
Abstract
We report here a semi-automated process by which mouse genome feature predictions and curated annotations (i.e., genes, pseudogenes, functional RNAs, etc.) from Ensembl, NCBI and Vertebrate Genome Annotation database (Vega) are reconciled with the genome features in the Mouse Genome Informatics (MGI) database (http://www.informatics.jax.org) into a comprehensive and non-redundant catalog. Our gene unification method employs an algorithm (fjoin—feature join) for efficient detection of genome coordinate overlaps among features represented in two annotation data sets. Following the analysis with fjoin, genome features are binned into six possible categories (1:1, 1:0, 0:1, 1:n, n:1, n:m) based on coordinate overlaps. These categories are subsequently prioritized for assessment of annotation equivalencies and differences. The version of the unified catalog reported here contains more than 59,000 entries, including 22,599 protein-coding coding genes, 12,455 pseudogenes, and 24,007 other feature types (e.g., microRNAs, lincRNAs, etc.). More than 23,000 of the entries in the MGI gene catalog have equivalent gene models in the annotation files obtained from NCBI, Vega, and Ensembl. 12,719 of the features are unique to NCBI relative to Ensembl/Vega; 11,957 are unique to Ensembl/Vega relative to NCBI, and 3095 are unique to MGI. More than 4000 genome features fall into categories that require manual inspection to resolve structural differences in the gene models from different annotation sources. Using the MGI unified gene catalog, researchers can easily generate a comprehensive report of mouse genome features from a single source and compare the details of gene and transcript structure using MGI’s mouse genome browser.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Generating lists of genes and other genome features in specific chromosomal regions of the reference mouse genome is a common task among biomedical researchers. Although conceptually simple, generating a complete and non-redundant list of genome features can be challenging because there are multiple major independent genome annotation providers that use different methods for predicting genes. Each of these genome annotation processes generates a set of gene models in which some predictions are unique to a particular pipeline. Even when genes are predicted in common, there are often differences in exon structure and inconsistencies in nomenclature. The accession identifiers associated with the predictions also differ among the various providers. As a consequence, a list of genes downloaded from one source does not always match a gene list obtained from a different source. Further, there are a number of annotation projects that specialize in specific types of genome features such as regulatory regions (Yue et al. 2014) and functional RNAs (Chan and Lowe 2009; Kozomara and Griffiths-Jones 2014). These features are often not included in the predictions of the major annotation providers or are represented incompletely.
We describe here the methods we use to combine annotations from multiple sources into a single “unified gene catalog” for the laboratory mouse reference genome. The Mouse Genome Informatics (MGI) unified gene catalog process does not simply append the different sources of mouse genome feature predictions/annotations together; rather, equivalent genome features from different sources are mapped to a single, unique accession identifier and assigned official standardized nomenclature. Genome features from specialty annotation databases such as miRBase for miRNAs (Kozomara and Griffiths-Jones 2014), Rfam for rRNAs (Burge et al. 2013), and gtRNAdb for tRNAs (Chan and Lowe 2009) are also integrated into in the MGI gene catalog. In this report, we focus on the integration of gene models and curated annotations from the three major genome annotation providers: NCBI, Ensembl, and Vertebrate Genome Annotation database (Vega).
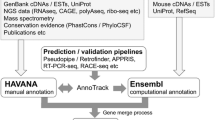
The MGI gene catalog is generated using a semi-automated, scalable analysis pipeline called GU (for “gene unification”) that estimates the equivalency of genome features based on genome coordinate overlap. At the heart of this pipeline, there is an algorithm called fjoin (feature join) (Richardson 2006). While a trivial-nested loop can find all pairs of overlapping features in two inputs files, the running time grows geometrically with the file sizes. Fjoin performs the same computation far more efficiently; comparison of two genome annotation files with hundreds of thousands of annotated features takes only minutes to perform. Genome features with overlapping coordinates form bipartite graphs, which are separated and categorized according to the number of participating top-level features. The categories are labeled by cardinality: 1:1, 1:0, 0:1, 1:n, n:1, and n:m (Fig. 1). These groupings make it easier to target gene sets that require manual inspection to resolve annotation discrepancies. The 1:1 category includes instances where a feature in one annotation file overlaps one, and only one, genome feature in the second annotation file, and vice versa. The 1:0 and 0:1 categories include features that are unique to one of the annotation files. The 1:n and n:1 categories include instances where a feature in one annotation file overlaps more than one feature in the other annotation file. The n:m category reflects complex relationships involving multiple features from both annotation files.

An overview of the gene unification process. Following the comparison of gene predictions and curated annotations using fjoin, the coordinate-based overlap results are binned into six categories. Three of the categories (1:1, 0:1, 1:0) can be loaded into MGI with minimal manual assessment. The other three categories (1:n, n:1, n:m) require manual assessment followed by resolution of annotation discrepancies through communication with the annotation provider(s) or by changes in MGI
The primary inputs for the MGI unified gene catalog are the genome feature predictions and annotations for the laboratory mouse reference genome generated by three major genome annotation providers: Ensembl, NCBI, and Vega. Each of these annotation providers employs different computational and manual methods that result in similar, but not identical, sets of gene models. For mouse, NCBI’s Eukaryotic Genome Annotation Pipeline starts with the alignment of transcripts and proteins, especially sequences that included the curated RefSeq resource (Pruitt et al. 2014). Splign is used for transcript alignment (Kapustin et al. 2008) and ProSplign for protein alignment). The pipeline also incorporates results from an HMM-based gene prediction program (Gnomon) (Pruitt et al. 2014). The Vega database represents clone-by-clone manual curation of finished genomic sequences by the Human and Vertebrate Analysis and Annotation (HAVANA) group at the Sanger Institute (Wilming et al. 2008). Ensembl’s automatic gene annotation system relies on the alignments of mRNAs and protein sequences to the assembly (Flicek et al. 2013). In addition, the Ensembl genome annotation incorporates all of the genes manually annotated by HAVANA group. GU accepts as input annotation files in General Feature Format (GFF or GFF3; http://www.sequenceontology.org/gff3.shtml). Genome features are usually genes, but any entity with genome coordinates can be used as input. GU is highly configurable and the amount of coordinate overlap required to call two features equivalent can be adjusted; the types of genome features to be included or excluded in the analysis can be configured, and a requirement for features to be on the same strand can be turned on or off.
Materials and methods
Data sources
The following genome feature prediction and curated annotation sources were used for the analysis: NCBI v104, Ensembl v78, and Vega v58 (Table 1). The annotations in Ensembl included both the computational predictions from the Ensembl genome analysis pipeline and the HAVANA team’s manually curated annotations in the Vega database. The manually curated annotations represent a subset of all genome features predicted in the mouse genome. Regardless, annotations from Vega were analyzed as a separate annotation source as we have previously observed cases where some genome features in Vega are not represented in the combined Ensembl/Vega annotation file.
Genome feature predictions from NCBI v104 were based on the reference genome assembly GRCm38.p2 for the laboratory mouse (C57BL/6J). Predictions and annotations from Ensembl v78 and Vega v58 were based on assembly GRCm38.p3. Details regarding the differences in the assembly versions are available from the Genome Reference Consortium (GRC) web site (http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/) (Church et al. 2015). Annotation files from Ensembl and Vega were converted from GTF format to GFF3 prior to the GU analysis.
Gene unification (GU) using the fjoin algorithm
Annotation files in GFF3 format from two genome annotation sources were used as input to the GU process using the fjoin algorithm (Richardson 2006). Pairwise comparison of the annotations from NCBI, Ensembl, and Vega to each other and to the current MGI gene catalog was performed. The fjoin program was configured to consider a genome coordinate overlap of a single-nucleotide position on the same strand as sufficient for establishing the initial assertion of equivalency of two genome features. For protein-coding genes, the fjoin analysis was further constrained to consider coordinate overlaps between exons. We informally evaluated the results obtained from fjoin using different overlap values (e.g., 1, 10, 20, 50, 70, and 100 nt). Only minor differences were noted in the numbers of features in each of the fjoin categories. For example, small RNAs were often included in 0:1 or 1:0 categories when overlaps of more than 20 nt were required leading to false negative equivalency assertions. Changing overlap parameters did not significantly reduce the time needed for manual review of genome features in complex overlap categories (e.g., 1:n, n:1, n:m).
Processing fjoin categories
Genome features that were deemed to be equivalent to existing MGI genome features (1:1 category) were loaded into MGI without manual review. For genome features novel to MGI (0:1 category), new gene records were created and assigned official nomenclature according to the guidelines of the International Committee on Standardized Nomenclature for Mice.
The genome features that fell into the n:1, 1:n, and n:m categories were reviewed manually. When the manual review identified genome features needing changes to annotations in external resources, the cases were shared with curators at HAVANA and NCBI using a private, on-line Mouse Genome Annotation (MGA) issue tracking system. The MGA allows for coordinated analysis and resolution of annotation discrepancies thereby supporting ongoing refinement and improvement of mouse genome annotations. The MGA resource was implemented using JIRA (https://www.atlassian.com/) and is hosted by NCBI.
Biotype conflicts
In addition to genome features and their coordinates, the MGI gene catalog also includes the biotype annotations associated with the features in each of the annotation sources. Some of the prediction/annotation sources include biotype annotations for both genome features and the transcripts of those features. We used only genome feature level annotations to identify contradictory biotypes among features our unification process identified as being equivalent; transcript-level biotypes were not considered in the biotype conflict evaluation. As there is no single biotype vocabulary that all annotation groups share in common, we maintain a mapping among the various terminologies in a biotype thesaurus. For example, NCBI annotates all pseudogenes using the biotype term “pseudo.” In contrast, pseudogenes from Vega are classified into multiple subcategories: unitary_pseudogene, processed_pseudogene, translated_processed_pseudogene, transcribed_processed_pseudogene, unprocessed_pseudogene, translated_unprocessed_pseudogene, and transcribed_unprocessed_pseudogene. All of these terms are considered equivalent to the biotype of “pseudogene” in MGI. Similarly, Vega’s IG_pseudogene and TR_pseudogene biotypes and Ensembl’s IG_D_pseudogene, IG_C_pseudogene, IG_V_pseudogene TR_J_pseudogene, and TR_V_pseudogene are all considered equivalent to the term “pseudogenic gene segment” in MGI.
The MGI biotype thesaurus is updated as new biotype terms appear in the annotation files from the three major annotation providers. Within MGI, genome features are given biotype labels based on terms in the Sequence Ontology (Mungall et al. 2011).
Updating the unified gene catalog
The unified gene catalog is updated whenever a new annotation version is released by NCBI, Ensembl, or Vega. The updates include the addition of new genome features as described above as well as the discontinuation of genes that no longer have evidence to support them. When a new version of the reference mouse genome assembly is released, the coordinates in the unified catalog are converted to the new assembly coordinates using NCBI Remap (http://www.ncbi.nlm.nih.gov/genome/tools/remap).
Results
Pairwise comparison of annotations using fjoin
The results of the GU process summarized in Table 2 illustrate that the majority of genome features from the various input annotations had equivalent entries in all three sources of genome annotation. However, there were also many features that were unique to a particular source (Table 2). In general, protein-coding gene predictions are largely similar across the three sources whereas the representation of pseudogenes and non-protein-coding genome features is more variable (Fig. 2a, b).

Example of genome features in the 1:1, 1:0, and 0:1 categories generated by fjoin. a (i) The Arl8b and Edem1 genes have equivalent (1:1) predictions in NCBI and Ensembl, but these genes are not currently represented in the Vega database (1:0). (ii) The NCBI non-protein-coding RNA gene (GeneID:102638990) is unique to the predictions from NCBI (0:1). (iii) The MGI gene, 9430088B20Rik (MGI:2445127), is unique to MGI (0:1). b (i) The Olfr794 gene (MGI:3030628) has equivalent (1:1) predictions in NCBI and Ensembl, but not in Vega (1:0). (ii) The pseudogene, Olfr795-ps1 (MGI:3030629), is only annotated by NCBI. (iii) The miRNA gene Gm23252 (MGI:5453029) is predicted only by Ensembl
The 3095 genome features that are unique to MGI mostly consist of genes created from full-length cDNAs sequenced as part of the functional annotation of the mouse (FANTOM) initiative (Okazaki et al. 2002). The 12,719 genome features that are unique to NCBI are mostly long non-coding RNA genes and pseudogenes. These distinct features are likely linked to NCBI’s genome analysis for version 104 which incorporated RNA Seq alignments from projects represented in the Sequence Read Archive (Shumway et al. 2010) to assist in gene structure prediction. Similar to NCBI, the 11,957 Ensembl genome annotations not in NCBI are mostly long non-coding RNA genes and pseudogenes. Over 900 of the unique genes in the Ensembl/Vega annotations are located on chromosome Y, which was not well annotated in previous releases. The reconciliation of genome features from Ensembl, Vega, and NCBI with the previous version of the MGI unified gene catalog resulted in 8896 new genome features in MGI. New records in MGI were created for these features, and they were reviewed and assigned official gene nomenclature and biotype annotations.
Over 4000 genome features from each of the gene prediction and manual annotation providers (NCBI, Ensembl, and Vega) fell in the 1:n, n:1, and n:m categories that need further evaluation (Fig. 3). In many cases, the complex overlaps among features reflect differences in how gene concepts are represented in different databases. For example, Ugt2a1 (MGI:2149905) and Ugt2a2 (MGI:3576095) are considered different genes in MGI and NCBI but the Ensembl/Vega groups consider these to be a single gene with multiple transcripts (Fig. 4). MGI database users are alerted to the fact that a genome feature overlaps other genome features by alerts provided in the “Other database links” section of the gene detail page. In yet other cases, the evaluation of genome features in this category identified issues with the gene predictions that required action on the part of the annotation providers. These were shared with the curation groups using the MGA issue tracking system (Fig. 5). The features with complex coordinate overlaps due to differences in gene concepts that appear repeatedly are not reviewed after each new version of the MGI gene catalog is generated. Typically, only about 10 % (several hundred) of the features in the 1:n, n:1, and n:m categories represent new cases that need manual review when the gene catalog is updated.

Example of genome features in the 1:n and n:m categories generated by fjoin. a The lincRNA gene, Gm13853 (MGI:3649279), has a 1:n relationship with two NCBI genes (GeneID:102634942 and GeneID:102634837) based on coordinate overlap shown in the boxed regions. b The ENSEMBL gene models, Gbp8 (ENSMUSG00000034438) and Gbp9 (ENSMUSG00000029298) both have extended first exons that overlap the upstream gene, Gbp4 (ENSMUSG00000079363) (shown in the boxed regions) resulting in a n:m relationship with the NCBI gene Gbp4 (GeneID:17472)

Differences in gene definitions among genome annotation groups lead to ambiguity in determining equivalency of genome features. The cases illustrated in this figure reflect differences in how genes are defined rather than annotation errors and are excluded from further manual review. a NCBI and MGI represent Ugt2a1 (MGI:2149905) and Ugt2a2 (MGI:3576095) as two different genes while Ensembl and HAVANA represent the data as a single gene with multiple alternative transcripts. b NCBI’s mouse genome annotation contains separate entries for (i) Esp5 (MGI:5522708) and (ii) Esp6 (MGI:3643294)as well as the (iii) Esp6Esp5 (MGI:5529083) read through product. Ensembl lacks a specific genome annotation for Esp5, but does represent (ii*) Esp6 and (iii*) Esp6Esp5

Example of annotation improvements as the results of the collaboration among curators from MGI, NCBI, and Vega. a Vega annotation version 35 for the reference mouse genome (GRCm37) included two separate genes (OTTMUSG0000009560 and OTTMUSG0000009562) that overlapped a single gene in the MGI catalog (Gm853; MGI:2685699). This case was identified by the review of features in the 1:n category following a previous fjoin analysis. b Upon review of all of the evidence, the HAVANA curation team merged gene OTTMUSG0000009560 with OTTMUSG0000009562. The transcript that was previously used as evidence of a different genes is now represented as an alternative processed transcript of OTTMUSG0000009562
Biotype conflicts
There are currently 2086 genes in MGI with biotype conflict note. These cases are highlighted to by the presence of a “biotype conflict” icon displayed on the MGI gene detail page (Fig. 6). One example of a feature with a conflict is the amylase 2b gene (Amy2b; MGI:104547). Amy2b is a functional gene in YBR strain; it is reported to be a null allele in the A/J inbred mouse strain. (Gumucio et al. 1985; Strahler and Meisler 1982). Amy2b is annotated as a pseudogene on the reference genome assembly by both Vega and Ensembl. MGI also annotates Amy2b as a pseudogene as there is no direct experimental data for its coding potential in C57BL/6 J. In NCBI, this gene is annotated as a protein-coding gene because according to their annotation guidelines, the biotype “protein coding” applies even if the gene is protein coding in some strains and is a pseudogene in others. A complete list of markers with biotype conflicts is available from the MGI ftp site (ftp://ftp.informatics.jax.org/pub/reports/MGI_BioTypeConflict.rpt).

a The MGI biotype conflict note is shown for the pseudogene, Amy2b (MGI:104547), which is annotated as pseudogene by both Vega and Ensembl but as a protein-coding gene by NCBI. b There is also a Strain-Specific Marker notification displayed for this locus because Amy2b has been shown to be a functional gene in the YBR strain but a null allele in the A/J mouse strain
Accessing the MGI unified gene catalog
The code and documentation for fjoin is available from MGI’s ftp site (ftp://ftp.informatics.jax.org/pub/fjoin/). The MGI unified gene catalog is searchable via the MGI database (http://www.informatics.jax.org). The MGI gene catalog is also displayed in context of the annotations from Ensembl, Vega, and NCBI using MGI’s JBrowse-based genome browser (http://jbrowse.informatics.jax.org). Within JBrowse users can view the details of similarities and differences of gene structure details across different annotation sources. Each genome feature in the MGI catalog is an aggregate representation generated by combining the annotations from multiple predictions into a single model (see Fig. 7). The annotations are available for download as a tab-delimited file from the MGI ftp site (ftp://ftp.informatics.jax.org/pub/reports/index.html#seq) and as a GFF3 formatted file (ftp://ftp.informatics.jax.org/pub/mgigff/). Also available at the ftp site is the aggregate genome feature file (MGI.exome.gff3.gz) that is used as the basis for the MGI genome feature track in MGI’s genome browser.

Example of a genome feature in the 1:1 category following fjoin analysis. The Zfp951 (MGI:2441896) gene has equivalent representations in the annotation output from Ensembl, Vega, and NCBI. However, the structural details of the predictions differ because of how evidence from different transcripts was incorporated into the gene model. The model displayed in the MGI Genome Features track represents an aggregate representation of the gene model components from all three prediction/annotation resources. The arrows highlight features that are present in gene predictions from Ensembl and HAVANA/Vega but not from NCBI
Researchers who wish to report an issue or suggested correction for specific mouse genome annotations can submit a report using the public MGA web site (http://www.ncbi.nlm.nih.gov/genome/guide/mouse/MGAReport.shtml). Submissions from the web site results in a “ticket” in the tracking system shared by the mouse annotation curation groups at The Jackson Laboratory, The Sanger Institute, and NCBI.
Discussion
In this report, we describe the method by which we combine annotation outputs from multiple, independent genome analysis pipelines into a unified gene catalog for the mouse reference genome. As annotated genome assemblies for other mouse strains are generated, we will use the methods described in this report to generate additional strain-specific gene catalogs. The algorithm that drives the GU process, fjoin, is organism-agnostic and could be used to support similar annotation integration efforts for any organism for which there are multiple sources of genome feature predictions.
The MGI unified gene catalog effort has similarities to the Consensus CDS (CCDS) project (to which MGI is a contributing partner) at NCBI (Farrell et al. 2014). Similar to the CCDS initiative, the primary inputs for MGI’s gene catalog are genome annotations from Ensembl, NCBI, and Vega. The CCDS focuses on those annotations/predictions with consistently annotated full-length coding regions (i.e., those with an ATG and valid stop-codon) that can be translated using consensus splice sites without frameshifts. Where the goal of the CCDS is to identify the highest confidence protein-coding gene annotations only, MGI’s gene catalog includes all genome annotations, regardless of biotype. Gene models categorized as equivalent by our unification process are likely to be representations of the same gene or transcription unit. Equivalent gene models, however, are not necessarily identical in gene structure and our pipeline does not evaluate which gene model is likely to be the “best” representation.
The unified gene catalog serves as the foundation for the annotation of biological attributes (i.e., phenotype, function, expression, and pathway membership) of mouse genes by expert curators and bio-data analysts at the MGI (http://www.informatics.jax.org) database (Eppig et al. 2015). The MGI gene catalog also serves as the basis for mouse genome features represented at NCBI’s Gene resource (Brown et al. 2015; Sayers et al. 2012) and was a primary source of genes for the first phase of The International Knockout Mouse Project (KOMP) (Bradley et al. 2012; International Mouse Knockout et al. 2007).
The output from MGI’s unified gene catalog process systematically identifies gene models that are potentially problematic in their structural details as well as those that appear to be equivalent across different sources but have contradictory biotype annotations. The ongoing assessment of genome annotation issues at MGI in cooperation with the international mouse genome annotation community ensures that the biomedical community gains maximum benefit from the reference mouse genome.
References
Bradley A, Anastassiadis K, Ayadi A, Battey JF, Bell C, Birling MC, Bottomley J, Brown SD, Burger A, Bult CJ, Bushell W, Collins FS, Desaintes C, Doe B, Economides A, Eppig JT, Finnell RH, Fletcher C, Fray M, Frendewey D, Friedel RH, Grosveld FG, Hansen J, Herault Y, Hicks G, Horlein A, Houghton R, Hrabe de Angelis M, Huylebroeck D, Iyer V, de Jong PJ, Kadin JA, Kaloff C, Kennedy K, Koutsourakis M, Lloyd KC, Marschall S, Mason J, McKerlie C, McLeod MP, von Melchner H, Moore M, Mujica AO, Nagy A, Nefedov M, Nutter LM, Pavlovic G, Peterson JL, Pollock J, Ramirez-Solis R, Rancourt DE, Raspa M, Remacle JE, Ringwald M, Rosen B, Rosenthal N, Rossant J, Ruiz Noppinger P, Ryder E, Schick JZ, Schnutgen F, Schofield P, Seisenberger C, Selloum M, Simpson EM, Skarnes WC, Smedley D, Stanford WL, Stewart AF, Stone K, Swan K, Tadepally H, Teboul L, Tocchini-Valentini GP, Valenzuela D, West AP, Yamamura K, Yoshinaga Y, Wurst W (2012) The mammalian gene function resource: the International Knockout Mouse Consortium. Mamm Genome 23:580–586
Brown GR, Hem V, Katz KS, Ovetsky M, Wallin C, Ermolaeva O, Tolstoy I, Tatusova T, Pruitt KD, Maglott DR, Murphy TD (2015) Gene: a gene-centered information resource at NCBI. Nucleic Acids Res 43:D36–D42
Burge SW, Daub J, Eberhardt R, Tate J, Barquist L, Nawrocki EP, Eddy SR, Gardner PP, Bateman A (2013) Rfam 11.0: 10 years of RNA families. Nucleic Acids Res 41:D226–D232
Chan PP, Lowe TM (2009) GtRNAdb: a database of transfer RNA genes detected in genomic sequence. Nucleic Acids Res 37:D93–D97
Church DM, Schneider VA, Steinberg KM, Schatz MC, Quinlan AR, Chin CS, Kitts PA, Aken B, Marth GT, Hoffman MM, Herrero J, Mendoza ML, Durbin R, Flicek P (2015) Extending reference assembly models. Genome Biol 16:13
Eppig JT, Blake JA, Bult CJ, Kadin JA, Richardson JE, Mouse Genome Database G (2015) The Mouse Genome Database (MGD): facilitating mouse as a model for human biology and disease. Nucleic Acids Res 43:D726–D736
Farrell CM, O’Leary NA, Harte RA, Loveland JE, Wilming LG, Wallin C, Diekhans M, Barrell D, Searle SM, Aken B, Hiatt SM, Frankish A, Suner MM, Rajput B, Steward CA, Brown GR, Bennett R, Murphy M, Wu W, Kay MP, Hart J, Rajan J, Weber J, Snow C, Riddick LD, Hunt T, Webb D, Thomas M, Tamez P, Rangwala SH, McGarvey KM, Pujar S, Shkeda A, Mudge JM, Gonzalez JM, Gilbert JG, Trevanion SJ, Baertsch R, Harrow JL, Hubbard T, Ostell JM, Haussler D, Pruitt KD (2014) Current status and new features of the Consensus Coding Sequence database. Nucleic Acids Res 42:D865–D872
Flicek P, Ahmed I, Amode MR, Barrell D, Beal K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fairley S, Fitzgerald S, Gil L, Garcia-Giron C, Gordon L, Hourlier T, Hunt S, Juettemann T, Kahari AK, Keenan S, Komorowska M, Kulesha E, Longden I, Maurel T, McLaren WM, Muffato M, Nag R, Overduin B, Pignatelli M, Pritchard B, Pritchard E, Riat HS, Ritchie GR, Ruffier M, Schuster M, Sheppard D, Sobral D, Taylor K, Thormann A, Trevanion S, White S, Wilder SP, Aken BL, Birney E, Cunningham F, Dunham I, Harrow J, Herrero J, Hubbard TJ, Johnson N, Kinsella R, Parker A, Spudich G, Yates A, Zadissa A, Searle SM (2013) Ensembl 2013. Nucleic Acids Res 41:D48–D55
Gumucio DL, Wiebauer K, Dranginis A, Samuelson LC, Treisman LO, Caldwell RM, Antonucci TK, Meisler MH (1985) Evolution of the amylase multigene family. YBR/Ki mice express a pancreatic amylase gene which is silent in other strains. J Biol Chem 260:13483–13489
International Mouse Knockout C, Collins FS, Rossant J, Wurst W (2007) A mouse for all reasons. Cell 128:9–13
Kapustin Y, Souvorov A, Tatusova T, Lipman D (2008) Splign: algorithms for computing spliced alignments with identification of paralogs. Biol Direct 3:20
Kozomara A, Griffiths-Jones S (2014) miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res 42:D68–D73
Mungall CJ, Batchelor C, Eilbeck K (2011) Evolution of the Sequence Ontology terms and relationships. J Biomed Inform 44:87–93
Okazaki Y, Furuno M, Kasukawa T, Adachi J, Bono H, Kondo S, Nikaido I, Osato N, Saito R, Suzuki H, Yamanaka I, Kiyosawa H, Yagi K, Tomaru Y, Hasegawa Y, Nogami A, Schonbach C, Gojobori T, Baldarelli R, Hill DP, Bult C, Hume DA, Quackenbush J, Schriml LM, Kanapin A, Matsuda H, Batalov S, Beisel KW, Blake JA, Bradt D, Brusic V, Chothia C, LE Corbani, Cousins S, Dalla E, Dragani TA, Fletcher CF, Forrest A, Frazer KS, Gaasterland T, Gariboldi M, Gissi C, Godzik A, Gough J, Grimmond S, Gustincich S, Hirokawa N, Jackson IJ, Jarvis ED, Kanai A, Kawaji H, Kawasawa Y, Kedzierski RM, King BL, Konagaya A, Kurochkin IV, Lee Y, Lenhard B, Lyons PA, Maglott, Maltais L, Marchionni L, McKenzie L, Miki H, Nagashima T, Numata K, Okido T, Pavan WJ, Pertea G, Pesole G, Petrovsky N, Pillai R, Pontius JU, Qi D, Ramachandran S, Ravasi T, Reed JC, Reed DJ, Reid J, Ring BZ, Ringwald M, Sandelin A, Schneider C, Semple CA, Setou M, Shimada K, Sultana R, Takenaka Y, Taylor, Teasdale RD, Tomita M, Verardo R, Wagner L, Wahlestedt C, Wang Y, Watanabe Y, Wells C, Wilming LG, Wynshaw-Boris A, Yanagisawa M, Yang I, Yang L, Yuan Z, Zavolan M, Zhu Y, Zimmer A, Carninci P, Hayatsu N, Hirozane-Kishikawa T, Konno H, Nakamura M, Sakazume N, Sato K, Shiraki T, Shiraki T, Waki K, Kawai J, Aizawa K, Arakawa T, Fukuda S, Hara A, Hashizume W, Imotani K, Ishii Y, Itoh M, Kagawa I, Miyazaki A, Sakai K, Sasaki D, Shibata K, Shinagawa A, Yasunishi A, Yoshino M, Waterston R, Lander ES, Rogers J, Birney E, Hayashizaki Y, Consortium F, I RGERGP, Team II (2002) Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNAs. Nature 420:563–573
Pruitt KD, Brown GR, Hiatt SM, Thibaud-Nissen F, Astashyn A, Ermolaeva O, Farrell CM, Hart J, Landrum MJ, McGarvey KM, Murphy MR, O’Leary NA, Pujar S, Rajput B, Rangwala SH, Riddick LD, Shkeda A, Sun H, Tamez P, Tully RE, Wallin C, Webb D, Weber J, Wu W, DiCuccio M, Kitts P, Maglott DR, Murphy TD, Ostell JM (2014) RefSeq: an update on mammalian reference sequences. Nucleic Acids Res 42:D756–D763
Richardson JE (2006) fjoin: simple and efficient computation of feature overlaps. J Comput Biol 13:1457–1464
Sayers EW, Barrett T, Benson DA, Bolton E, Bryant SH, Canese K, Chetvernin V, Church DM, Dicuccio M, Federhen S, Feolo M, Fingerman IM, Geer LY, Helmberg W, Kapustin Y, Krasnov S, Landsman D, Lipman DJ, Lu Z, Madden TL, Madej T, Maglott DR, Marchler-Bauer A, Miller V, Karsch-Mizrachi I, Ostell J, Panchenko A, Phan L, Pruitt KD, Schuler GD, Sequeira E, Sherry ST, Shumway M, Sirotkin K, Slotta D, Souvorov A, Starchenko G, Tatusova TA, Wagner L, Wang Y, Wilbur WJ, Yaschenko E, Ye J (2012) Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 40:D13–D25
Shumway M, Cochrane G, Sugawara H (2010) Archiving next generation sequencing data. Nucleic Acids Res 38:D870–D871
Strahler JR, Meisler M (1982) Two distinct pancreatic amylase genes are active in YBR mice. Genetics 101:91–102
Wilming LG, Gilbert JG, Howe K, Trevanion S, Hubbard T, Harrow JL (2008) The vertebrate genome annotation (Vega) database. Nucleic Acids Res 36:D753–D760
Yue F, Cheng Y, Breschi A, Vierstra J, Wu W, Ryba T, Sandstrom R, Ma Z, Davis C, Pope BD, Shen Y, Pervouchine DD, Djebali S, Thurman RE, Kaul R, Rynes E, Kirilusha A, Marinov GK, Williams BA, Trout D, Amrhein H, Fisher-Aylor K, Antoshechkin I, DeSalvo G, See LH, Fastuca M, Drenkow J, Zaleski C, Dobin A, Prieto P, Lagarde J, Bussotti G, Tanzer A, Denas O, Li K, Bender MA, Zhang M, Byron R, Groudine MT, McCleary D, Pham L, Ye Z, Kuan S, Edsall L, Wu YC, Rasmussen MD, Bansal MS, Kellis M, Keller CA, Morrissey CS, Mishra T, Jain D, Dogan N, Harris RS, Cayting P, Kawli T, Boyle AP, Euskirchen G, Kundaje A, Lin S, Lin Y, Jansen C, Malladi VS, Cline MS, Erickson DT, Kirkup VM, Learned K, Sloan CA, Rosenbloom KR, Lacerda de Sousa B, Beal K, Pignatelli M, Flicek P, Lian J, Kahveci T, Lee D, Kent WJ, Ramalho Santos M, Herrero J, Notredame C, Johnson A, Vong S, Lee K, Bates D, Neri F, Diegel M, Canfield T, Sabo PJ, Wilken MS, Reh TA, Giste E, Shafer A, Kutyavin T, Haugen E, Dunn D, Reynolds AP, Neph S, Humbert R, Hansen RS, De Bruijn M, Selleri L, Rudensky A, Josefowicz S, Samstein R, Eichler EE, Orkin SH, Levasseur D, Papayannopoulou T, Chang KH, Skoultchi A, Gosh S, Disteche C, Treuting P, Wang Y, Weiss MJ, Blobel GA, Cao X, Zhong S, Wang T, Good PJ, Lowdon RF, Adams LB, Zhou XQ, Pazin MJ, Feingold EA, Wold B, Taylor J, Mortazavi A, Weissman SM, Stamatoyannopoulos JA, Snyder MP, Guigo R, Gingeras TR, Gilbert DM, Hardison RC, Beer MA, Ren B, Mouse EC (2014) A comparative encyclopedia of DNA elements in the mouse genome. Nature 515:355–364
Acknowledgments
The development of the fjoin algorithm and its application to building and maintaining a unified catalog of mouse genes was supported by NIH HG00330-P1. The authors gratefully acknowledge the ongoing contribution of all of the members of the MGI consortium at The Jackson Laboratory to maintaining and functionally annotating the reference mouse genome feature catalog. The authors thank all of the curators at NCBI and HAVANA for their collaborative efforts to improve annotations for the reference mouse genome. The authors thank Dr. Joel Graber for his critical review of an early version of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
J. E. Richardson and Y. Zhu have contributed equally to this work.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Zhu, Y., Richardson, J.E., Hale, P. et al. A unified gene catalog for the laboratory mouse reference genome. Mamm Genome 26, 295–304 (2015). https://doi.org/10.1007/s00335-015-9571-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00335-015-9571-1