
Abstract
Emerging infectious diseases have caused a global effort for development of fast and accurate detection techniques. The rapidly mutating nature of viruses presents a major difficulty, highlighting the need for specific detection of genetically diverse strains. One such infectious agent is SARS-associated coronavirus (SARS-CoV), which emerged in 2003. This study aimed to develop a real-time RT-PCR detection assay specific for SARS-CoV, taking into account its intrinsic polymorphic nature due to genetic drift and recombination and the possibility of continuous and multiple introductions of genetically non-identical strains into the human population, by using mismatch-tolerant molecular beacons designed to specifically detect the SARS-CoV S, E, M and N genes. These were applied in simple, reproducible duplex and multiplex real-time PCR assays on 25 post-mortem samples and constructed RNA controls, and they demonstrated high target detection ability and specificity. This assay can readily be adapted for detection of other emerging and rapidly mutating pathogens.
Similar content being viewed by others


Introduction
Emerging viruses with a high rate of genetic drift cause setbacks in the global scientific effort to develop fast and accurate diagnostic detection methods. A novel virus, SARS-associated coronavirus (SARS-CoV), which spread throughout the world in 2003, was quickly characterised [13, 21] and linked to severe acute respiratory syndrome (SARS) [2, 11, 15]. Human coronaviruses are confined mainly to the upper respiratory tract, causing simple illnesses, whereas in animals, they have been associated with severe diseases [16]. There is still an ongoing possibility of SARS-CoV re-emergence, since evidence suggests that it arose from an animal coronavirus [5, 25]. Poor clinical definition and lack of known prevention methods and treatments [10, 22] mean that early diagnosis is crucial. There has been a huge effort to develop diagnostics [1, 9, 12, 17, 18, 26, 27], nevertheless, molecular detection tests suffer from speed and sensitivity limitations and are not standardised [14, 22]. Accurate and prompt detection of the virus is, therefore, a priority.
Current molecular assays fail to consider the intrinsically polymorphic nature of coronaviruses, as seen in the pathogenic SARS-CoV Tor2 and Urbani strains, and the possibility of continuous introductions of genetically non-identical strains into human populations. This study aimed to develop a real-time RT-PCR assay using mismatch-tolerant molecular beacons [6, 7, 24] to detect SARS-CoV and to distinguish between pathogenic and non-pathogenic strains. The assay incorporates four beacons, targeting the S, E, M and N genes in addition to an internal positive control (IPC), and it was validated using clinical samples.
Materials and methods
Molecular beacon and primer design
Primers and molecular beacons were designed de novo for this study (Table 1). Small regions (95-107 nucleotides long) of the SARS-CoV S, E, M and N genes were targeted for detection (Fig. 1). The oligonucleotide design was based on multiple alignments of sequences of these genes available in the GenBank database (http://www.ncbi.nlm.nih.gov/Genbank) and thermodynamic compatibility. Careful choice of beacons and primers by use of the Integrated DNA Technologies (http://eu.idtdna.com) dimerisation tool ensured the avoidance of potentially hazardous primer-primer or molecular beacon-primer dimerisation and the thermodynamic compatibility of the beacons with their application in a five-amplicon multiplex assay. The molecular beacons, target oligonucleotides and primers were synthesised at MWG-Biotech AG Ltd (Ebersberg, Germany). To facilitate formation of stem structures, the ends of each molecular beacon were designed to have a high GC content and to be complementary to each other. All beacons were labelled with DABCYL, (4′-(4′-dimethylaminophenylazo) benzoic acid) at the 3’ end and TET (tetrachloro-6′-carbofluorescein) at the 5’ end, except MBIPC, the IPC beacon, which was labelled with FAM (fluorescein). To form the loop element, each beacon was designed to contain a 24-26-nucleotide-long probe sequence complementary to part of the gene fragment to be amplified. The beacons were designed to form long, stable hybrids, giving them the ability to be mismatch-tolerant, meaning that the beacons can still detect their target amplicons despite any polymorphic substitutions arising as a consequence of genetic drift of the virus or viral recombination. The GC content of the beacon was kept high to ensure that, under the PCR cycling conditions, the molecular beacon would remain in a closed conformation in the absence of its target. In order to identify the best target sequences within each viral gene that would discriminate the SARS-Urbani and SARS-Tor2 strains from other non-pathogenic human and animal coronavirus strains, multiple sequence alignments of available coronavirus gene sequences deposited in GenBank were carried out (Fig. 2). DNA sequences of SARS-CoV genes of SARS-Urbani (GenBank accession number AY278741) and SARS-Tor2 (AY274119) were compared with those from reference viruses representing each species in the three known groups of coronaviruses. Reference sequences from group 1 (G1) included human coronavirus 229E (HCoV-229E), AF304460; porcine epidemic diarrhea virus (PEDV), AF353511; transmissible gastroenteritis virus (TGEV), AJ271965; canine coronavirus (CCoV), D13096; feline coronavirus (FCoV), AY204704; porcine respiratory coronavirus (PRCoV), Z24675. Those from group 2 (G2) were bovine coronavirus (BCoV), AF220295; murine hepatitis virus (MHV), AF201929; human coronavirus OC43 (HCoV-OC43), M76373; porcine hemagglutinating encephalomyelitis virus (HEV), AY078417; rat coronavirus (RtCoV), AF207551; and the references virus for group 3 (G3) was infectious bronchitis virus (IBV), M95169. Sequence alignments were performed with ClustalW [23] to identify genomic regions with the highest genetic distance between SARS-CoV and other coronavirus strains, with special emphasis on probe-target sequences that encompass gaps or deletions in the SARS-CoV genome. Like the probe sequences, primer selection was also based on the ability for amplification of the SARS-CoV targets that would form primer-target and probe-target hybrids with a theoretical melting temperature that would maximize the ability of the primer to bind to the target even if nucleotide substitutions are present to achieve mismatch tolerance. Finally, primer sequences were selected that enable the amplicons formed to be approximately the same length, about 100 nucleotides.

Assay design. Schematic representation of the SARS-associated coronavirus (SARS-CoV) genome highlighting the main protein-encoding genes, which are also targeted in this assay (A), and magnified to indicate the start and end locations of these genes and the known open reading frames using nucleotide positions (B). Further magnification of the target genes (C) shows the amplicons of each of the four genes (S, E, M and N) incorporated in the assay. Target sequences of the amplicons are shown in boxes, whereas primers and molecular beacons are shown in bold. Numbers indicate the nucleotide positions of the start and end locations of the target amplicon that correspond to forward primer, probe and reverse primer, using the SARS-Tor2 strain (GenBank accession no. AY274119)

Sequence alignments of the target genes. DNA sequence alignments of the gene regions targeted in the real-time PCR assay to compare SARS-CoV-specific genes from SARS-Tor2 and SARS-Urbani strains to those of other non-pathogenic human and animal reference strains representing each species in the three known groups of coronaviruses. Group 1 (G1): human coronavirus 229E (HCoV–229E); porcine epidemic diarrhoea virus (PEDV); transmissible gastroenteritis virus (TGEV); canine coronavirus (CCoV); feline coronavirus (FCoV); porcine respiratory coronavirus (PRCoV). Group 2 (G2): bovine coronavirus (BCoV); murine hepatitis virus (MHV); human coronavirus OC43 (HCoV–OC43); porcine hemagglutinating encephalomyelitis virus (HEV); rat coronavirus (RtCoV). Group 3 (G3): infectious bronchitis virus (IBV). GenBank accession numbers are AY278741, AY274119, AF304460, AF353511, AJ271965, D13096, AY204704, Z24675, AF220295, AF201929, M76373, AY078417, AF207551 and M95169, respectively. Optimal target sequences that discriminate the SARS-associated pathogenic strains from the rest were identified. Numbers indicate the nucleotide position according to the SARS-Tor2 genome (GenBank accession no. AY274119). The primers and molecular beacon and their target sequences are highlighted in the alignments
Internal positive control
A synthetic oligonucleotide was designed to be amplified by the forward S gene and reverse E gene primers (Table 1) and used as an internal positive control (IPC) during amplification.
Thermal profiles of molecular beacons
The thermodynamic characteristics of the molecular beacons were assessed by melting curve analyses performed in a 7700 Prism Spectrofluorometric Thermal Cycler (Applied Biosystems, Foster City, CA, USA) and validated using the 7900 HT Fast Real-Time PCR System (Applied Biosystems). Different concentrations of beacons were used to find one at which fluorescence was neither too low nor saturating. The 25-μl reaction consisted of 1X Platinum ® PCR Supermix (Invitrogen), 1 μl of the beacon in various concentrations, with or without 100 pmol of a single-stranded complementary target. The cycling parameters were as follows: 2 min at 95°C, followed by 50 cycles of data collection for 30 s and a second step for 10 s, starting at 80°C and decreasing by 1°C per cycle. Fluorescence was measured at 490 nm and recorded at each cycle.
RNA standards
RNA transcripts corresponding to all targets were prepared by in vitro transcription using a MEGAscript T7 transcription kit (Ambion, Houston, TX) according to the manufacturer’s instructions. DNA templates were generated by PCR using forward primers (Table 1) with the bacteriophage T7 RNA polymerase promoter recognition sequence (TAATACGACTCACTATAGG) incorporated at their 5’ end and reverse primers integrating the gene-specific reverse transcriptase primer-binding site at their 3’ end (22165-RT, 5’-AGGCTGTAAGAACCAAGAGGCAACTTAAAAATAGGTTTC-3’; 26227-RT, 5’-TATTGCAGCAGTACAAGCGCAGTAAGGATGGCTA-3’; 26651-RT, 5’-AAGCAACGAAGTAGCAAGCCATTGCAATCGCAATC-3’; or 28492-RT, 5’-GCCTTCTTTGTTAGCGTAG GGAAGTGAAGCTTC-3’; the gene-specific reverse transcriptase binding sites are underlined).
The PCR reaction contained 1X PCR buffer (QIAGEN, Mississauga, Canada), 0.5 mmol/L dNTPs (Gibco BRL), 0.4 μmol/L of each primer and 2.5 U of HotStarTaq DNA polymerase (QIAGEN). The amplification conditions were as follows: 95°C for 15 min, 40 cycles of 94°C for 30 s, 55°C for 30 s and 72°C for 80 s, followed by 72°C for 5 min. The eluted RNA transcripts were purified and measured with a UV spectrophotometer (NanoDrop Technologies, USA).
Clinical samples
The samples included in this study were collected from patients who met the CDC/WHO case definition of probable SARS in Toronto, Canada, in 2003. Fifty post-mortem tissue samples (Table 2) were collected from 17 patients who died of probable SARS. Twenty-one post-mortem and two pre-mortem tissue samples from eight individuals who died from causes other than SARS during the outbreak were included as controls. Samples collected at the time of autopsy were snap-frozen in ethanol and dry ice and stored at −70°C. Samples were coded and processed independently. This study was approved by the research ethics boards of the Mount Sinai Hospital and the University Health Network, Toronto, and the Chief Coroner’s Office of Ontario, Canada.
For RT-PCR preparation, 30 mg of each sample was thawed and homogenized in 600 μl RLT lysis buffer (QIAGEN) using disposable tissue grinders (Kendall PrecisionTM, Mansfield, USA). The homogenate was passed through QIAshredder columns (QIAGEN) prior to RNA isolation using an RNeasy Mini Kit (QIAGEN).
Reverse transcription and real-time RT-PCR
Samples were analysed in duplicate in duplex and multiplex RT-PCR reactions. For reverse transcription, 2 μl of RNA served as a template in a reaction with 20 pmol IPC RT primer (E-RT; 5’-TATTGCAGCAGTAC-3’) and 20 pmol of one of the other RT primers (S-RT; 5’-AGGCTGTAAGAA-3’, E-RT, M-RT; 5’-AAGCAACGAA GTAG-3’ or N-RT; 5’-GCCTTCTTTGTTAG-3’; the same RT primer, E-RT, was used for both gene E and IPC), using an Omniscript Reverse Transcription Kit (Qiagen, Hilden, Germany). Reactions were incubated at 37°C for 60 min followed by inactivation at 95°C for 5 min.
Real-time RT-PCR was carried out on a LightCycler real-time PCR platform (Roche Diagnostics, Laval, Canada) using a Platinum® Taq Polymerase PCR Kit (Invitrogen). Each 20-μl reaction contained 2 μl template cDNA, 10 pmol each of the beacon, the MBIPC and the primers, 87.5 pmol MgCl2 and 1X PCR buffer. Cycling conditions were 95°C for 30 s, 40 cycles of 95°C for 20 s, 45°C for 20 s (data collection) and 72°C for 20 s. Non-template controls were also run. In the multiplex protocol, the same cycling conditions and reagents were applied, and the beacons and primers (Table 1) of all four SARS-CoV genes (10 pmol each) were added in each reaction.
A similar analysis was performed on the synthetic RNA standards. Reverse transcription was carried out as described above. Duplex real-time RT-PCR was carried out using a 7700 Prism spectrofluorometric thermal cycler (Applied Biosystems) and confirmed on a 7900 HT Fast Real-Time PCR System (Applied Biosystems). Each 25-μl duplex PCR reaction contained 5 μl cDNA, 1X Platinum® Quantitative Supermix-UDG (Invitrogen), 10 pmol MBIPC, 10 pmol molecular beacon (Table 1) and 20 pmol each of the target and IPC primers (Table 1). Initial denaturation was carried out at 95°C for 10 min, followed by 50 cycles at 95°C for 15 s, 45°C for 30 s (data collection step) and 72°C for 30 s.
Statistical evaluation of the real-time PCR assay
To evaluate the specificity and target detection ability of the assay, 95% confidence intervals were calculated using a Wilson score interval.
Results
Thermal denaturation characteristics of molecular beacons
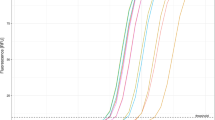
Normalised fluorescence signals for beacon and beacon-target hybrids were plotted against temperature to give thermal denaturation profiles (Fig. 3) and to determine the optimal hybridisation temperature between the beacon and its target. The beacons alone exhibited a melting temperature above 60°C. In the 31-55°C temperature range, the probe-target hybrids elicited significantly stronger fluorescence than the probe alone, allowing clear detection. A temperature of 45°C was chosen as optimal for the real-time PCR reactions. The optimal concentration for all beacons was found to be 10 pmol/μl.

Thermal denaturation profiles of the molecular beacons. Thermal denaturation profiles of the molecular beacons used in this study as established by melting curve analysis (described in Materials and methods). The figure shows normalised fluorescence thermal transitions of molecular beacons (bottom part of plot) and beacon-target complexes (top part of plot)
Real-time RT-PCR evaluation
The assay was tested on synthetic RNA standards and clinical samples (Table 2). All samples were investigated in duplicate with duplex and multiplex assays to detect the IPC and the S, E, M and N genes. Positive results had CT values of 5-15 for lung tissue and 10-25 for intestinal tissue. All 50 specimens from the 17 SARS-CoV-infected cases were found positive for the four targets, indicating a detection ability of 100% (95%CI = 81.6-100%). The specificity was also 100% (95%CI = 67.6-100%), since all 21 control specimens were negative (CT > 45). Non-template controls were included in all assays and exhibited undetectable amplification (CT > 45). Detection of both the IPC and the SARS-CoV-specific genes were clear in all SARS-CoV-infected samples, whereas only the IPC amplification was detected in the controls.
The results show that the real-time RT-PCR accurately detects SARS-CoV RNA and is both accurate and specific enough to distinguish all SARS-CoV-infected samples from control samples. The assay gives the same results on any of three different PCR platforms used (Lightcycler, ABI7700 and ABI7900).
The technology described in this study is part of US Patent No. 7,709,188 B2, issued 4 May 2010.
Discussion
A future re-emergence of SARS-CoV is possible, since viruses are difficult to eradicate and are able to survive in intermediate hosts. A fast, accurate detection assay for routine diagnostic tests may be crucial if therapies are to be applied efficiently and effectively. Furthermore, such assays may be adapted for other mutating infectious agents, which also pose a public health hazard.
Despite being a coronavirus by morphology [11], SARS-CoV strains Tor2 [13] and Urbani [21] have a distant relationship to other known coronaviruses. Phylogenetic analysis revealed regions of very low similarity between the SARS-CoV strains and other coronaviruses. These genetically distant regions have been used here for the design of primers and molecular beacons (Figure 2). The lack of a significant phylogenetic relationship between SARS-CoV and other coronaviruses in the S, E, M and N proteins has been reported previously [19]. The same study also demonstrated no recombinant N or S protein in the SARS-CoV lineage. This and the fact that they are transcribed in large quantities were reasons for choosing the N and S genes as targets.
The existing SARS detection methodology fails to consider the polymorphic nature of coronaviruses and to account for the possibility of introductions of genetically non-identical strains into human populations. The present assay tackles this first by using multiple genetic sequences as targets. The reason for using four different molecular beacons, each targeting a different gene, is to minimise the likelihood of not detecting SARS-CoV due to continuous evolutionary changes in the virus. Also, by simultaneously detecting four targets with multiplex single-fluorophore detection, detection sensitivity is enhanced compared to the detection of just one target. SARS-CoV viral load is extremely low at disease onset, raising a major challenge for early detection [20]. Secondly, mismatch-tolerant molecular beacons have been designed to achieve a compromise between specificity and mismatch tolerance. Traditionally, targets are chosen to be perfectly complementary to the probes, making binding extremely specific [24]. Mismatch-tolerant beacons form longer probe-target hybrids, allowing the duplex to contain mismatched base-pairs [3]. Thus, the beacons designed here bind only to gene regions of the selected pathogenic strains and at the same time could tolerate unpredicted base substitutions.
The assay exhibited target detection ability and specificity of 100%. The detection limit of the assay was also investigated by constructing standard curves using in vitro RNA transcripts, which were linear over a 6-log range of target concentrations, and the lower limit of detection was 5 copies per reaction (data not shown). All SARS-CoV-infected clinical samples were positive, and all control samples were negative (Table 2). Low CT values were observed in lung samples where the SARS-CoV viral load has been reported to be highest amongst human organs [4]. Furthermore, these results support reports of viral dissemination in the lobes of the lung [14] and multiorgan viral dissemination [4, 8]. Potential false negative results were avoided by including an internal positive control.
This is the first report of a real-time RT-PCR SARS-CoV detection assay with molecular beacons that can tolerate mutations attributed to genetic drift and recombination, can distinguish between SARS-CoV and non-pathogenic coronavirus strains and includes an internal positive control. This assay provides a fast and simple way to detect SARS-CoV in diagnostic tests, and, importantly, provides a template for fast molecular detection assays for emerging mutating pathogens.
References
Bressler AM, Nolte FS (2004) Preclinical evaluation of two real-time, reverse transcription-PCR assays for detection of the severe acute respiratory syndrome coronavirus. J Clin Microbiol 42:987–991
Drosten C, Gunther S, Preiser W, van der Werf S, Brodt HR, Becker S, Rabenau H, Panning M, Kolesnikova L, Fouchier RA, Berger A, Burguiere AM, Cinatl J, Eickmann M, Escriou N, Grywna K, Kramme S, Manuguerra JC, Muller S, Rickerts V, Sturmer M, Vieth S, Klenk HD, Osterhaus AD, Schmitz H, Doerr HW (2003) Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med 348:1967–1976
El-Hajj HH, Marras SA, Tyagi S, Shashkina E, Kamboj M, Kiehn TE, Glickman MS, Kramer FR, Alland D (2009) Use of sloppy molecular beacon probes for identification of mycobacterial species. J Clin Microbiol 47:1190–1198
Farcas GA, Poutanen SM, Mazzulli T, Willey BM, Butany J, Asa SL, Faure P, Akhavan P, Low DE, Kain KC (2005) Fatal severe acute respiratory syndrome is associated with multiorgan involvement by coronavirus. J Infect Dis 191:193–197
Guan Y, Zheng BJ, He YQ, Liu XL, Zhuang ZX, Cheung CL, Luo SW, Li PH, Zhang LJ, Guan YJ, Butt KM, Wong KL, Chan KW, Lim W, Shortridge KF, Yuen KY, Peiris JS, Poon LL (2003) Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science 302:276–278
Hadjinicolaou AV, Demetriou VL, Emmanuel MA, Kakoyiannis CK, Kostrikis LG (2009) Molecular beacon-based real-time PCR detection of primary isolates of Salmonella Typhimurium and Salmonella Enteritidis in environmental and clinical samples. BMC Microbiol 9:97
Hadjinicolaou AV, Demetriou VL, Hezka J, Beyer W, Hadfield TL, Kostrikis LG (2009) Use of molecular beacons and multi-allelic real-time PCR for detection of and discrimination between virulent Bacillus anthracis and other Bacillus isolates. J Microbiol Methods 78:45–53
Hung EC, Chim SS, Chan PK, Tong YK, Ng EK, Chiu RW, Leung CB, Sung JJ, Tam JS, Lo YM (2003) Detection of SARS coronavirus RNA in the cerebrospinal fluid of a patient with severe acute respiratory syndrome. Clin Chem 49:2108–2109
Jiang SS, Chen TC, Yang JY, Hsiung CA, Su IJ, Liu YL, Chen PC, Juang JL (2004) Sensitive and quantitative detection of severe acute respiratory syndrome coronavirus infection by real-time nested polymerase chain reaction. Clin Infect Dis 38:293–296
Keightley MC, Sillekens P, Schippers W, Rinaldo C, George KS (2005) Real-time NASBA detection of SARS-associated coronavirus and comparison with real-time reverse transcription-PCR. J Med Virol 77:602–608
Ksiazek TG, Erdman D, Goldsmith CS, Zaki SR, Peret T, Emery S, Tong S, Urbani C, Comer JA, Lim W, Rollin PE, Dowell SF, Ling AE, Humphrey CD, Shieh WJ, Guarner J, Paddock CD, Rota P, Fields B, DeRisi J, Yang JY, Cox N, Hughes JM, LeDuc JW, Bellini WJ, Anderson LJ (2003) A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med 348:1953–1966
Mahony JB, Petrich A, Louie L, Song X, Chong S, Smieja M, Chernesky M, Loeb M, Richardson S (2004) Performance and cost evaluation of one commercial and six in-house conventional and real-time reverse transcription-pcr assays for detection of severe acute respiratory syndrome coronavirus. J Clin Microbiol 42:1471–1476
Marra MA, Jones SJ, Astell CR, Holt RA, Brooks-Wilson A, Butterfield YS, Khattra J, Asano JK, Barber SA, Chan SY, Cloutier A, Coughlin SM, Freeman D, Girn N, Griffith OL, Leach SR, Mayo M, McDonald H, Montgomery SB, Pandoh PK, Petrescu AS, Robertson AG, Schein JE, Siddiqui A, Smailus DE, Stott JM, Yang GS, Plummer F, Andonov A, Artsob H, Bastien N, Bernard K, Booth TF, Bowness D, Czub M, Drebot M, Fernando L, Flick R, Garbutt M, Gray M, Grolla A, Jones S, Feldmann H, Meyers A, Kabani A, Li Y, Normand S, Stroher U, Tipples GA, Tyler S, Vogrig R, Ward D, Watson B, Brunham RC, Krajden M, Petric M, Skowronski DM, Upton C, Roper RL (2003) The Genome sequence of the SARS-associated coronavirus. Science 300:1399–1404
Mazzulli T, Farcas GA, Poutanen SM, Willey BM, Low DE, Butany J, Asa SL, Kain KC (2004) Severe acute respiratory syndrome-associated coronavirus in lung tissue. Emerg Infect Dis 10:20–24
Peiris JS, Lai ST, Poon LL, Guan Y, Yam LY, Lim W, Nicholls J, Yee WK, Yan WW, Cheung MT, Cheng VC, Chan KH, Tsang DN, Yung RW, Ng TK, Yuen KY (2003) Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet 361:1319–1325
Peiris JS, Yuen KY, Osterhaus AD, Stohr K (2003) The severe acute respiratory syndrome. N Engl J Med 349:2431–2441
Poon LL, Chan KH, Wong OK, Yam WC, Yuen KY, Guan Y, Lo YM, Peiris JS (2003) Early diagnosis of SARS coronavirus infection by real time RT-PCR. J Clin Virol 28:233–238
Poon LL, Chan KH, Wong OK, Cheung TK, Ng I, Zheng B, Seto WH, Yuen KY, Guan Y, Peiris JS (2004) Detection of SARS coronavirus in patients with severe acute respiratory syndrome by conventional and real-time quantitative reverse transcription-PCR assays. Clin Chem 50:67–72
Rest JS, Mindell DP (2003) SARS associated coronavirus has a recombinant polymerase and coronaviruses have a history of host-shifting. Infect Genet Evol 3:219–225
Rosling L, Rosling M (2003) Pneumonia causes panic in Guangdong province. BMJ 326:416
Rota PA, Oberste MS, Monroe SS, Nix WA, Campagnoli R, Icenogle JP, Penaranda S, Bankamp B, Maher K, Chen MH, Tong S, Tamin A, Lowe L, Frace M, DeRisi JL, Chen Q, Wang D, Erdman DD, Peret TC, Burns C, Ksiazek TG, Rollin PE, Sanchez A, Liffick S, Holloway B, Limor J, McCaustland K, Olsen-Rasmussen M, Fouchier R, Gunther S, Osterhaus AD, Drosten C, Pallansch MA, Anderson LJ, Bellini WJ (2003) Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science 300:1394–1399
Suresh MR, Bhatnagar PK, Das D (2008) Molecular targets for diagnostics and therapeutics of severe acute respiratory syndrome (SARS-CoV). J Pharm Pharm Sci 11:1s–13s
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Tyagi S, Kramer FR (1996) Molecular beacons: probes that fluoresce upon hybridization. Nat Biotechnol 14:303–308
Wang LF, Shi Z, Zhang S, Field H, Daszak P, Eaton BT (2006) Review of bats and SARS. Emerg Infect Dis 12:1834–1840
Weidmann M, Zanotto PM, Weber F, Spiegel M, Brodt HR, Hufert FT (2004) High-efficiency detection of severe acute respiratory syndrome virus genetic material. J Clin Microbiol 42:2771–2773
Wu Q, Xu Z, Wei T, Zeng H, Li J, Gang H, Sun M, Jiang F, Wang X, Dong W, Yang L, Wang J (2004) Development of Taqman RT-nested PCR system for clinical SARS-CoV detection. J Virol Methods 119:17–23
Acknowledgments
This work was supported by research funds from Birch Biomedical Research LLC (LGK). This study was funded in part by a CIHR Team Grant in Malaria (KCK), CIHR MT-13721 (KCK), Genome Canada through the Ontario Genomics Institute (KCK) and CIHR Canada Research Chair (KCK). The funding agencies had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript. The authors thank Fred Kramer and Salvatore Marras (Public Health Research Institute Center, New Jersey Medical School, University of Medicine and Dentistry New Jersey, Newark, New Jersey, USA) for experimental support during the early phase of the project.
Conflict of interest
The authors have no conflict of interests to declare.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hadjinicolaou, A.V., Farcas, G.A., Demetriou, V.L. et al. Development of a molecular-beacon-based multi-allelic real-time RT-PCR assay for the detection of human coronavirus causing severe acute respiratory syndrome (SARS-CoV): a general methodology for detecting rapidly mutating viruses. Arch Virol 156, 671–680 (2011). https://doi.org/10.1007/s00705-010-0906-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-010-0906-7