
Abstract
The red-crowned crane is one of the rarest crane species, and its population is decreasing due to loss of habitat, poisoning, and infections. Using a viral metagenomics approach, we analyzed the virome of feces from wild and captive red-crowned cranes, which were pooled separately. Vertebrate viruses belonging to the families Picornaviridae, Parvoviridae, Circoviridae, and Caliciviridae were detected. Among the members of the family Picornaviridae, we found three that appear to represent new genera. Six nearly complete genomes from members of the family Parvoviridae were also obtained, including four new members of the proposed genus “Chapparvovirus”, and two members of the genus Aveparvovirus. Six small circular DNA genomes were also characterized. One nearly complete genome showing a low level of sequence identity to caliciviruses was also characterized. Numerous viruses believed to infect insects, plants, and crustaceans were also identified, which were probably derived from the diet of red-crowned cranes. This study increases our understanding of the enteric virome of red-crowned cranes and provides a baseline for comparison to those of other birds or following disease outbreaks.
Similar content being viewed by others


Introduction
The red-crowned crane is one of the rarest crane species, and it has been designated as “endangered” in the Red List of the International Union for Conservation of Nature (IUCN, 2014). The current population is estimated to be 2,750 individuals (BirdLife International, 2015), most of which migrate seasonally. In the spring and summer, migratory populations of red-crowned cranes breed in Siberia (eastern Russia), northeastern China, and occasionally in northeastern Mongolia. In the fall, they migrate in flocks to Korea and east-central China to spend the winter. They are omnivores that feed on small fish, worms, insects, frogs, and plants.
The number of red-crowned cranes is decreasing, mainly due to the loss of habitat [1], and health threats, including food poisoning [2], nutritional metabolic disease [3], and infectious diseases [4]. So far, few virological studies have been conducted on red-crowned cranes [5].
Viral metagenomics is a new research tool that enables the discovery of putative novel pathogens [6]. It has been used recently in numerous animal virus discoveries, providing information on the diversity of animal viromes, helping to determine the etiology of diarrheal disease in animals, and identifying potential zoonotic and emerging viruses.
The objective of this study was to investigate the intestinal virome of red-crowned cranes using a viral metagenomics approach. Fecal samples from red-crowned cranes in Yancheng Biosphere Reserve, China, were collected, and viral nucleic acid sequence information was generated and analyzed.
Materials and methods
Sample collection and preparation
Ninety-three fecal samples were collected from individual red-crowned cranes in Yancheng Biosphere Reserve in Yancheng, Jiangsu Province, China, from January 2014 to May 2015. All samples were shipped on dry ice. Of the 93 fecal samples, 41 were collected from wild red-crowned cranes at a food provision site in Yancheng Biosphere Reserve, 20 were collected at a captive breeding center, and the remaining 32 were collected from ornamental animals. In order to avoid contamination, fresh samples were collected carefully by a specialized breeder able to distinguish the feces of red-crowned cranes from those of other birds. Fecal samples were resuspended individually in ten volumes of phosphate-buffered saline (PBS) and vigorously vortexed for 5 min. After centrifugation (10 min, 15,000×g), the supernatant was then collected by pipette and stored at -80 °C before nucleic acid extraction.
Viral metagenomic analysis
The supernatant of 500 μl of each sample was filtered through a 0.45-μm filter (Millipore) to remove eukaryotic and bacterial cell-sized particles. The filtrates were then treated with DNase and RNase at 37 °C for 60 min to digest unprotected nucleic acid [7, 8]. The remaining total nucleic acid was then extracted using a QiaAmp Viral RNA Mini Kit (QIAGEN) according to the manufacturer’s protocol. Six libraries were then constructed using a Nextera XT DNA Sample Preparation Kit (Illumina) and sequenced using the Illumina MiSeq platform with 250 base-paired ends with dual barcoding for each pool. The information for each library is shown in Supplementary Table S1. For bioinformatics analysis, paired-end reads of 250 bp generated by MiSeq were debarcoded using vendor software from Illumina. An in-house analysis pipeline running on a 32-node Linux cluster was used to process the data. Clonal reads were removed, and low-sequencing-quality tails were trimmed using a Phred quality score of ten as the threshold. Adaptors were trimmed using the default parameters of VecScreen, which was NCBI BLASTn [9] with specialized parameters designed for adaptor removal. The cleaned reads were assembled de novo using SOAPdenovo2 version r240 with a kmer size of 63 with default settings [10]. The assembled contigs, along with singlets, were aligned to an in-house viral proteome database using BLASTx with an E-value cutoff of < 10−5 [11].
PCR screening and genome sequencing
PCR screening was performed for the picornaviruses, parvoviruses, circoviruses and caliciviruses in the fecal samples. Inverse PCR was used to generate the complete genome sequence of the novel circoviruses. Sequence gaps were bridged by RT-PCR or PCR to acquire the complete coding sequences of the picornaviruses and parvoviruses in the fecal samples. The Sanger method was used for sequencing the amplicons.
Phylogenetic analysis
Phylogenetic analysis was performed based on the deduced amino acid sequences from the present study, their closest BLASTx matches in GenBank (those with an E-value cutoff of < 10−5 and sharing the highest identity with our sequences were selected as reference strains), and representative members of related viral species or genera. Sequence alignment was performed using CLUSTAL W with the default settings. Phylogenetic trees with 500 bootstrap resamples of the alignment data sets were generated using the maximum-likelihood method in MEGA5.0 [12]. Bootstrap values (based on 500 replicates) for each node are given. Putative ORFs in the genome were predicted using a combination of Geneious 8.1 software and NCBI ORF finder. Putative exons and introns were predicted by Netgenes2 at http://www.cbs.dtu.dk/services/NetGene2/, and the stem-loop structures of circoviruses were predicted by Mfold at http://unafold.rna.albany.edu/?q=mfold.
Results
Viral metagenomic overview
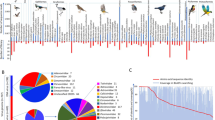
Six libraries were constructed, and the total raw sequence reads generated by the Illumina MiSeq 2x250 base runs are shown in Supplementary Table S1. Raw sequence reads were binned by barcode and quality-filtered, leaving high-quality reads, which were assembled de novo within each barcode. The resulting sequence contigs and singlets were compared with the viral reference database and the GenBank non-redundant protein database using a BLASTx search with an E value cutoff of < 10−5. Deduced amino acid sequences similar to those of known or putative eukaryotic viral proteins are summarized in Supplementary Table S2. A total of 227,383 sequence reads had the best matches with viral proteins, accounting for 11.72% of the total number of unique reads. Among them, 40,017 reads belonged to unclassified viruses, accounting for 2.06% of the total number of unique reads. 78.96% of the virus-like sequences were related to mammalian and avian viruses, belonging to seven virus families: Adenoviridae, Astroviridae, Caliciviridae, Circoviridae, Hepeviridae, Parvoviridae, and Picornaviridae. Sequence reads from members of the family Picornaviridae accounted for the largest proportion (49.50%), followed by Parvoviridae (26.15%), Circoviridae (2.29%), and Caliciviridae (1.06%) (Fig. 1). We further characterized 18 complete or nearly complete genomes of vertebrate viruses.

The composition of the intestinal virome in red-crowned cranes. The percentage of virus sequence reads including bacteriophage, plant viruses, insect viruses, mammalian viruses and other unidentified viruses is shown on the left as a pie chart). The percentage of virus sequence reads belonging to different eukaryotic virus families is shown on the right as a pie chart). Different colors in the pie charts indicate different virus types or virus families
Three new putative genera in the family Picornaviridae
Picornaviruses are small, non-enveloped, positive single-stranded RNA viruses with a genome of 7-9 kb in size, the member of which cause different diseases in a variety of vertebrate hosts.
The family Picornaviridae currently consists of 94 species grouped into 40 genera. In this study, a picornavirus was found in the feces of wild red-crowned crane (Grus japonensis), and tentatively named “gapovirus” (Grus japonensisvirus). We successfully amplified the complete genome of gapovirus, which included a 571-bp 5’ UTR, a 6696-bp polyprotein ORF and a 163-bp 3’ UTR. Like other picornaviruses, the polyprotein of gapovirus could theoretically be cleaved into VP1-4, 2A-2C, and 3A-3D (Fig. 2d). The hypothetical cleavage map of the gapovirus polyprotein was derived from a comparison with other known viruses in the genus Hepatovirus. The P1 polypeptide (793 aa) belongs to the rhv-like superfamily and shares 39.8% amino acid sequence identity to that of bat hepatovirus (GenBank no. KT452729), the highest similarity among members in the genus Hepatovirus. The P1 was assumed to be cleaved at VP4/VP2 (36I/37T), VP2/VP3 (260T/261M) and VP3/VP1 (512Q/513A). The P2 polypeptide of 642 aa contains non-structural proteins and is speculated to be cleaved at 2A/2B (1004D/1005V) and 2B/2C (1108Q/1109S). BLASTp showed that the P2 region had 37% amino acid sequence identity to that of bat hepatovirus (GenBank no. KT452729), Similar to members in the genus Hepatovirus, the P3 polypeptide contains proteins 3A, 3B, 3Cpro (protease) and 3Dpol (RNA-dependent RNA polymerase). The P3 is 796 aa in length and is speculated to be cleaved at 3A/3B (1499A/1500K), 3B/3C (1527Q/1528S) and 3C/3D (1746G/1747R). A BLASTp analysis showed that the P3 polyprotein of gapovirus shared 43% amino acid sequence identity with the rodent virus Tupaia hepatovirus A (GenBank no. NC_028981), the highest similarity among members in the genus Hepatovirus. Phylogenetic analysis was performed on the P2 and P3 regions of gapovirus and representative strains of 35 genera of the family Picornaviridae. The result indicated that gapovirus was not closely related to members of any previously recognized or proposed genera (Fig. 2a and Supplementary Fig. S1), and it formed a separate branch distinguishing it from members of the genera Hepatovirus and Tremovirus (Fig. 2b). The P1 region of gapovirus shares 39.8% amino acid sequence identity to bat hepatovirus, but members of the genus Hepatovirus share over 70% sequence identity with one another (Fig. 2c). According to the International Committee on Taxonomy of Viruses (http://www.picornastudygroup.com/definitions/genus_definition.htm), the members of a picornavirus genus should share > 40%, > 40%, and > 50% amino acid sequence identity in their P1, P2, and P3 region, respectively. Our data indicate that gapovirus qualifies as a member of a novel genus in the family Picornaviridae.

Sequence comparison, genomic organization, and phylogenetic analysis of the novel picornaviruses identified in red-crowned cranes. (a) Phylogenetic analysis based on the complete amino acid sequence of P3 proteins of gapovirus, grusavirus, and 35 representative strains of all 35 genera of the family Picornaviridae. The virus indicated by a solid black circle was from the wild group, and the one marked with an unfilled black circle was from the ornamental group. (b) Phylogenetic analysis based on the complete amino acid sequences of P1 proteins of gapovirus, grusavirus, and the representative members of the genera Hepatovirus and Tremovirus. (c) Pairwise comparison of the novel picornavirus with representative members of the genera Hepatovirus and Tremovirus. (d) Genome organization of gapovirus
Another novel picornavirus was detected in the feces from an ornamental red-crowned crane and was tentatively named “grusavirus” (Grus japonensisstool associated picornavirus). A nearly complete genome sequence of 7073 bp was determined, including a 591-bp 5’ URT, the complete P1 and P2 regions, and part of the P3 region. The hypothetical cleavage map of the grusavirus polyprotein was derived from alignments with other known viruses of the genus Hepatovirus. The P1 region of grusavirus is assumed to be cleaved at VP4/VP2 (22Q/23D), VP2/VP3 (242N/243A) and VP3/VP1 (495Q/496G). The P1 polypeptide is 772 aa in length, belongs to the rhv-like superfamily, and has the best BLASTp match to phopivirus in genus Hepatovirus (GenBank no. NC_027818). The latter was identified in a seal, sharing 43% amino acid sequence identity in P1 to grusavirus. The P2 region of grusavirus is 636 aa in length and contains nonstructural proteins with cleavage at 2A/2B (E973/T974), and 2B/2C (S1081/I1082). BLASTp analysis showed that the P2 region had 32% amino acid sequence identity to Tupaia hepatovirus A (GenBank no. NC_028981), the highest similarity among members of the genus Hepatovirus. The partial P3 region is 752 aa in length, including complete 3A, 3B, 3C, and partial 3D, and is assumed to be cleaved at 3A/3B (F1463/K1464), 3B/3C (H1489/R1490), and 3C/3D (H1707/R1708). The partial P3 region shared 41% amino acid sequence identity with phopivirus (GenBank no. NC_027818), the highest among members of the genus Hepatovirus. The gapovirus and grusavirus amino acid sequence are 34%, 30%, and 36% identical in P1, P2 and P3, respectively. Phylogenetic analysis based on P1 and P3 showed that grusavirus belongs to a different clade from the members of the genus Hepatovirus, and therefore represents a new genus in the family Picornaviridae (Fig. 2a and 2b).
Four large contigs (> 4000 bp) whose putative protein products shared the highest amino acid sequence similarity with duck hepatitis A virus (DHAV) were also detected, including three from wild cranes and one from ornamental red-crowned cranes. Three complete genome sequences and one nearly complete genome sequence were then determined using RT-PCR to bridge gaps between contigs, followed by direct Sanger sequencing, and were tentatively named GrHAV. The polyproteins of those four GrHAVs were 2214 aa, 2125 aa, 2377 aa, and 2178 aa, respectively (Fig. 3a). Hypothetical maps of those four GrHAV polyproteins were derived by comparison with the polyproteins of DHAV strains (Fig. 3a and Supplementary Table S3), although some cleavage sites within the region of VP1 to 2A2 were uncertain because of high variation among GrHAVs and DHAVs. The similarity between the four GrHAVs and DHAVs in pairwise comparison of the VP1-2A2 regions ranged from 16% to 25% (Supplementary Fig. S2). The length of the VP1-2A1 region of the four GrHAVs varied in length, (476 aa, 501 aa, 608 aa and 630 aa). Phylogenetic analysis based on the P1 region showed that these four GrHAVs clustered separately from their closest relatives in the genera Avisivirus and Avihepatovirus (Fig. 3b). Pairwise sequence similarity comparison of the P1 regions of these four GrHAVs and the representative strains of the genus Avisivirus and Avihepatovirus showed that the four GrHAVs shared 32.4%-35.9% amino acid sequence identity with different strains of the genus Avihepatovirus and 29.9%-32% amino acid sequence identity with the representative strain of the genus Avisivirus (Fig. 3c). Based on the phylogenetic analysis of P1 region and genetic distance calculations, the four GrHAVs should be assigned to a novel genus in the family Picornaviridae.

Sequence comparisons, genomic organization, and phylogenetic analysis of the four novel picornaviruses of the genus Avihepatovirus identified in red-crowned cranes in this study. (a) Alignment of the encoded protein of GrHAV1-4 with that of the representative strain (NC_008250). (b) Phylogenetic analysis based on the amino acid sequence of the P1 region of GrHAVs, three DHAVs, and representative members of each of the 35 genera in the family Picornaviridae. (c) Pairwise comparison of GrHAV1-4 with representative strains of DHAV1-3 and a representative strain of the genus Avisivirus based on the P1 region
Four new parvoviruses in the proposed genus “Chapparvovirus “
Parvoviruses are icosahedral, non-enveloped viruses with single-stranded DNA genomes of about 5 kb in length. The family Parvoviridae is divided into two subfamilies, Parvovirinae and Densovirinae, which include viruses infecting vertebrates and invertebrates, respectively. The subfamily Parvovirinae includes eight genera: Dependoparvovirus, Copiparvovirus, Bocaparvovirus, Amdoparvovirus, Aveparvovirus, Protoparvovirus, Tetraparvovirus, and Erythroparvovirus [13]. Recently, two new genera, “Marinoparvovirus” and “Chapparvovirus” were proposed [14].
In this study, nearly complete genome sequences of four novel parvoviruses were acquired by combing de novo assembly and PCR to bridge sequence gaps from three different libraries, and these were temporarily named Grus japonensisassociated-parvoviruses (GAPVs). The nearly complete genomes of GAPV1-4 were 4305 bp, 4354 bp, 4274 bp, and 4534 bp in length, respectively, all of which included two ORFs encoded one nonstructural protein (NS) and one structural protein (VP). The length of the NS protein is 663 aa for GAPV1, 660 aa for GAPV2, 657 aa for GAPV3, and 662 aa for GAPV4. The conserved motif “GPSNTGKS” associated with ATPase is present in NS1. The VP protein of the different GAPVs is variable in length. The shortest VP is in GAPV3 (483 aa), followed by GAPV1 (514 aa), GAPV4 (529 aa), and GAPV2 (537 aa), all of which are much shorter than those of the other members of the subfamily Parvovirinae (approximately 700 aa on average). The conserved phospholipase A2 (PLA2) motif that is often present in members of other genera was absent in the VP protein of GAPVs. Phylogenetic analysis of the NS1 amino acid sequences showed that GAPV1-4 clustered with TP1 from a turkey (GenBank no. KF925531) and HK_2014 from a chicken (GenBank no. KM254174) (Fig. 4c), forming an avian chapparvovirus clade. Pairwise comparison of NS1 proteins of GAPV1-4 revealed 41.7-44.3% amino acid sequence identity with that of TP1. The NS1 amino acid identity among the four GAPVs is 40.9%-45.2%. According to ICTV guidelines, parvoviruses sharing > 85% amino acid sequence identity in the NS1 protein belong to the same species. Based on this criterion, the GAPVs identified here belong to four different novel species within the proposed genus “Chapparvovirus”.

Sequence comparison, genomic organization, and phylogenetic analysis four novel parvoviruses (GAPV1-4) identified in red-crowned cranes. (a) Genome organization of GAPV1-4. (b) Pairwise comparison of GAPV1-4 with other members of the proposed genus “Chapparvovirus”. (c) Phylogenetic analysis based on the complete amino acid sequence of NS1 of GAPV1-4 and the other members of the proposed genus “Chapparvovirus”. The virus indicated by a solid black circle was from the wild group, the one marked with an unfilled black circle was from the ornamental group, and the ones marked with unfilled triangles were from the breeding group
A new parvovirus in the genus Aveparvovirus
Enteric disease in poultry is an ongoing problem worldwide [15]. Many enteric viruses have been identified in turkeys and chickens, including avian astroviruses [16], rotaviruses [17], reoviruses [18], and coronaviruses [18]. More recently, some reports have revealed that parvoviruses in the genus Aveparvovirus were associated with enteric disease in poultry [19].
In this study, two large contigs with lengths of 3,685 bp and 5,231 bp sharing the highest amino acid sequence similarity with turkey parvovirus 1078 (GenBank no. GU214705) were found in two different libraries. Further amplification to fill the gaps between different sequence segments resulted in two nearly complete virus genome sequences. These viruses were named RCPV1 (red-crowned crane parvovirus) and RCPV2, respectively. The genomes of RCPV1 and RCPV2 were 5,515 bp and 5,456 bp in length, and both contain three ORFs (Fig. 5a). The left ORF encoded a non-structural protein (NS) and was 680 aa in length in both RCPVs. The right ORF encoded a viral capsid protein (VP) including VP1 and VP2, where the VP1 proteins of RCPV1 and RCPV2 were identical in length, 672 aa, and the VP2 protein was 531 aa long in both RCPVs. Both RCPVs had a middle ORF encoding a 161-aa protein. Pairwise comparison showed that the two RCPVs shared 99.3% amino acid sequence similarity in the NS1 protein and 82% based in VP1 protein. RCPVs shared the highest NS1 amino acid sequence similarity (58% identity) with NS1 of chicken parvovirus ABU-P1 (GenBank no. GU214704), which was detected in a fecal sample [19]. Like chicken parvovirus ABU-P1, the RCPVs possessed a conserved genome structure and protein motifs including the helicase superfamily domains, the ATP- or GTP-binding Walker loop motifs 451GPANTGKT458, 491VLWWEECTMK500, and 531TPMVITSNN539, and two replication initiator motifs, 167S/NGKIHFHVLF176 and 232RGYSPKLGKSIPQP245. Phylogenetic analysis based on the NS1 protein sequence showed that RCPVs clustered with chicken parvovirus (GenBank no. GU214704) and turkey parvovirus (GenBank no. GU214706) in a clade corresponding to the genus Aveparvovirus, which is genetically distant from the other clades formed by members of the subfamily Parvoviridae (Fig. 5b). The ICTV recently proposed that parvoviruses sharing > 85% amino acid sequence identity in NS1 belong to the same species. Based on this criterion, the RCPVs identified here belong to a single novel species in the genus Aveparvovirus.

Genomic organization and phylogenetic analysis of two novel parvoviruses belonging to the genus Aveparvovirus identified in red-crowned cranes. (a) Genome organization of RCPV1-2. (b) Phylogenetic analysis based on the complete amino acid sequence of NS1 of RCPV1-2 and those of representative members of all 10 genera of the family Parvoviridae
New members of the circo-like viruses and Genomoviridae
Members of the genus Gemycircularvirus, family Genomoviridae, have a ~ 2 to 2.4-kb circular ssDNA genome encoding at least two proteins, CP and Rep, where the Rep is encoded on the single-stranded genome in viral particles, while the CP protein is encoded on the complementary strand [20]. Gemycircularvirus was first identified in the fungus S. sclerotiorum and subsequently found in insects, humans and animals, in a sewage oxidation pond, and in plants [21,22,23,24].
In this study, a large number of gemycircularvirus sequence reads were detected in the feces samples of red-crowned cranes. Two complete genome sequences of gemycircularvirus, named Grus japonensisassociated gemycircularvirus (GaGmCV) were assembled from two libraries, one of which was from wild red-crowned cranes and the other one was from breeding red-crowned cranes. The genomes of the two GaGmCVs were 2,138 bp and 2,277 bp in length, respectively. Genome analysis indicated that GaGmCVs have two main ORFs that separately encode CP and Rep protein that are 297 aa and 309 aa in length, respectively, when including a putative intron (Fig. 6b and c). The conserved PCR motifs I, II, and III were present in Rep, where PCR motif I was identified as L(F/L)TYSQ, PCR motif II was TH(L/Y)H(C/A), and PCR motif III was YA(I/T)K. The conserved motifs of the superfamily 3 helicases were not obvious through alignment with sequences of other gemycircularviruses. The nonanucleotide sequences of GaGmCVs were atypical, including only eight nucleotides in this motif, “GGACGAAA” and “GAAACTTA”, respectively (Fig. 6h). BLASTp analysis based on the Rep of GaGmCVs showed that GaGmCV1 shared the highest amino acid sequence similarity (48% identity) with Pacific flying fox feces associated gemycircularvirus 11 (GenBank no. KT732807), and GaGmCV2 shared the highest amino acid sequence similarity (69% identity) with dragonfly-associated circular virus 3 (GenBank no. NC_023870). Phylogenetic analysis based on the amino acid sequence of Rep protein showed that GaGmCVs were located on two separate branches, with GaGmCV2 clustering with dragonfly-associated circular virus 3 (GenBank no. NC_023870), while GaGmCV1 clustered with FaGmV-4 (GenBank no. KF371638) identified in fur seal [25] (Fig. 6a).

Phylogenetic analysis and genomic organization of novel circo-like viruses and gemycircularviruses identified in red-crowned cranes. (a) Phylogenetic analysis was performed based on the amino acid sequence of the Rep protein. The sequence alignments included two gemycircularviruses and four novel circo-like viruses identified here, their best BLASTp matches in GenBank based on the Rep proteins, and representative strains of gemycircularviruses and circoviruses. The host or source of the viruses included in the phylogenetic analysis are indicated on the branches. (b and c) The genomic organization of the gemycircularviruses identified in red-crowned cranes. (d, e, f, and g) The genomic organization of GaCV1-4 identified in red-crowned cranes. (h) The stem-loop structures of circo-like viruses identified in red-crowned cranes
The family Circoviridae consists of non-enveloped viruses with a circular single-stranded DNA genome of ~ 2 kb and is currently divided into two genera, Circovirus and Cyclovirus, [26]. The genus circovirus comprises 29 species, members of which have been identified in birds, dogs, minks, pigs, bats and fish [27,28,29,30,31,32]. Circoviruses were originally isolated from various birds. Some Circovirus strains have been associated with a variety of diseases in birds and pigs, including respiratory and enteric disease, dermatitis, and reproductive problems [33, 34]. Here, four genome sequences were assembled from sequences present in red-crowned crane feces. They were temporarily named GaCV1-4. The complete genome sizes were 1,852 bp for GaCV1, 2,288 bp for GaCV2, 1,686 bp for GaCV3, and 1830 bp for GaCV4, all of which encoded two major proteins: CP and Rep (Fig. 6d-g). A BLASTp search based on the Rep proteins indicated that GaCV1-4 shared the highest amino acid similarity of 41%, 41%, 56%, and 45%, respectively, with their most closely related viruses. Phylogenetic analysis over the amino acid sequence of Rep protein confirmed that those four novel circo-like viruses did not belong to the genus Gemycircularvirus or Circovirus (Fig. 6a). GaCV1 and GaCV2 clustered with the Lake Sarah-associated circular virus 1 isolate LSaCV-1-LSCO-2013 (GenBank no. NC_029607), forming a separate branch. GaCV3 clustered with Odonata-associated circular virus 19 isolate OdasCV-19-US-1594LM1-12 (GenBank no.KM598404) forming a separate branch. GaCV4 clustered with Lake Sarah-associated circular virus 50 isolate LSaCV-50-LSCO-2013 (GenBank no. NC_029627), forming a separate branch (Fig. 6a). The stem-loop structures of GaCVs were non-canonical compared with other circoviruses (Fig. 6h).
A novel calicivirus
Caliciviruses are non-enveloped icosahedral viruses with a single-stranded, positive-sense, polyadenylated RNA genome of ~ 6.4 to ~ 8.5 kb that contains two or three major open reading frames [35]. Caliciviruses can infect humans and various animals and induce associated diseases. Human caliciviruses cause nonbacterial gastroenteritis in humans of all ages [36], while animal caliciviruses are associated with a wide spectrum of diseases, including gastroenteritis, vesicular lesions, reproductive failure, respiratory infections, and fatal hemorrhagic disease [37, 38]. The family Caliciviridae is divided into five genera: Norovirus, Sapovirus, Lagovirus, Vesivirus and Nebovirus.
In this study, a library contained 2274 sequence reads related to calicivirus that could be assembled into an 8,327-bp contig. Sequence analysis indicated that this 8,327-bp sequence included a 551-bp 5’UTR, a complete ORF1 (7,182 bp), and a partial ORF2 (594 bp). This calicivirus sequence was tentatively named RaCV. Based on an alignment of the amino acid sequences of RaCV and its relatives in GenBank, five potential cleavage sites were found in ORF1, 447Q/S448, 790Q/A791, 1055Q/S1056, 1133Q/N1134, and 1795E/G1796, respectively, which divided the ORF1 polyprotein into Nterm (49.98 kDa), NTPase (37.9 kDa), p29 protein (28.99 kDa), VPg (8.9 kDa), Pro-Pol (73.1 kDa), and VP1 (62.9 kDa) (Fig. 7a). Some conserved CV amino acid motifs within ORF1 were identified, including the NTPase motif 599GPPGIGKT606, the 3C protease motif 1248GDCGLP1253, and the RNA-dependent RNA polymerase motifs 1586GLPSG1590 and 1634YGDD1637. Based on the ORF1 polyprotein, RaCV shared the highest amino acid sequence similarity (61% identity) to turkey calicivirus (GenBank no. JQ347522), and shared < 30% identity with other members of the family Caliciviridae. Phylogenetically, RaCV clustered with turkey calicivirus (GenBank no. JQ347522) and goose calicivirus (GenBank no. KJ473715), indicating that RaCV might belong to a novel species in the proposed genus “Nacovirus” (Fig. 7b)

Genomic organization and phylogenetic analysis of the novel calicivirus identified in red-crowned cranes. (a) Genome organization of RaCV1. The potential cleavage site and encodes proteins are shown. (b) Phylogenetic analysis based on the complete amino acid sequence of the ORF1 protein of RaCV1 and representative members of the family Caliciviridae
Other mammalian viruses
In addition to the genomes described in detail above, less-complete genome sequences of other viruses were also assembled. Fifty-six sequence reads related to adenoviruses in two libraries of the ornamental group could be assembled into four contigs. A BLASTx search showed that this adenovirus shared the highest amino acid sequence similarity (71% identity) with skua adenovirus 1 (GenBank no. NC_016437), which was isolated from the kidney of a south polar skua bird [39]. In one library from the ornamental group, 24 sequence reads related to astroviruses were found and assembled into three contigs. A BLASTx search showed that this astrovirus shared the highest amino acid sequence similarity (46% identity) with the nonstructural polyprotein of the avian avastrovirus 3 (GenBank no. EU143843). We also found 27 sequences reads related to members of the family Hepeviridae in a library from the wild group, including a 647-bp contig showing the highest amino acid sequence similarity (34% identity) to the nonstructural polyprotein of bat hepevirus (GenBank no. KJ562187).
Insect, crustacean shellfish and plant viruses
About 1.16% (2,636 sequence reads) and 0.05% (124 sequence reads) of the red-crowned crane fecal virome was related to insect and crustacean shellfish viruses, respectively (Fig. 1a). Detection of these viral sequences in fecal samples may be due to insect and aquatic animal consumption. Among these insect viruses, RNA viruses were dominant, making up 92% of the total insect viral sequences, and 81% of the RNA sequences were related to those of members of the family Dicistroviridae. BLASTx showed contigs with a variable degree of similarity to known insect viruses Solenopsis invicta virus 1, Israeli acute paralysis virus, Homalodisca coagulate virus 1, Anopheles C virus, Nilaparvata lungens C virus, cricket paralysis virus, and acute bee paralysis virus. Insect DNA viruses mainly included members of the families Iridoviridae and Bidnaviridae, making up about 8% of insect viral sequences. Aquatic animal viruses were mainly composed of members of the families of Nodaviridae and Reoviridae.
About 2.13% of the red-crowned crane fecal virome (4,835 sequence reads) was related to plant viruses (Fig. 1a), 68% of which were DNA viruses of the families Phycodnaviridae, Geminiviridae, and Nanoviridae, and 32% were single-stranded RNA viruses of the family Tombusviridae, followed by the families Secoviridae, Luteoviridae, Marnaviridae, Partitiviridae, Bromoviridae, and Virgaviridae.
Discussion
Red-crowned cranes, the rarest cranes, continue to suffer from habitat loss, environmental pollution and various diseases including parasitic, bacterial and viral diseases. Although some countries have strengthened protective measures, the number of red-crowned cranes continues to decrease. Viral metagenomics can be used to characterize viral nucleic acids enriched from a variety of sources without cell culture amplification and has been used in numerous animal virus discoveries [31, 40, 41]. We describe here the fecal virome of red-crowned cranes from three different sources. The fecal virome of red-crowned cranes included a large numbers of vertebrate, plant, insect, and aquatic animal viruses. Plant, aquatic and insect viral sequences may reflect the omnivorous diet of red-crowned cranes. Vertebrate viral sequences predominated in the total number of reads but varied from 45.2 to 97.7% of total viral reads among different groups of animals. Parvoviridae reads dominated in wild and breeding groups, while the largest percentage of viral reads in the ornamental group were from members of the family Picornaviridae. This difference may be due to the susceptibility or exposure of the different groups and/or simply the stochastic nature of infections in these groups.
Caliciviruses include many important human and veterinary pathogens [42]. The feces from the wild birds contained a novel calicivirus of the genus Nacovirus.
Picornaviruses showed a high level of diversity, including members of three different (novel) genera, two of which included viruses related to members of the genus Hepatovirus. The other genus included four different strains isolated from the wild and ornamental groups that were related to members of the genus Avihepatovirus, which cause fatal duck hepatitis characterized primarily by liver necrosis and hemorrhage [43]. Whether infected cranes suffer from hepatitis will require analysis of liver tissues for evidence of viral replication. The high variation of VP1-2A in the different GrHAVs may also reflect different biological properties.
Parvovirus sequence reads were an important component of the fecal virome of both the wild and breeding groups and were the second most common viral reads (after picornaviruses) in the ornamental group. Parvoviruses are commonly detected DNA viruses that have been associated with a variety of animal diseases [44, 45]. Here, we characterized six genomes of novel parvoviruses belonging to two different genera. Four of them clustered with turkey parvovirus 1 and chicken parvovirus in the proposed genus “Chapparvovirus”. We also characterized two novel, closely related genomes of parvoviruses in the genus Aveparvovirus from the wild and ornamental groups.
Six small, circular genomes were characterized, with two belonging to gemycircularviruses (family Genomoviridae) and four to novel circo-like viruses. Gemycircularviruses have been detected in diverse organisms and environments, but the host species of these viruses is only known for one virus infecting fungi [46]. The two gemycircularviruses might therefore also originate from fungi in these birds’ diet. The four novel circo-like viruses were all from the wild bird group, but the identity of their cellular host currently remains uncertain.
Our study provides an overview of the intestinal virome of red-crowned cranes and significantly increases the diversity of viruses known to infect this endangered species, which is considered a symbol of luck, longevity and fidelity in China. This information may help in monitoring changes in the enteric virome of these bird populations and provide viral genome sequences for future studies of their pathogenic potential.
Nucleotide sequence accession numbers
The viral genomes described in detail here were deposited in GenBank under the accession numbers KY312540-KY312558. The raw sequence reads from the metagenomic library were deposited in the Shirt Read Archive of GenBank database under accession number SRR7285959.
References
Wang H, Gao J, Pu R, Ren L, Kong Y, Li H, Li L (2014) Natural and anthropogenic influences on a red-crowned crane habitat in the Yellow River Delta Natural Reserve, 1992–2008. Environ Monit Assess 186:4013–4028
Liu DW, Liu HY, Zhang HB, Cao MC, Sun Y, Wu WD, Lu CH (2016) Potential natural exposure of endangered red-crowned crane (Grus japonensis) to mycotoxins aflatoxin B1, deoxynivalenol, zearalenone, T-2 toxin, and ochratoxin A. J Zhejiang Univ Sci B 17:158–168
Luo J, Ye Y, Gao Z, Wang W, Hartup BK (2016) Lead in the Red-Crowned Cranes (Grus japonensis) in Zhalong Wetland, Northeastern China: a report. Bull Environ Contam Toxicol 97:177–183
Xie Y, Xia P, Wang H, Yu H, Giesy JP, Zhang Y, Mora MA, Zhang X (2016) Effects of captivity and artificial breeding on microbiota in feces of the red-crowned crane (Grus japonensis). Sci Rep 6:33350
Lian X, Ming X, Xu J, Cheng W, Zhang X, Chen H, Ding C, Jung YS, Qian Y (2018) First molecular detection and characterization of Marek’s disease virus in red-crowned cranes (Grus japonensis): a case report. BMC Vet Res 14:122
Delwart EL (2007) Viral metagenomics. Rev Med Virol 17:115–131
Yang S, Wang Y, Li W, Fan Z, Jiang L, Lin Y, Fu X, Shen Q, Sun Z, Wang X, Deng X, Zhang W, Delwart E (2016) A novel bocavirus from domestic mink, China. Virus Genes 52:887–890
Zhang W, Li L, Deng X, Blumel J, Nubling CM, Hunfeld A, Baylis SA, Delwart E (2016) Viral nucleic acids in human plasma pools. Transfusion 56:2248–2255
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, He G, Chen Y, Pan Q, Liu Y, Tang J, Wu G, Zhang H, Shi Y, Liu Y, Yu C, Wang B, Lu Y, Han C, Cheung DW, Yiu SM, Peng S, Xiaoqian Z, Liu G, Liao X, Li Y, Yang H, Wang J, Lam TW, Wang J (2015) Erratum: SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience 4:30
Deng X, Naccache SN, Ng T, Federman S, Li L, Chiu CY, Delwart EL (2015) An ensemble strategy that significantly improves de novo assembly of microbial genomes from metagenomic next-generation sequencing data. Nucleic Acids Res 43:e46
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Cotmore SF, Agbandje-McKenna M, Chiorini JA, Mukha DV, Pintel DJ, Qiu J, Soderlund-Venermo M, Tattersall P, Tijssen P, Gatherer D, Davison AJ (2014) The family Parvoviridae. Arch Virol 159:1239–1247
Phan TG, Gulland F, Simeone C, Deng X, Delwart E (2015) Sesavirus: prototype of a new parvovirus genus in feces of a sea lion. Virus Genes 50:134–136
Koo BS, Lee HR, Jeon EO, Han MS, Min KC, Lee SB, Mo IP (2013) Molecular survey of enteric viruses in commercial chicken farms in Korea with a history of enteritis. Poult Sci 92:2876–2885
Da Silva SE, Bonetti AM, Petrocelli AT, Ferrari HF, Luvizotto MC, Cardoso TC (2008) Detection of Turkey astrovirus in young poults affected with poult enteritis complex in Brazil. J Vet Med Sci 70:629–631
McNulty MS, Allan GM, Todd D, McFerran JB (1979) Isolation and cell culture propagation of rotaviruses from turkeys and chickens. Arch Virol 61:13–21
Spackman E, Kapczynski D, Sellers H (2005) Multiplex real-time reverse transcription-polymerase chain reaction for the detection of three viruses associated with poult enteritis complex: turkey astrovirus, turkey coronavirus, and turkey reovirus. Avian Dis 49:86–91
Day JM, Zsak L (2010) Determination and analysis of the full-length chicken parvovirus genome. Virology 399:59–64
Krupovic M, Ghabrial SA, Jiang D, Varsani A (2016) Genomoviridae: a new family of widespread single-stranded DNA viruses. Arch Virol 161:2633–2643
Rosario K, Marinov M, Stainton D, Kraberger S, Wiltshire EJ, Collings DA, Walters M, Martin DP, Breitbart M, Varsani A (2011) Dragonfly cyclovirus, a novel single-stranded DNA virus discovered in dragonflies (Odonata: Anisoptera). J Gen Virol 92:1302–1308
Li L, Kapoor A, Slikas B, Bamidele OS, Wang C, Shaukat S, Masroor MA, Wilson ML, Ndjango JB, Peeters M, Gross-Camp ND, Muller MN, Hahn BH, Wolfe ND, Triki H, Bartkus J, Zaidi SZ, Delwart E (2010) Multiple diverse circoviruses infect farm animals and are commonly found in human and chimpanzee feces. J Virol 84:1674–1682
Kraberger S, Arguello-Astorga GR, Greenfield LG, Galilee C, Law D, Martin DP, Varsani A (2015) Characterisation of a diverse range of circular replication-associated protein encoding DNA viruses recovered from a sewage treatment oxidation pond. Infect Genet Evolut 31:73–86
Du Z, Tang Y, Zhang S, She X, Lan G, Varsani A, He Z (2014) Identification and molecular characterization of a single-stranded circular DNA virus with similarities to Sclerotinia sclerotiorum hypovirulence-associated DNA virus 1. Arch Virol 159:1527–1531
Sikorski A, Massaro M, Kraberger S, Young LM, Smalley D, Martin DP, Varsani A (2013) Novel myco-like DNA viruses discovered in the faecal matter of various animals. Virus Res 177:209–216
Li L, Shan T, Soji OB, Alam MM, Kunz TH, Zaidi SZ, Delwart E (2011) Possible cross-species transmission of circoviruses and cycloviruses among farm animals. J Gen Virol 92:768–772
Todd D, Scott AN, Fringuelli E, Shivraprasad HL, Gavier-Widen D, Smyth JA (2007) Molecular characterization of novel circoviruses from finch and gull. Avian Pathol 36:75–81
Decaro N, Martella V, Desario C, Lanave G, Circella E, Cavalli A, Elia G, Camero M, Buonavoglia C (2014) Genomic characterization of a circovirus associated with fatal hemorrhagic enteritis in dog, Italy. PloS One 9:e105909
Lian H, Liu Y, Li N, Wang Y, Zhang S, Hu R (2014) Novel circovirus from mink, China. Emerg Infect Dis 20:1548–1550
Gagnon CA, Music N, Fontaine G, Tremblay D, Harel J (2010) Emergence of a new type of porcine circovirus in swine (PCV): a type 1 and type 2 PCV recombinant. Vet Microbiol 144:18–23
Wu Z, Yang L, Ren X, He G, Zhang J, Yang J, Qian Z, Dong J, Sun L, Zhu Y, Du J, Yang F, Zhang S, Jin Q (2016) Deciphering the bat virome catalog to better understand the ecological diversity of bat viruses and the bat origin of emerging infectious diseases. ISME J 10:609–620
Lorincz M, Dan A, Lang M, Csaba G, Toth AG, Szekely C, Csagola A, Tuboly T (2012) Novel circovirus in European catfish (Silurus glanis). Arch Virol 157:1173–1176
Todd D (2004) Avian circovirus diseases: lessons for the study of PMWS. Vet Microbiol 98:169–174
Opriessnig T, Halbur PG (2012) Concurrent infections are important for expression of porcine circovirus associated disease. Virus Res 164:20–32
Guo M, Chang KO, Hardy ME, Zhang Q, Parwani AV, Saif LJ (1999) Molecular characterization of a porcine enteric calicivirus genetically related to Sapporo-like human caliciviruses. J Virol 73:9625–9631
Bank-Wolf BR, Konig M, Thiel HJ (2010) Zoonotic aspects of infections with noroviruses and sapoviruses. Vet Microbiol 140:204–212
Carter MJ, Milton ID, Meanger J, Bennett M, Gaskell RM, Turner PC (1992) The complete nucleotide sequence of a feline calicivirus. Virology 190:443–448
Meyers G, Wirblich C, Thiel HJ (1991) Rabbit hemorrhagic disease virus–molecular cloning and nucleotide sequencing of a calicivirus genome. Virology 184:664–676
Park YM, Kim JH, Gu SH, Lee SY, Lee MG, Kang YK, Kang SH, Kim HJ, Song JW (2012) Full genome analysis of a novel adenovirus from the South Polar skua (Catharacta maccormicki) in Antarctica. Virology 422:144–150
Li L, Shan T, Wang C, Cote C, Kolman J, Onions D, Gulland FM, Delwart E (2011) The fecal viral flora of California sea lions. J Virol 85:9909–9917
Scheel TK, Simmonds P, Kapoor A (2015) Surveying the global virome: identification and characterization of HCV-related animal hepaciviruses. Antiviral Res 115:83–93
Farkas T, Dufour J, Jiang X, Sestak K (2010) Detection of norovirus-, sapovirus- and rhesus enteric calicivirus-specific antibodies in captive juvenile macaques. J Gen Virol 91:734–738
Soliman M, Alfajaro MM, Lee MH, Jeong YJ, Kim DS, Son KY, Kwon J, Choi JS, Lim JS, Choi JS, Lee TU, Cho KO, Kang MI (2015) The prevalence of duck hepatitis A virus types 1 and 3 on Korean duck farms. Arch Virol 160:493–498
Zsak L, Strother KO, Kisary J (2008) Partial genome sequence analysis of parvoviruses associated with enteric disease in poultry. Avian Pathol 37:435–441
Opriessnig T, Xiao CT, Gerber PF, Halbur PG (2014) Identification of recently described porcine parvoviruses in archived North American samples from 1996 and association with porcine circovirus associated disease. Vet Microbiol 173:9–16
Yu X, Li B, Fu Y, Jiang D, Ghabrial SA, Li G, Peng Y, Xie J, Cheng J, Huang J, Yi X (2010) A geminivirus-related DNA mycovirus that confers hypovirulence to a plant pathogenic fungus. Proc Natl Acad Sci USA 107:8387–8392
Acknowledgements
This work was partly supported by National Key Research and Development Programs of China No. 2017YFC1200201, National Natural Science Foundation of China No. 81741062, Jiangsu Provincial Key Research and Development Projects No. BE2017693, the Professional Research Foundation for Advanced Talents of Jiangsu University No. 12JDG085 and 13JDG087, the Postdoctoral Foundation of Jiangsu Province No. 1302057C and 1302058C, China Postdoctoral Foundation No. 2014M561597.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This study did not include experiments with human participants or animals performed by any of the authors.
Additional information
Handling Editor: Chan-Shing Lin.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Wang, Y., Yang, S., Liu, D. et al. The fecal virome of red-crowned cranes. Arch Virol 164, 3–16 (2019). https://doi.org/10.1007/s00705-018-4037-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-018-4037-x