron microscopy (Zeichhardt et al. 1985) and human adenoviruses, which replicate and produce progeny virions within the nucleus of an infected cell. Parvoviruses, which are among the smallest animal DNA viruses also enter cells via the CCP. The infection by canine parvovirus (CPV) and adeno-associated virus type 2 (AAV2) was inhibited by over-expression of a dominant negative mutant (K44A) of dynamin 2, and by treatment with lysosomotropic agents, including ammonium chloride and bafilomycin A, suggesting the involvement of the CCP pathway to late endosomes/lysosomes (Bartlett et al. 2000; Basak and Turner 1992; Parker and Parrish 2000). Shortly after uptake, CPV capsids colocalize with transferrin in perinuclear recycling endosomes (Parker and Parrish 2000). The incoming viruses are exposed to the acidic milieu of endosomes within minutes and many respond to the pH drop by undergoing changes that lead to acid-dependent disruption of the endosomal membrane. In some cases, such as for the Ebola virus, the SARS coronavirus, and the nonenveloped mammalian reoviruses, acidic pH alone is not sufficient to induce fusion. In these cases, proteolytic cleavage of viral proteins by endosomal proteases is also required to make the virus penetration-competent (Chandran et al. 2005; Ebert et al. 2002).

Entry of a pH dependent enveloped virus. Many viruses are endocytosed via a clathrin-dependent pathway into clathrin coated pits. The clathrin coat is rapidly lost inside the cell and the vesicle matures into an early endosome. The acidic pH of the endosome brings about a conformational change in the viral envelope protein leading to fusion of the viral and endosomal membranes and capsid release into the cytosol. Genome release often occurs close to the viral replication centre. After capsid disassembly the viral RNA can initiate the replication cycle
Caveolar Pathway
The caveolar pathway is a well characterized entry portal for the non-enveloped simian virus 40 (SV40) (Pelkmans and Helenius 2002), mouse polyomavirus (Richterova et al. 2001) and EV-1, a picornavirus that binds to integrins (Pietiainen et al. 2004). Though this endocytic route is dynamin and cholesterol dependent, compared to clathrin-mediated entry, internalization of caveolae is much slower and the resulting vesicles do not become acidified. The caveolar pathway takes the majority of internalized viruses to pH neutral organelles called caveosomes from where cargo is sorted depending upon specific cues (Pelkmans et al. 2004).
Caveolae are also known to be the major signal initiating centres within the cell (Ceresa and Schmid 2000). As a result viruses that utilize this pathway also activate signaling in cells. For example, EV-1 utilizes integrins α2β1 as the entry receptor. Virus binding leads to cross-linking of the integrin complexes, its re-localization to caveolae and subsequent internalization in a protein kinase c-dependent manner (Marjomaki et al. 2002; Upla et al. 2004). Exogenously added glycosphingolipids can activate c-Src resulting in caveolin-1 phosphorylation and increased caveolar dynamics. Viruses like SV40 can stimulate c-Src at the site of glycosphingolipid crosslinking, by locally increasing the concentration of GM1. Thus c-Src plays an important role for SV40 internalization and infection (Pelkmans et al. 2005). RNAi silencing screens for the entire human kinome have demonstrated the involvement of nearly 80 kinases in caveolar endocytosis in HeLa cells (Pelkmans et al. 2005). Many of these kinases are also known to regulate integrin signaling and actin dynamics. Only a partial overlap was observed between the kinases involved in caveolar endocytosis versus those involved in clathrin mediated uptake of VSV, highlighting the complexity of these processes.
Clathrin and dynamin Independent pathways
Recent studies on the mechanisms of endocytosis have revealed a startling diversity of ways by which membrane proteins and lipids are internalized from the surface of eukaryotic cells. In addition to the classical receptor-dependent endocytic mechanisms, many pathways that do not utilize the clathrin/caveolar-coat or the ubiquitous dynamin GTPAse for vesicle detachment have been discovered (Nichols and Lippincott-Schwartz 2001; Sabharanjak et al. 2002). The entry of many viruses is implicated to occur through these pathways, based mainly on the presence of viruses in non-coated vesicles by electron microscopy (Marsh and Helenius 2006). In addition, many viruses can exploit several endocytic pathways, enabling them to infect a wide range of cells under various conditions, the well studied examples of this kind being SV40 (Damm et al. 2005), herpes simplex virus (Nicola and Straus 2004) and the influenza virus (Sieczkarski and Whittaker 2002). Direct visualization in a virus tracking experiment has shown the entry of influenza viruses simultaneously in two pathways. Whereas most viruses are internalized through clathrin-mediated endocytosis by promoting the de novo formation of clathrin-coated pits at the viral binding site, the remaining virus particles enter through a clathrin- and caveolin-independent pathway (Rust et al. 2004).
A novel endocytic pathway involved in the endocytosis of GPI-anchored proteins (GPI-APs) and a bulk of the fluid phase in the cell has been described (Kalia et al. 2006; Sabharanjak et al. 2002). Endosomes of GPI-APs and fluid, also called GEECs (GPI-AP enriched early endosomal compartments) are generated from primary endocytic events and are more acidic than sorting endosomes derived from the clathrin-coated pit pathway. The GEECs ultimately deliver their contents to sorting endosomes. This pathway is also the major route of uptake of cholera toxin by the cell (Kalia et al. 2006). Studies suggest that the GEEC pathway could represent a key pathway for diverse functions across phyla. It would be interesting to see if this pathway is also exploited by viruses for entry into cells. A recent study showed that a majority of the echoviruses enters via a fluid phase pathway (Karjalainen et al. 2008), but the molecular players of the pathway remain to be characterized. Our initial studies using fluorescently labelled hepatitis E virus like particles have shown that a majority of the particles co-localize with fluid endosomes, but show no overlap with sorting endosomes marked with transferrin, indicating internalization via a clathrin-independent pathway (MK, unpublished data).
Macropinocytosis
Macropinocytosis is generally considered to be a non-specific mechanism for internalization that is primarily driven by actin. It occurs in response to cell stimulation when large folds of the plasma membrane fuse back to form huge endosomes. The finding that macropinosomes can become acidified and intersect with sorting vesicles (Hewlett et al. 1994), makes them possible routes of entry for a wide variety of viruses. Vaccinia virus can activate p21-activated kinase 1 (PAK1) leading to macropinocytosis and viral entry (Mercer and Helenius 2008). A recent study on adenovirus has shown integrin-dependent virus binding leading to activation of macropinocytosis and virus internalization into macropinosomes (Amstutz et al. 2008). Macropinocytic uptake of HIV-1 into macrophages has been demonstrated, however most of the virions internalized this way were found to be degraded (Marechal et al. 2001).
The presence of multiple pathways and previously unobserved endocytic organelles challenges established assumptions about the entry of many viruses. It was recently observed that influenza virus, which was thought to enter by the clathrin-coated pit pathway (Matlin et al. 1981), can also infect cells in which the clathrin-coated vesicle transport was blocked (Sieczkarski and Whittaker 2002). The electron microscopic visualization of many viruses in non-coated vesicles is also indicative of the presence of non-clathrin pathways. The GM1-associated virus, SV40 is also efficiently internalized in a dynamin and clathrin-independent manner in fibroblasts derived from caveolin-1 knockout mice (Damm et al. 2005). Studies have also shown that polyomavirus can enter cells via a non-clathrin, non-caveolin and dynamin independent pathway (Gilbert and Benjamin 2000). More studies will reveal whether the clathrin-independent pathways used by influenza virus, SV40 and polyomavirus are similar or different in terms of their regulation. Recent studies have revisited poliovirus and have shown that clathrin-mediated endocytosis is not essential for its entry into cells. Live cell imaging studies in HeLa cells found poliovirus to enter cells through a clathrin-, caveolin-, flotillin-, and microtubule-independent, but tyrosine kinase- and actin-dependent endocytic mechanism (Brandenburg et al. 2007). This study demonstrates that uncoating of the poliovirus capsid does not occur on the cell surface as previously believed, but within the cell. In another study, poliovirus entry into human brain microvascular endothelial cells found that though uncoating was found to occur inside the cell, virus entry depended on both caveolin and dynamin, and the virus was detected in caveolin-containing vesicles within the cytoplasm (Coyne et al. 2007). It thus seems likely that poliovirus uses different mechanisms as it enters different cell types. As the cellular entry of more viruses is characterized, this may be more a rule than an exception. The cellular context is likely to play a determining role in the entry pathway a virus takes in a given cell type.
Genome release for enveloped and non-enveloped viruses
The mechanisms of genome release are widely different between enveloped and non-enveloped viruses because of different surface compositions. Enveloped viruses fuse with the plasma or endosome membranes, thereby exposing the genome or capsid to the cytosol, whereas non-enveloped viruses partially disrupt membranes to release viral nucleic acids. Many viruses require the low pH environment in the endosome to trigger a conformational change in the viral glycoprotein that leads to the exposure and insertion of the fusion peptide into the cellular membrane.
Viral Glycoprotein mediated membrane fusion
Emerging single-virus tracking experiments have also aided in understanding the fusion mechanisms of enveloped viruses in live cells. Enveloped animal viruses employ membrane fusion to enter the cytoplasm, and this process is mediated by the viral surface proteins. For many viruses the envelope glycoproteins are synthesized as “inactive” precursors that undergo proteolytic cleavage to become fully active. After binding to the receptor, a conformational change, sometimes pH triggered, is necessary to expose a hydrophobic “fusion-peptide”, which inserts into the cell membrane and mediates the fusion of viral and cellular membranes (Jahn et al. 2003; Peisajovich and Shai 2002). Viral glycoproteins are thus classified as pH independent or pH dependent based on the trigger required to activate their membrane fusion potential. Enveloped viruses like herpes simplex virus 1 (HSV-1), Sendai virus and retroviruses like HIV-1 have pH independent fusion proteins and can penetrate cells by direct fusion at the plasma membrane. The primed and triggered conformational changes in the fusion proteins result in close apposition of the viral and cellular bilayers, membrane merger, and cytoplasmic delivery of the viral nucleocapsid. Well-studied proteins of this category are the influenza virus hemagglutinin (HA), and the tick-borne encephalitis virus (TBEV) E protein (Skehel and Wiley 2000) and the Ebola virus glycoprotein (Chandran et al. 2005). Fusion appears to be driven, in many cases, by a coiled-coiled structure intimately involved with the conformational changes that accompany the process (Matthews et al. 2000). A number of studies have been carried out on viral envelope glycoproteins, including their X-ray crystal structure determination (Heldwein et al. 2006; Roche et al. 2006). Amongst the best characterized membrane fusion mechanisms are those of the influenza virus whose integral membrane protein M2 forms pH-gated proton channels in the viral lipid envelope (Pinto et al. 1992). The low pH of an endosme activates the M2 channel before haemagglutinin-mediated fusion. Proton influx leads to acidification of the viral interior facilitating dissociation of the matrix protein from the viral nucleoproteins—a step required for unpacking of the viral genome (Helenius 1992). Inhibiting the proton conductance of M2 using the anti-viral drugs amantadine or rimantadine inhibits viral replication (Pinto et al. 1992). The structure of the tetrameric M2 channel in complex with rimantadine, an anti-viral drug that inhibits proton conductance of M2, has recently been determined by NMR (Schnell and Chou 2008). In the closed state, four tightly packed transmembrane helices define a narrow proton channel with a ‘tryptophan gate’ locked by intermolecular interactions with aspartic acid. The transmembrane helical packing destabilizes on lowering the pH and opens to allow proton influx through water. The NMR study suggests that rimantadine binds at four equivalent sites near the gate and stabilizes the closed conformation of the pore and thus works as an anti-viral drug. The discovery of an external drug-binding site was unexpected since earlier studies had suggested that drug-binding site is lined by residues that are mutated in drug-resistant viruses (Grambas et al. 1992; Pinto and Lamb 1995). The authors suggest that drug-resitant mutants either increase the hydrophilicity of the pore or weaken helix-helix packing, that leads to channel opening. However, in sharp contrast, another X-ray crystallography study on the M2 channel in the presence and absence of the drug amantadine shows the channel to be open with helices spreading out on the cytoplasmic side to widen the Tryptophan gate, and a single amantadine molecule plugging the open pore (Stouffer et al. 2008). In the open structure, four critical amino-acid residues project side chains into the pore near the amantadine binding site, and it is predicted that these substitutions will impare drug binding and hence inhibition.
Penetration by non-enveloped viruses
Whereas a detailed understanding of the entry process and fusion mechanisms are available for many enveloped viruses, much less is know about the entry of non-enveloped viruses. Because these viruses do not have a membrane, the entry mechanism cannot involve membrane fusion, but, capsid-dependent mechanisms for penetrating the cell membrane or lysing endosomes are likely to be involved. Many studies indicate that penetration of non-enveloped viruses also involves cooperative changes in virus particles triggered by receptor binding or low pH (Hogle 2002; Rossmann et al. 2002). Most non-enveloped virus capsid proteins have short, membrane altering, amphiphatic or hydrophobic sequences that mediate membrane penetration (Banerjee and Johnson 2008). These “penetration proteins” may undergo primed and triggered conformational transitions that allow them to interact with membranes. Adenovirus particles exposed to low pH undergo a conformational change in the penton base that renders the particle hydrophobic and capable of permeabilizing liposomes (Blumenthal et al. 1986).
The entry mechanism of poliovirus, the best-characterized member of the family Picornaviridae, has been examined in much detail. On receptor binding, the poliovirus capsid undergoes conformational rearrangements, exposing the N-termius of the capsid protein VP1 and the myristoylated autoclevage peptide VP4, which can insert into liposomes (Belnap et al. 2000; Fricks and Hogle 1990). These newly exposed sequences are then thought to form a transmembrane pore through which the genomic RNA may be extruded into the cytoplasm (Hogle 2002). The reovirus protein μ1 is found in virions mostly as autolytic fragments μ1N and μ1C, which participate directly in membrane penetration during virus entry into cells (Agosto et al. 2006; Chandran et al. 2002). The bluetongue virus, another member of the non-enveloped Reoviridae family, has two capsid proteins, a receptor-binding protein, VP2, and a second protein, VP5, which shares structural features with class I fusion proteins of enveloped viruses. The VP5 can undergo pH-dependent conformational changes that render it capable of interacting with cellular membranes (Forzan et al. 2004). Non-enveloped Nodaviruses, like the flockhouse virus have a unique capsid protein associated to a gamma peptide of 44 residues, which results from the self-clevage of a capsid protein precursor (Wery et al. 1994). The gamma peptide has a lipid-binding domain and can permeabilize biological membranes allowing genome translocation through the membrane (Banerjee and Johnson 2008; Bong et al. 1999). One of the four structural peptides, pep46, a 46-amino acid amphiphilic peptide of the infectious bursal disease virus deforms synthetic membranes and induces pores with a diameter of less than 10 nm, which are visualized by cryoelectron microscopy (Galloux et al. 2007).
Virus induced signaling
To prepare a cell for the invasion, virus particles can trigger intracellular events as soon as they bind to the plasma membrane. This generally involves the binding to and the cross-linking of cell-surface molecules such as glycosphingolipids, receptor tyrosine kinases and integrins. Initiation of signaling at the cell surface leads to the propogation of a downstream cascade from a single site on the plasma membrane. Signaling can lead to sequestration of the virus particle to an endocytic hotspot from where it is internalized and can reorganise the actin cytoskeleton to form membrane ruffles and increase macropinocytosis. Signaling events have been studied extensively in the entry of SV40 and adenoviruses. The SV40 binds to GM1 and activates tyrosine kinases leading to endocytosis via the normally dormant caveolae (Pelkmans et al. 2002). A second signal is induced in the caveosome to target SV40 to the ER (Pelkmans and Helenius 2003). Adenovirus 2 binds and clusters integrins leading to activation of a variety of protein kinases, phosphatidylinositol-3-OH kinase (PI3K), and small GTPAses (Greber 2002). The activation of focal adhesion kinase (FAK) impacts downstream molecules such as the ERK1/ERK2 mitogen-activated protein kinases. Major changes occur in cell surface dynamics, and in microtubule-mediated transport, which promote internalization, penetration and trafficking of the incoming virus. Activation of PI3K also results in the inhibition of apoptosis (an innate defense mechanism) and increased survival of host cells, which allows completion of the virus replication cycle and production of progeny virions. The human cytomegalovirus, a member of the family Herpesviridae, activates several signaling pathways through the interaction between its envelope glycoprotein B and the epidermal growth-factor receptor (Wang et al. 2003). Poliovirus entry into HeLa cells requires tyrosine kinase activity (Brandenburg et al. 2007), and it is likely that receptor-induced signals will prove to be important for infection in a variety of cell types. Virus induced signaling events thus affect host innate immune responses as well as cell survival and cell proliferation, all of which contribute positively to the the formation of new virions.
Perspectives
Despite being simple in structure, viruses are remarkably versatile in their quest to infect and multiply. During infection, many cellular processes are subjected to manipulation by viruses. Therefore, unravelling viral pathogenesis and the spread of infection will require a detailed understanding of how viruses exploit the cellular machinery of the host. The first contact of a virus with the target cell, which includes binding and penetration of the plasma membrane is perhaps the most important decision point in the infection cycle. Our current understanding of the complex phenomenon of viral infection would not have been possible without technical developments in the field of light and electron microscopy. Viral entry events can now be analysed with increased spatial and temporal resolution. Single-virus imaging in live cells has readdressed and redefined many aspects of viral entry. These have also been complemented with excellent protein crystallography and ultrastructural studies. Future work will certainly see detailed characterization of novel endocytic pathways that have surfaced as viral entry portals. This will require high-resolution time-lapse imaging of the dynamics of multiple pathway components combined with computational image analysis. The plasticity observed in many cellular pathways, and the observation that viruses can make use of multiple internalization pathways suggests that direct inhibition of virus entry may not be feasible. The increased level of complexity demands investigations of viral infection at a systems biology level. Future challenges lie in getting a clear understanding of the link between viral entry and cellular signaling. This is necessary for the development of new antiviral drugs directed against cellular targets, such as signaling intermediates and/or regulators, rather than viral ones. Another important avenue will be to establish that the pathways observed in tissue culture experiments in vitro also operate in vivo during a productive infection. With the rapid development of in vivo imaging methods it should become possible to track virus particles in live tissues and animals. Thus, despite being sometimes lethal biological warriors, viruses continue to enthral us, as we use them as powerful tools to unravel cellular mysteries.
References
Agosto MA, Ivanovic T, Nibert ML (2006) Mammalian reovirus, a nonfusogenic nonenveloped virus, forms size-selective pores in a model membrane. Proc Natl Acad Sci USA 103:16496–16501
Amstutz B, Gastaldelli M, Kalin S, Imelli N, Boucke K, Wandeler E, Mercer J, Hemmi S, Greber UF (2008) Subversion of CtBP1-controlled macropinocytosis by human adenovirus serotype 3. EMBO J 27:956–969
Arthos J, Cicala C, Martinelli E, Macleod K, Van Ryk D, Wei D, Xiao Z, Veenstra TD, Conrad TP, Lempicki RA et al (2008) HIV-1 envelope protein binds to and signals through integrin alpha4beta7, the gut mucosal homing receptor for peripheral T cells. Nat Immunol 9:301–309
Banerjee M, Johnson JE (2008) Activation, exposure and penetration of virally encoded, membrane-active polypeptides during non-enveloped virus entry. Curr Protein Pept Sci 9:16–27
Barth H, Schafer C, Adah MI, Zhang F, Linhardt RJ, Toyoda H, Kinoshita-Toyoda A, Toida T, Van Kuppevelt TH, Depla E et al (2003) Cellular binding of hepatitis C virus envelope glycoprotein E2 requires cell surface heparan sulfate. J Biol Chem 278:41003–41012
Bartlett JS, Wilcher R, Samulski RJ (2000) Infectious entry pathway of adeno-associated virus and adeno-associated virus vectors. J Virol 74:2777–2785
Basak S, Turner H (1992) Infectious entry pathway for canine parvovirus. Virology 186:368–376
Belnap DM, Filman DJ, Trus BL, Cheng N, Booy FP, Conway JF, Curry S, Hiremath CN, Tsang SK, Steven AC, Hogle JM (2000) Molecular tectonic model of virus structural transitions: the putative cell entry states of poliovirus. J Virol 74:1342–1354
Bernfield M, Gotte M, Park PW, Reizes O, Fitzgerald ML, Lincecum J, Zako M (1999) Functions of cell surface heparan sulfate proteoglycans. Annu Rev Biochem 68:729–777
Bewley MC, Springer K, Zhang YB, Freimuth P, Flanagan JM (1999) Structural analysis of the mechanism of adenovirus binding to its human cellular receptor, CAR. Science 286:1579–1583
Blumenthal R, Seth P, Willingham MC, Pastan I (1986) pH-dependent lysis of liposomes by adenovirus. Biochemistry 25:2231–2237
Bong DT, Steinem C, Janshoff A, Johnson JE, Reza Ghadiri M (1999) A highly membrane-active peptide in Flock House virus: implications for the mechanism of nodavirus infection. Chem Biol 6:473–481
Brandenburg B, Lee LY, Lakadamyali M, Rust MJ, Zhuang X, Hogle JM (2007) Imaging poliovirus entry in live cells. PLoS Biol 5:e183
Brodsky FM, Chen CY, Knuehl C, Towler MC, Wakeham DE (2001) Biological basket weaving: formation and function of clathrin-coated vesicles. Annu Rev Cell Dev Biol 17:517–568
Cantin R, Methot S, Tremblay MJ (2005) Plunder and stowaways: incorporation of cellular proteins by enveloped viruses. J Virol 79:6577–6587
Ceresa BP, Schmid SL (2000) Regulation of signal transduction by endocytosis. Curr Opin Cell Biol 12:204–210
Chandran K, Farsetta DL, Nibert ML (2002) Strategy for nonenveloped virus entry: a hydrophobic conformer of the reovirus membrane penetration protein micro 1 mediates membrane disruption. J Virol 76:9920–9933
Chandran K, Sullivan NJ, Felbor U, Whelan SP, Cunningham JM (2005) Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science 308:1643–1645
Chen Y, Maguire T, Hileman RE, Fromm JR, Esko JD, Linhardt RJ, Marks RM (1997) Dengue virus infectivity depends on envelope protein binding to target cell heparan sulfate. Nat Med 3:866–871
Chung CS, Hsiao JC, Chang YS, Chang W (1998) A27L protein mediates vaccinia virus interaction with cell surface heparan sulfate. J Virol 72:1577–1585
Coyne CB, Kim KS, Bergelson JM (2007) Poliovirus entry into human brain microvascular cells requires receptor-induced activation of SHP-2. EMBO J 26:4016–4028
Damm EM, Pelkmans L, Kartenbeck J, Mezzacasa A, Kurzchalia T, Helenius A (2005) Clathrin- and caveolin-1-independent endocytosis: entry of simian virus 40 into cells devoid of caveolae. J Cell Biol 168:477–488
Dechecchi MC, Tamanini A, Bonizzato A, Cabrini G (2000) Heparan sulfate glycosaminoglycans are involved in adenovirus type 5 and 2-host cell interactions. Virology 268:382–390
Ebert DH, Deussing J, Peters C, Dermody TS (2002) Cathepsin L and cathepsin B mediate reovirus disassembly in murine fibroblast cells. J Biol Chem 277:24609–24617
Evans DJ, Almond JW (1998) Cell receptors for picornaviruses as determinants of cell tropism and pathogenesis. Trends Microbiol 6:198–202
Feinberg H, Mitchell DA, Drickamer K, Weis WI (2001) Structural basis for selective recognition of oligosaccharides by DC-SIGN and DC-SIGNR. Science 294:2163–2166
Feire AL, Koss H, Compton T (2004) Cellular integrins function as entry receptors for human cytomegalovirus via a highly conserved disintegrin-like domain. Proc Natl Acad Sci USA 101:15470–15475
Forzan M, Wirblich C, Roy P (2004) A capsid protein of nonenveloped Bluetongue virus exhibits membrane fusion activity. Proc Natl Acad Sci USA 101:2100–2105
Fricks CE, Hogle JM (1990) Cell-induced conformational change in poliovirus: externalization of the amino terminus of VP1 is responsible for liposome binding. J Virol 64:1934–1945
Furuta RA, Wild CT, Weng Y, Weiss CD (1998) Capture of an early fusion-active conformation of HIV-1 gp41. Nat Struct Biol 5:276–279
Galloux M, Libersou S, Morellet N, Bouaziz S, Da Costa B, Ouldali M, Lepault J, Delmas B (2007) Infectious bursal disease virus, a non-enveloped virus, possesses a capsid-associated peptide that deforms and perforates biological membranes. J Biol Chem 282:20774–20784
Gaudin Y, Ruigrok RW, Brunner J (1995) Low-pH induced conformational changes in viral fusion proteins: implications for the fusion mechanism. J Gen Virol 76(Pt 7):1541–1556
Gavrilovskaya IN, Shepley M, Shaw R, Ginsberg MH, Mackow ER (1998) beta3 Integrins mediate the cellular entry of hantaviruses that cause respiratory failure. Proc Natl Acad Sci USA 95:7074–7079
Geijtenbeek TB, Kwon DS, Torensma R, van Vliet SJ, van Duijnhoven GC, Middel J, Cornelissen IL, Nottet HS, KewalRamani VN, Littman DR et al (2000) DC-SIGN, a dendritic cell-specific HIV-1-binding protein that enhances trans-infection of T cells. Cell 100:587–597
Gilbert JM, Benjamin TL (2000) Early steps of polyomavirus entry into cells. J Virol 74:8582–8588
Giroglou T, Florin L, Schafer F, Streeck RE, Sapp M (2001) Human papillomavirus infection requires cell surface heparan sulfate. J Virol 75:1565–1570
Graham KL, Fleming FE, Halasz P, Hewish MJ, Nagesha HS, Holmes IH, Takada Y, Coulson BS (2005) Rotaviruses interact with alpha4beta7 and alpha4beta1 integrins by binding the same integrin domains as natural ligands. J Gen Virol 86:3397–3408
Grambas S, Bennett MS, Hay AJ (1992) Influence of amantadine resistance mutations on the pH regulatory function of the M2 protein of influenza A viruses. Virology 191:541–549
Greber UF (2002) Signalling in viral entry. Cell Mol Life Sci 59:608–626
Guglielmi KM, Johnson EM, Stehle T, Dermody TS (2006) Attachment and cell entry of mammalian orthoreovirus. Curr Top Microbiol Immunol 309:1–38
Heldwein EE, Lou H, Bender FC, Cohen GH, Eisenberg RJ, Harrison SC (2006) Crystal structure of glycoprotein B from herpes simplex virus 1. Science 313:217–220
Helenius A (1992) Unpacking the incoming influenza virus. Cell 69:577–578
Hewlett LJ, Prescott AR, Watts C (1994) The coated pit and macropinocytic pathways serve distinct endosome populations. J Cell Biol 124:689–703
Hogle JM (2002) Poliovirus cell entry: common structural themes in viral cell entry pathways. Annu Rev Microbiol 56:677–702
Isa P, Arias CF, Lopez S (2006) Role of sialic acids in rotavirus infection. Glycoconj J 23:27–37
Jahn R, Lang T, Sudhof TC (2003) Membrane fusion. Cell 112:519–533
Kalia M, Kumari S, Chadda R, Hill MM, Parton RG, Mayor S (2006) Arf6-independent GPI-anchored protein-enriched early endosomal compartments fuse with sorting endosomes via a Rab5/phosphatidylinositol-3’-kinase-dependent machinery. Mol Biol Cell 17:3689–3704
Kalia M, Chandra V, Rahman SA, Sehgal D, Jameel S (2009) Heparan sulfate proteoglycans are required for cellular binding of the hepatitis E virus ORF2 capsid protein and for viral infection. J Virol [Epub ahead of print]
Karjalainen M, Kakkonen E, Upla P, Paloranta H, Kankaanpaa P, Liberali P, Renkema GH, Hyypia T, Heino J, Marjomaki V (2008) A Raft-derived, Pak1-regulated entry participates in alpha2beta1 integrin-dependent sorting to caveosomes. Mol Biol Cell 19:2857–2869
Kwong PD, Wyatt R, Robinson J, Sweet RW, Sodroski J, Hendrickson WA (1998) Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 393:648–659
Lindahl U, Kusche-Gullberg M, Kjellen L (1998) Regulated diversity of heparan sulfate. J Biol Chem 273:24979–24982
Lonberg-Holm K, Philipson L (1974) Early interaction between animal viruses and cells. Monogr Virol 9:1–148
Lyon M, Gallagher JT (1998) Bio-specific sequences and domains in heparan sulphate and the regulation of cell growth and adhesion. Matrix Biol 17:485–493
Marechal V, Prevost MC, Petit C, Perret E, Heard JM, Schwartz O (2001) Human immunodeficiency virus type 1 entry into macrophages mediated by macropinocytosis. J Virol 75:11166–11177
Marjomaki V, Pietiainen V, Matilainen H, Upla P, Ivaska J, Nissinen L, Reunanen H, Huttunen P, Hyypia T, Heino J (2002) Internalization of echovirus 1 in caveolae. J Virol 76:1856–1865
Marsh M, Helenius A (2006) Virus entry: open sesame. Cell 124:729–740
Matlin KS, Reggio H, Helenius A, Simons K (1981) Infectious entry pathway of influenza virus in a canine kidney cell line. J Cell Biol 91:601–613
Matthews JM, Young TF, Tucker SP, Mackay JP (2000) The core of the respiratory syncytial virus fusion protein is a trimeric coiled coil. J Virol 74:5911–5920
Mercer J, Helenius A (2008) Vaccinia virus uses macropinocytosis and apoptotic mimicry to enter host cells. Science 320:531–535
Merrifield CJ, Feldman ME, Wan L, Almers W (2002) Imaging actin and dynamin recruitment during invagination of single clathrin-coated pits. Nat Cell Biol 4:691–698
Mondor I, Ugolini S, Sattentau QJ (1998) Human immunodeficiency virus type 1 attachment to HeLa CD4 cells is CD4 independent and gp120 dependent and requires cell surface heparans. J Virol 72:3623–3634
Nichols BJ, Lippincott-Schwartz J (2001) Endocytosis without clathrin coats. Trends Cell Biol 11:406–412
Nicola AV, Straus SE (2004) Cellular and viral requirements for rapid endocytic entry of herpes simplex virus. J Virol 78:7508–7517
Olofsson S, Bergstrom T (2005) Glycoconjugate glycans as viral receptors. Ann Med 37:154–172
Parker JS, Parrish CR (2000) Cellular uptake and infection by canine parvovirus involves rapid dynamin-regulated clathrin-mediated endocytosis, followed by slower intracellular trafficking. J Virol 74:1919–1930
Peisajovich SG, Shai Y (2002) New insights into the mechanism of virus-induced membrane fusion. Trends Biochem Sci 27:183–190
Pelkmans L, Helenius A (2002) Endocytosis via caveolae. Traffic 3:311–320
Pelkmans L, Helenius A (2003) Insider information: what viruses tell us about endocytosis? Curr Opin Cell Biol 15:414–422
Pelkmans L, Puntener D, Helenius A (2002) Local actin polymerization and dynamin recruitment in SV40-induced internalization of caveolae. Science 296:535–539
Pelkmans L, Burli T, Zerial M, Helenius A (2004) Caveolin-stabilized membrane domains as multifunctional transport and sorting devices in endocytic membrane traffic. Cell 118:767–780
Pelkmans L, Fava E, Grabner H, Hannus M, Habermann B, Krausz E, Zerial M (2005) Genome-wide analysis of human kinases in clathrin- and caveolae/raft-mediated endocytosis. Nature 436:78–86
Pietiainen V, Marjomaki V, Upla P, Pelkmans L, Helenius A, Hyypia T (2004) Echovirus 1 endocytosis into caveosomes requires lipid rafts, dynamin II, and signaling events. Mol Biol Cell 15:4911–4925
Pinon JD, Klasse PJ, Jassal SR, Welson S, Weber J, Brighty DW, Sattentau QJ (2003) Human T-cell leukemia virus type 1 envelope glycoprotein gp46 interacts with cell surface heparan sulfate proteoglycans. J Virol 77:9922–9930
Pinto LH, Lamb RA (1995) Understanding the mechanism of action of the anti-influenza virus drug amantadine. Trends Microbiol 3:271
Pinto LH, Holsinger LJ, Lamb RA (1992) Influenza virus M2 protein has ion channel activity. Cell 69:517–528
Pohlmann S, Zhang J, Baribaud F, Chen Z, Leslie GJ, Lin G, Granelli-Piperno A, Doms RW, Rice CM, McKeating JA (2003) Hepatitis C virus glycoproteins interact with DC-SIGN and DC-SIGNR. J Virol 77:4070–4080
Richterova Z, Liebl D, Horak M, Palkova Z, Stokrova J, Hozak P, Korb J, Forstova J (2001) Caveolae are involved in the trafficking of mouse polyomavirus virions and artificial VP1 pseudocapsids toward cell nuclei. J Virol 75:10880–10891
Roche S, Bressanelli S, Rey FA, Gaudin Y (2006) Crystal structure of the low-pH form of the vesicular stomatitis virus glycoprotein G. Science 313:187–191
Rossmann MG, He Y, Kuhn RJ (2002) Picornavirus-receptor interactions. Trends Microbiol 10:324–331
Rust MJ, Lakadamyali M, Zhang F, Zhuang X (2004) Assembly of endocytic machinery around individual influenza viruses during viral entry. Nat Struct Mol Biol 11:567–573
Sabharanjak S, Sharma P, Parton RG, Mayor S (2002) GPI-anchored proteins are delivered to recycling endosomes via a distinct cdc42-regulated, clathrin-independent pinocytic pathway. Dev Cell 2:411–423
Schnell JR, Chou JJ (2008) Structure and mechanism of the M2 proton channel of influenza A virus. Nature 451:591–595
Schwegmann-Wessels C, Herrler G (2006) Sialic acids as receptor determinants for coronaviruses. Glycoconj J 23:51–58
Shukla D, Liu J, Blaiklock P, Shworak NW, Bai X, Esko JD, Cohen GH, Eisenberg RJ, Rosenberg RD, Spear PG (1999) A novel role for 3-O-sulfated heparan sulfate in herpes simplex virus 1 entry. Cell 99:13–22
Sieczkarski SB, Whittaker GR (2002) Influenza virus can enter and infect cells in the absence of clathrin-mediated endocytosis. J Virol 76:10455–10464
Simmons G, Reeves JD, Grogan CC, Vandenberghe LH, Baribaud F, Whitbeck JC, Burke E, Buchmeier MJ, Soilleux EJ, Riley JL et al (2003) DC-SIGN and DC-SIGNR bind ebola glycoproteins and enhance infection of macrophages and endothelial cells. Virology 305:115–123
Skehel JJ, Wiley DC (2000) Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu Rev Biochem 69:531–569
Sorkin A (2004) Cargo recognition during clathrin-mediated endocytosis: a team effort. Curr Opin Cell Biol 16:392–399
Stewart PL, Nemerow GR (2007) Cell integrins: commonly used receptors for diverse viral pathogens. Trends Microbiol 15:500–507
Stewart PL, Dermody TS, Nemerow GR (2003) Structural basis of nonenveloped virus cell entry. Adv Protein Chem 64:455–491
Stouffer AL, Acharya R, Salom D, Levine AS, Di Costanzo L, Soto CS, Tereshko V, Nanda V, Stayrook S, DeGrado WF (2008) Structural basis for the function and inhibition of an influenza virus proton channel. Nature 451:596–599
Tamura M, Natori K, Kobayashi M, Miyamura T, Takeda N (2004) Genogroup II noroviruses efficiently bind to heparan sulfate proteoglycan associated with the cellular membrane. J Virol 78:3817–3826
Tsai B, Gilbert JM, Stehle T, Lencer W, Benjamin TL, Rapoport TA (2003) Gangliosides are receptors for murine polyoma virus and SV40. EMBO J 22:4346–4355
Upla P, Marjomaki V, Kankaanpaa P, Ivaska J, Hyypia T, Van Der Goot FG, Heino J (2004) Clustering induces a lateral redistribution of alpha 2 beta 1 integrin from membrane rafts to caveolae and subsequent protein kinase C-dependent internalization. Mol Biol Cell 15:625–636
Vlasak M, Goesler I, Blaas D (2005) Human rhinovirus type 89 variants use heparan sulfate proteoglycan for cell attachment. J Virol 79:5963–5970
Wang X, Huong SM, Chiu ML, Raab-Traub N, Huang ES (2003) Epidermal growth factor receptor is a cellular receptor for human cytomegalovirus. Nature 424:456–461
Wery JP, Reddy VS, Hosur MV, Johnson JE (1994) The refined three-dimensional structure of an insect virus at 2.8 A resolution. J Mol Biol 235:565–586
Whittaker GR, Helenius A (1998) Nuclear import and export of viruses and virus genomes. Virology 246:1–23
Wickham TJ, Filardo EJ, Cheresh DA, Nemerow GR (1994) Integrin alpha v beta 5 selectively promotes adenovirus mediated cell membrane permeabilization. J Cell Biol 127:257–264
WuDunn D, Spear PG (1989) Initial interaction of herpes simplex virus with cells is binding to heparan sulfate. J Virol 63:52–58
Zeichhardt H, Wetz K, Willingmann P, Habermehl KO (1985) Entry of poliovirus type 1 and Mouse Elberfeld (ME) virus into HEp-2 cells: receptor-mediated endocytosis and endosomal or lysosomal uncoating. J Gen Virol 66(Pt 3):483–492
Acknowledgments
MK is supported by “Innovative Young Biotechnologist Award” from Department of Biotechnology, Government of India. We apologize to researchers whose work has not been directly cited due to limitations of space.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kalia, M., Jameel, S. Virus entry paradigms. Amino Acids 41, 1147–1157 (2011). https://doi.org/10.1007/s00726-009-0363-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-009-0363-3