
Abstract
Mouse genetics is a powerful approach for discovering genes and other genome features influencing human pain sensitivity. Genetic mapping studies have historically been limited by low mapping resolution of conventional mouse crosses, resulting in pain-related quantitative trait loci (QTL) spanning several megabases and containing hundreds of candidate genes. The recently developed Diversity Outbred (DO) population is derived from the same eight inbred founder strains as the Collaborative Cross, including three wild-derived strains. DO mice offer increased genetic heterozygosity and allelic diversity compared to crosses involving standard mouse strains. The high rate of recombinatorial precision afforded by DO mice makes them an ideal resource for high-resolution genetic mapping, allowing the circumvention of costly fine-mapping studies. We utilized a cohort of ~300 DO mice to map a 3.8 Mbp QTL on chromosome 8 associated with acute thermal pain sensitivity, which we have tentatively named Tpnr6. We used haplotype block partitioning to narrow Tpnr6 to a width of ~230 Kbp, reducing the number of putative candidate genes from 44 to 3. The plausibility of each candidate gene’s role in pain response was assessed using an integrative bioinformatics approach, combining data related to protein domain, biological annotation, gene expression pattern, and protein functional interaction. Our results reveal a novel, putative role for the protein-coding gene, Hydin, in thermal pain response, possibly through the gene’s role in ciliary motility in the choroid plexus–cerebrospinal fluid system of the brain. Real-time quantitative-PCR analysis showed no expression differences in Hydin transcript levels between pain-sensitive and pain-resistant mice, suggesting that Hydin may influence hot-plate behavior through other biological mechanisms.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Human pain sensitivity varies widely between individuals, and the significant influence of genetic factors on pain sensitivity is now widely appreciated (Mogil et al. 1999a). Genetic differences in underlying nociceptive and central pain processing mechanisms are partially responsible for observed variation in pain sensitivity. Although acute and chronic pain are considered clinically distinct phenomena, they are understood to exhibit some degree of overlap at the physiological and molecular genetic levels (Mogil et al. 1999b, 2005b). A genetic understanding of variability in pain sensitivity is essential to developing improved prevention and treatment methods for both acute and chronic pain.
The application of complex trait analysis to human pain genetics research has facilitated the discovery of genes that underlie variability in pain sensitivity and analgesic response (Chou et al. 2006; Compton et al. 2003; Diatchenko et al. 2005, 2006; Fillingim et al. 2005; Indo et al. 1996; Janicki et al. 2006; Klepstad et al. 2004; Nackley et al. 2006; Poulsen et al. 1996; Rakvag et al. 2005; Reyes-Gibby et al. 2007; Sindrup et al. 1990; Tsao et al. 2011; Zubieta et al. 2003). However, human complex trait analyses typically require tens of thousands of individuals to achieve adequate statistical power, in part due to the inherent difficulty of controlling gene–environment interactions in human cohorts (Flint and Eskin 2012). Control over environmental variables is of paramount importance in genetic studies of complex traits. Even when adequate sample size and statistical power are present, the sum of the identified genetic effects in human genetic studies of complex traits comprises only a fraction of the estimated trait heritability, usually less than half (Stranger et al. 2011). Inbred mouse strains facilitate partitioning of trait variance into genetic and environmental components—a nearly impossible task in human studies of pain (Mogil and Grisel 1998). Pain genetics studies using mouse models require only a few hundred animals to identify loci that explain 50 % or more of the phenotypic variance for a particular trait (Flint and Eskin 2012).
Genetic linkage mapping, a technique commonly used to map regions of the genome associated with a phenotype of interest, has facilitated the identification of at least 14 pain-related quantitative trait loci (QTL) in the laboratory mouse to date (Devor et al. 2005, 2007; Furuse et al. 2003; Honda and Takano 2009; LaCroix-Fralish et al. 2009; Mogil et al. 1997, 2005b, 2006; Nair et al. 2011; Nissenbaum et al. 2010; Seltzer et al. 2001; Wilson et al. 2002). Most current mapping populations are produced using traditional two-strain breeding schemes, resulting in QTL spanning an average of ~30 Mbp and containing hundreds of potential candidate genes. The low genetic recombination density and resulting lack of mapping precision afforded by historical mapping crosses necessitates years of additional fine mapping to elucidate the underlying candidate gene(s). Identification of the Pain1 candidate gene Cacng2 (MGI:1316660; calcium channel, voltage-dependent, gamma subunit 2) involved 3 years of additional fine mapping by three different laboratories (Devor et al. 2005, 2007; Nissenbaum et al. 2010; Seltzer et al. 2001). The 50 Mbp thermal pain QTL Tpnr4 required the generation of two F2 hybrid crosses and a congenic strain to identify the candidate gene Calca (MGI:2151253; calcitonin/calcitonin-related polypeptide, alpha) (Mogil et al. 2005b). In the case of Mc1r (MGI:99456; melanocortin 1 receptor), narrowing of the original 24 Mbp QTL and confirmation of its female-specific role in pain response required an additional 2 years of work (Mogil et al. 2003, 2005a).
Emerging mouse populations such as the Diversity Outbred (DO) stock (Churchill et al. 2012) offer increased heterozygosity and allelic diversity compared to conventional mapping populations. DO mice are produced by the repeated random outcrossing of non-siblings from the Oak Ridge National Laboratory (ORNL) Collaborative Cross (CC) colony (Chesler et al. 2008), a genetically defined and reproducible panel of recombinant inbred strains derived from a set of eight genetically diverse parental strains (A/J, C57BL/6J, 129S1/SvImJ, NOD/ShiLtJ, NZO/HlLtJ, CAST/EiJ, PWK/PhJ, and WSB/EiJ). The DO mouse population captures the same set of natural allelic variants derived from a common set of eight founder strains. Due to a randomized outbreeding strategy, each mouse is a unique individual with one of an effectively limitless combination of the segregating alleles. DO mice have approximately 45 million segregating SNPs—four times the number found in classical laboratory mouse strains—making them an ideal resource for high-resolution genetic mapping (Svenson et al. 2012).
We present here the first application of DO mice to pain genetics research. Utilizing a statistical algorithm recently developed for genetic linkage mapping in CC and DO mice (Svenson et al. 2012), we were able to map a 3.8 Mbp QTL containing 44 candidate genes in a single study using ~300 DO mice. We narrowed the QTL region, which we have tentatively named Tpnr6 (thermal pain response 6), by partitioning the QTL into intervals using haplotype blocks. The narrowed Tpnr6 region is ~230 Kbp in size and contains three putative candidate genes: (1) the protein-coding gene Hydin (axonemal central pair apparatus protein; MGI:2389007), (2) the lincRNA gene Gm26832 (predicted gene, 26832; MGI:5477326), and (3) the snRNA gene Gm23627 (predicted gene, 23627; MGI:5453404). We assessed the plausibility of each candidate gene’s role in pain response using an integrative bioinformatics approach, combining data related to protein domain, biological annotation, gene expression pattern, and protein functional interaction. Our results reveal a novel, putative role for the protein-coding gene Hydin in thermal pain response, possibly through its role in ciliary motility in the choroid plexus–cerebrospinal fluid (CP–CSF) system of the brain (Fig. 1).

Study Workflow. The above illustration depicts the power of precise genetic mapping combined with integrative bioinformatics to elucidate candidate genes involved in complex traits. The application of genetic and genomic techniques facilitated the quick identification of a single candidate gene for hot-plate response, the protein-coding gene, Hydin
Materials and methods
Diversity Outbred mice
Incipient CC breeding lines used to establish the DO population at The Jackson Laboratory were received from ORNL and were of generation G2:Fn (n ≤ 12). Male and female DO mice (n = 283; J:DO, JAX stock number 009376) from generations 4 and 5 (G4 and G5) of outcrossing were obtained from The Jackson Laboratory at 6 weeks of age and transferred to the housing facility via wheeled cart. Mice were acclimated to the vivarium for at least 2 weeks prior to testing at 13–17 weeks of age. Mice were housed in duplex polycarbonate cages with a Shepherd Shack on ventilated racks providing 99.997 % HEPA filtered air to each cage in a climate-controlled room under a standard 12:12 light–dark cycle (lights on at 0600 h). Pine cob bedding was changed weekly and mice were provided ad libitum access to food (NIH31 5K52 chow, LabDiet/PMI Nutrition, St. Louis, MO, USA) and acidified water. Initially, all mice were housed in a cage density of five males or females. During the course of the study, ∼20 % of G4 and 46 % of G5 pens of male mice were separated into smaller groups (1–4) due to aggressive behaviors. All procedures and protocols were approved by The Jackson Laboratory Animal Care and Use Committee (Protocol #EJC10-06), and were conducted in compliance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals.
Genotyping
DNA was prepared from tail biopsies and genotyping was outsourced to GeneSeek (http://www.neogen.com/GeneSeek) for analysis using the Mouse Universal Genotyping Array (MUGA; GeneSeek, Lincoln, NE, USA). Built on the Illumina Infinium platform (San Diego, CA, USA), the MUGA contains 7,851 SNP markers distributed throughout the mouse genome (average spacing of 325 Kb, standard deviation of 191 Kb) (Neogen 2010). The array’s SNP panel was optimized for the eight laboratory mouse strains that are founders of the DO mapping population, making the MUGA optimal for detecting heterozygous regions and discriminating between haplotypes in homozygous regions (Neogen 2010). The MUGA marker panel provides an average effective sampling sensitivity of just over 1 Mb, allowing any of the CC founders to be identified within a window of four to five SNPs.
Experimental design
A total of 283, 13–17 week old, DO mice (139 male, 144 female) were phenotyped using the hot-plate assay of acute thermal nociception. The DO mice utilized in this study were part of a larger phenotyping initiative described by Logan et al. (2013), in which the mice were subject to a battery of noninvasive behavioral tests to assess activity, anxiety, and response to novelty. Assays were performed in order of increasing perceived stressfulness in an effort to minimize the impact of carry-over effects as follows: day 1, open-field; day 3, light–dark box; day 4, visual-cliff avoidance; day 5, hot-plate; and day 9, tail-suspension test. Mice were randomly assigned to testing groups, such that an equal number of male and female mice were tested each day (n = ~24 per sex). Groups 1 through 4 were comprised of G4 individuals; groups 5 and 6 were of generation G5. Mice were between 12 and 16 weeks of age on the first day of testing. For the open-field, light–dark box, and visual-cliff tests, mice were habituated to the testing room for 1 h prior to testing, and 30 min was used for the hot-plate and tail-suspension tests. For each assay, mice were removed by the tail then returned to the clean side of a duplex home cage until each cage mate had completed testing. Several experimenters participated in the testing, but a single experimenter handled the mice for each test and the same individuals were in the room during all sessions of a particular test.
Hot-plate assay of acute thermal nociception
A modified version of the technique of Eddy and Leimbach (1953) [originally described by Woolfe and MacDonald (1944)] was used to perform the hot-plate assay. Mice were brought to the testing room at 9:00 a.m. and allowed to acclimate for 30 min. Pain reflexes in response to a thermal stimulus were measured using a Hot Plate Analgesia Meter (IITC Life Science Inc., cat #39, Woodland Hills, CA, USA) as previously described (Bannon and Malmberg 2007; Mogil et al. 1999a; Wilson and Mogil 2001). The surface of the hot plate (11 cm × 10.5 cm) was preheated to a constant temperature of 55 °C. Each mouse was individually placed on the hot plate surrounded by a clear acrylic cylinder (15 cm tall, 10 cm internal diameter, open top with clear acrylic lid) to constrain locomotion of the subject. Each mouse remained on the plate until it performed either of two behaviors regarded as indicative of nociception: hindpaw lick or hindpaw shake/flutter (Espejo and Mir 1993; Hammond 1989). Latency to respond to the heat stimulus was measured to the nearest 0.01 s. Each mouse was immediately removed from the hot plate as soon as it performed either of the two nociceptive responses. If a mouse did not respond within 30 s, the test was terminated and the mouse removed from the hot plate. Animals were tested individually and were not habituated to the apparatus prior to testing. Only hindpaw responses were used as endpoint criteria, as forepaw licking and lifting are components of normal grooming behavior (Mogil et al. 1999a). Jumping was not included as a phenotypic endpoint due to uncertainty as to whether jumping behavior is indicative of a nociceptive or activity-related response. Logan et al. (2013) noted that the inclusion of wild-derived alleles in the DO can result in atypical or “inappropriate” behaviors in a subset of mice during certain behavioral tests. While behaviors such as repeated jumping, wall climbing, and alternate paw lifting are interesting variants amenable to genetic mapping, they require careful analysis and interpretation and were not included as part of this study. Fourteen of the 283 phenotyped mice were excluded from the genetic linkage mapping analysis because they exhibited such behaviors.
Genetic linkage mapping
Eight DO samples were excluded from mapping analysis due to missing genotype calls, bringing the total number of mice to 261. QTL mapping was carried out as described by Svenson et al. (2012). Founder haplotypes were reconstructed using a Hidden Markov Model (HMM) that produced a matrix of 36 genotype probabilities for each sample at each SNP. Genotype probabilities at each SNP were then collapsed to an eight-state allele dosage matrix by summing the probabilities contributed by each founder. Phenotypic data were normalized by natural log transformation prior to linkage mapping analysis. Mapping was performed using QTLRel software (http://www.palmerlab.org/software) (Cheng 2011). A mixed model was fit with sex and experimental group as additive covariates and a random effect was included to account for kinship. Regression coefficients for additive effects of founder alleles were estimated at each genomic location. Significance thresholds were obtained by performing 1,000 permutations of the genome scans with phenotype data being shuffled among individuals and 2-logarithm of odds (LOD) support intervals from the linear model were determined for significant (p ≤ 0.05), suggestive (p ≤ 0.10), and trending (p ≤ 0.63) QTL peaks. All phenotype and genotype data have been made publicly available through the Mouse Phenome Database (MPD, http://phenome.jax.org/; MPD:470) and the QTL Archive (http://qtlarchive.org/; Recla_2013).
Identification of candidate genes
The genomic elements (e.g., genes, transposable elements, intergenic regions) associated with the QTL detected by genetic linkage mapping were identified using the MGI mouse genome browser (http://gbrowse.informatics.jax.org/cgi-bin/gbrowse/mouse_current/; NCBI Build 37; analysis conducted August 2011). All genes within the QTL (coding and non-coding) were considered putative candidate genes. SNPs within each gene were examined for the presence of at least one inter-strain polymorphism matching the allelic effect pattern of the genetic mapping results (Churchill et al. 2012). SNP data were obtained from dbSNP (Build 128, http://www.ncbi.nlm.nih.gov/projects/SNP/) and the Wellcome Trust Sanger Institute (http://www.sanger.ac.uk/cgi-bin/modelorgs/mousegenomes/snps.pl).
Plausibility analysis of candidate genes
In order to begin to explore and understand the biological processes and molecular pathways underlying a complex trait such as pain sensitivity, it is important to understand that while proteins carry out their functions by interacting with each other, they do not necessarily interact physically (Doerr 2010). The integration of heterogeneous genome-wide data can facilitate the development of functional relationship networks revealing the collaborative roles between proteins in carrying out a biological process (Guan et al. 2008). Genes involved in a complex trait are likely to participate in similar or overlapping pathways and should thus be more likely to share protein domains, biological annotations, and patterns of gene expression. Using these paradigms, the plausibility of each candidate gene’s role in pain response was assessed by compiling heterogeneous genomic evidence from a variety of electronic databases.
In order to examine the known functional relationships between each candidate gene and known pain genes, a list of experimentally validated pain-related genes was compiled by querying the online databases described in Table 1 using the terms “pain” and “nociception.” Ingenuity Pathway Analysis (IPA, http://www.ingenuity.com/), a bioinformatics system based on a manually curated knowledgebase, was used to map the known IPA functional interactions between candidate and known pain genes.
MouseMap (http://sonorus.princeton.edu/mousemap/applet.shtml) is a web-based tool that provides access to tissue-specific predicted functional relationships between mouse genes (Guan et al. 2012). A list of brain ventricular zone functional relationships predicted by MouseMap was obtained from Dr. Yuanfang Guan (University of Michigan Medical Center, Ann Arbor, Michigan) and analyzed for direct relationships with the candidate gene Hydin with edge weight scores ≥0.9. The functional relationship networks generated by MouseMap and IPA were assessed for enrichment of Gene Ontology (GO) (Ashburner et al. 2000) and Mammalian Phenotype (MP) (Smith and Eppig 2009) terms using VLAD (http://proto.informatics.jax.org/prototypes/vlad-1.0.3/), a web-based tool for statistical term enrichment (annotation date: November 2, 2011).
For each candidate gene, gene expression annotations were collected from the Allen Brain Atlas (Lein et al. 2007), Gene Expression Omnibus (GEO) (Barrett et al. 2013), IPA, and the Mouse Genome Database (MGD) (Eppig et al. 2012). Serial Pattern of Expression Levels Locator [SPELL (Hibbs et al. 2007)], a query-driven search engine for large gene expression microarray compendia, was used to predict genes that share spatial and temporal expression patterns with the candidate genes. InterPro protein domain (Hunter et al. 2012) and MP annotations were obtained from MGD. Pain-related phenotype data were collected from MGD, IPA, PainGenesdb, and GeneWeaver, a web-based tool that allows users to integrate phenotype-centered gene sets across species, tissues, and experimental platform (Baker et al. 2012). GO terms were obtained from MGD and AmiGO (Carbon et al. 2009), and IPA canonical pathways were obtained from IPA. All queries were performed July–November of 2011. A summary of the electronic databases mined for biological annotations during this analysis is provided in Table 2.
Real-time quantitative PCR analysis of the candidate gene Hydin
Whole brains from male and female pain-sensitive (C57BL/6J) and pain-resistant (A/J) mice (n = 5/group) were dissected and stored in RNAlater ® (Life Technologies, Carlsbad, CA, USA) prior to homogenization in TRIzol®. Total RNA was extracted using the PureLink™ RNA Mini Kit (Life Technologies, Carlsbad, CA, USA) as per manufacturer’s protocol. Samples were further purified with on-column DNase I treatment (Qiagen, Venlo, Netherlands). Extracted RNA samples were assessed for integrity and concentration using the Agilent Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA) and Nanodrop (ThermoFisher Scientific, Waltham, MA, USA), respectively. Samples with RNA integrity numbers (RIN) of 7 and above were used for downstream applications. cDNA was synthesized from extracted RNA using the MessageSensor™ RT Kit (Life Technologies, Carlsbad, CA, USA). Hydin expression levels were measured using diluted cDNA with the TaqMan Gene Expression system (Life Technologies, Carlsbad, CA, USA) and the Applied Biosystems Prism 7900HT real-time quantitative PCR system (Life Technologies, Carlsbad, CA, USA) with a pre-designed target candidate gene assay (Mm00556723_m1; Life Technologies, Carlsbad, CA, USA). Cycle conditions consisted of 2 min at 50 °C and 10 min at 95 °C, followed by 40 cycles of 15 s at 95 °C, 1 min at 60 °C. Relative fold change in expression was normalized to Gapdh expression using the well-established delta–delta Ct method (Livak and Schmittgen 2001). All assays were performed in triplicate. Hydin expression level was reported as mean ± SE and a linear regression model was used to investigate the effects of sex, strain, and their interaction (significant differences declared at p ≤ 0.05). Statistical comparisons were performed using the JMP 10.0 software package.
Results
Genetic linkage mapping
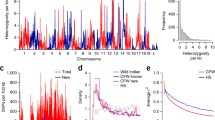
Linkage mapping identified one QTL peak nearing genome-wide significance located on chromosome 8, which has been tentatively named thermal pain response 6 [Tpnr6; MGI:5294244 (provisional)] (Fig. 2a). The peak of the Tpnr6 QTL is located at chr8:113017713 (rs31242180, NCBI Build 37) and has a LOD score of 6.1. An approximate “confidence interval” for Tpnr6 was calculated using a 2-LOD drop, resulting in an interval width of 3.8 Mbp (5′ flanking SNP UNC080383989, Chr 8: 110437369; 3′ flanking SNP rs31329971, Chr 8:114226254).

Hot-plate latency has a significant QTL on chromosome 8. a Genome-wide scan for hot-plate latency reveals a QTL with a peak LOD score of 6.1. Permutation-derived significance thresholds are marked by horizontal lines: 0.63 (bottom), 0.1 (middle), 0.05 (top). b The eight coefficients of the QTL model show the effects on hot-plate latency contributed by each founder haplotype on chromosome 8. Colored lines represent the phenotypic effect of each DO founder strain’s allelic contribution at each SNP locus across the chromosome. c QTL plot for the chromosome 8 locus
The allelic effect plot shown in Fig. 2b illustrates the phenotypic effect of each DO founder strain’s genetic contribution at each genomic position along chromosome 8. Within Tpnr6, the allelic contribution from A/J is reported to condition high hot-plate latency (low sensitivity to acute thermal pain), while allelic contributions from CAST/EiJ and C57BL/6J are reported to condition low hot-plate latency (high sensitivity to acute thermal pain). The remaining five DO founder strains are reported to have allelic contributions that condition responses to acute thermal pain that fall between A/J and C57BL/6J in the following order:
-
(1)
A/J (least sensitive)
-
(2)
WSB/EiJ and 129S1/SvlmJ
-
(3)
NOD/ShiLtJ and PWK/PhJ
-
(4)
NZO/HlLtJ
-
(5)
CAST/EiJ and C57BL/6J (most sensitive)
Hot-plate data from inbred DO founder strains have been generated by four independent labs: Jaxwest (2013), Mogil et al. (2013), Koide et al. (2013), and Chesler et al. (2013). All four datasets recapitulate the inbred strain sensitivity ranking predicted by the allelic contributions at the Tpnr6 locus (the rankings of NOD/ShiLtJ and C57BL/6J are reversed in the Chesler study).
Candidate gene identification
The 3.8 Mbp Tpnr6 QTL contains 44 protein-coding and non-coding genes (NCBI Build 37; MGI Genes and Markers query performed July 2011, Feature Type “gene”). We further narrowed the Tpnr6 interval by identifying haplotype blocks within Tpnr6 that differ between A/J and C57BL/6J | CAST/EiJ and the remaining five DO founder strains. This method of partitioning QTL into intervals that can be summarized by a single phylogenetic tree among progenitors is analogous to the approach demonstrated by Goodson et al. (2012) using anxiety phenotypes.
We used the University of North Carolina’s Mouse Phylogeny Viewer (MPV, http://msub.csbio.unc.edu) to identify one haplotype block region within Tpnr6 that differed between A/J and C57BL/6J (Chr 8:112802216.113013057). The MPV does not incorporate haplotype data from the three wild-derived DO founder strains (WSB/EiJ, PWK/PhJ, CAST/EiJ). To circumvent this limitation, we examined SNP data from the Wellcome Trust Sanger Institute (REL-1211; NCBI build 37) and searched for haplotype regions within Tpnr6 matching the A/J / C57BL/6J | CAST/EiJ allelic effect pattern. We identified a SNP exclusive to A/J near the 5′ end of the haplotype block reported by the MPV, classified by Sanger as “upstream_genetic_variant” and “downstream_genetic_variant” (no rsID; chr8:112787281). We observed several CAST/EiJ-exclusive SNPs within the Tpnr6 locus, particularly near the 3′ end of the candidate gene Hydin. A single CAST/EiJ-exclusive SNP classified as “upstream_genetic_variant” was present near the reported end of the MPV haplotype block (rs245877262; chr8:113018557), just downstream of the 3′ end of Hydin. There were also several regions within Tpnr6 where the C57BL/6J haplotype was distinct from the remaining strains, including large regions between the A/J- and CAST/EiJ-exclusive SNPs. We interpret our observations to be consistent with the allelic effect pattern observed at the Tpnr6 locus. It is likely that there are at least two SNPs conditioning the observed allelic effect; one private SNP between the five inbred strains (A/J-exclusive), and one private SNP between the inbred strains and CAST/EiJ (CAST/EiJ-exclusive). We used these haplotype data to further narrow the Tpnr6 locus. The narrowed haplotype region is ~230 kbp wide and contains three putative candidate genes:
-
1.
Hydin (axonemal central pair apparatus protein; MGI:2389007); protein-coding gene.
-
2.
Gm26832 (predicted gene, 26832; MGI:5477326); lincRNA gene.
-
3.
Gm23627 (predicted gene, 23627; MGI:5453404); snRNA gene.
The narrowed Tpnr6 interval contains four validated SNPs that differ between A/J and C57BL/6J: rs32704439, rs13479984, rs33400842, rs33170521 according to dbSNP at NCBI and SNP data from the Mouse Genomes program at the Wellcome Trust Sanger Institute. All four SNPs are located within intronic sequence of the protein-coding gene Hydin. Two of the SNPs (rs32704439 and rs13479984) are present as the same allelic variant in C57BL/6J and CAST/EiJ, matching the allelic effect pattern indicated by the genetic mapping results (CAST/EiJ data for rs33400842 and rs33170521 not available). Thus, of the three candidate genes present within the narrowed Tpnr6 locus, only Hydin is reported to contain inter-strain polymorphisms that match the effect pattern of our genetic mapping results, suggesting Hydin as the most plausible candidate gene.
Plausibility analysis of candidate genes’ role in pain response
The biological annotations, pathways, and functional interaction networks associated with each of the three Tpnr6 candidate genes were identified as described in “Plausibility analysis of candidate genes” section. At the time of this writing, there were no biological annotations or functional data for the predicted genes Gm26832 or Gm23627. Hydin is known to be expressed exclusively in motile secondary cilia, which are present in only a few distinct anatomical regions in humans and mice: respiratory epithelium, luminal surface of the oviduct, efferent ducts of the testis, inner ear, and most relevant to pain, the ependymal surface of the brain ventricles (Davy 2004). GeneWeaver reports an association between Hydin and three pain-related phenotypes: acetaminophen interaction, response to morphine, and increased inflammatory response (GS169774, GS127240, GS36729, GS35547). SPELL analysis revealed no predicted co-expressed genes for Hydin, and IPA returned no known functional interactions between Hydin and known pain genes. MouseMap predicted 2,661 genes having direct functional relationships with Hydin in the brain ventricular zone. VLAD analysis of the gene set revealed a high enrichment for GO terms related to reproduction, organismal development, and pattern specification—observations supported by experimental evidence for Hydin’s role in developmental processes (Davy 2004) and the widely acknowledged importance of motile cilia in tissue pattern specification during development (Ibanez-Tallon et al. 2003). VLAD results also revealed a highly significant enrichment for pain-related GO and MP terms, including terms related to pain threshold, behavioral pain response, thermoception, and thermal nociception. Approximately 15 % of the genes in the set had pain-related annotations, a far greater percentage than would be expected by random chance (data not shown). A significant enrichment for the pain-related GO terms “catecholamine uptake involved in synaptic transmission” (GO:0051940; p val = 4.93E−03) and “voltage-gated cation channel activity” (GO:0022843; p val = 1.53E−06) were also observed.
Real-time qPCR analysis of Hydin
In order to test for differences in Hydin transcript levels between A/J and C57BL/6J mice, total RNA was extracted from whole brain tissue and reverse transcribed for real-time qPCR analysis of gene expression. Linear regression analysis identified strain as the only significant model term with C57BL/6J mice exhibiting significantly higher Hydin transcript levels compared to A/J (1.16 ± 0.056 vs. 0.99 ± 0.053, p = 0.0451).
The brains of C57BL/6J mice are, on average, slightly larger than those of A/J mice (Wahlsten et al. 2006), which may account for the observed differences in Hydin transcript levels. In the brain, Hydin is expressed solely in the motile cilia of the CP tissue lining the brain ventricles. CP tissue is difficult to dissect completely and exclusively. We chose to examine RNA extracted from whole brains, as we expect the amount of Hydin transcript from a whole brain sample to be similar to the amount observed in a perfectly dissected CP sample. While this assumption holds true for each individual sample to the best of current biological knowledge, it does not account for the fact that a mouse with a larger brain may have a greater surface area of CP tissue, and thus perhaps a greater number of motile cilia expressing Hydin. Therefore, we expect a lower concentration of Hydin in mice with larger brains. By normalizing RNA concentration to brain size, we account for the possibility that observed Hydin expression differences may be due to greater CP tissue volume alone. In an effort to adjust for CP tissue volume differences between samples, we normalized Hydin expression based on sex- and strain-specific mean brain weight (g) (Wahlsten et al. 2006; Wahlsten and Crabbe 2013). After normalizing to brain weight, we found that no model factors (sex, strain, or their interaction) were returned as significant (sex by strain interaction, p = 0.93; strain, p = 0.97; sex, p = 0.81).
Discussion
We have presented here the first application of DO mice to pain genetics research. Genetic linkage mapping in DO mice produced a much more precise and efficient mapping result than conventional mapping populations. The ability to map a 3.8 Mbp QTL is a marked improvement over conventional two-strain mapping cross studies, which typically result in pain-related confidence intervals of ~30 Mbp. Precise genetic mapping combined with integrative bioinformatic techniques allowed us to identify a single candidate pain gene, Hydin, in a single 6 month study—an endeavor that typically requires multiple mapping and fine-mapping studies over a period of years.
The suite of bioinformatics tools described in this report outlines a systematic strategy for QTL refinement. The time- and labor-intensive nature of functional characterization studies indicates the need for computational assessment of QTL validity prior to experimental follow-up. We evaluated the genomic sufficiency of the Tpnr6 locus by examining the SNP variants and allelic effect patterns present within the interval, allowing us to narrow Tpnr6 to the candidate gene Hydin. We then assessed Hydin’s functional sufficiency by compiling functional annotation data summarizing Hydin’s current relevance to pain response. While none of the tools described here should be used exhaustively or in an exclusionary manner, all were helpful in assessing the pain-related validity of the Tpnr6 locus.
While inbred laboratory strains exhibit sufficient genetic and phenotypic variation for behavioral QTL detection, it has been speculated that historical inbreeding processes selected for ease of handling. As previously reported by Philip et al. (2011), domestication of inbred strains, including the eight DO progenitors, likely operated on multiple loci throughout the genome. The different docility and wildness alleles present at these loci may have influenced the hot-plate endpoint behaviors measured during this study, raising the question of whether DO mice are amenable to behavioral testing. In 2011, Koide et al. (2011) reported that wild-derived inbred lines are amenable to standard behavioral testing. Since mice exhibiting atypical or “inappropriate” behaviors during behavioral testing such as repeated jumping, wall climbing, and alternate paw lifting were excluded from the present study, results from DO mice do not indicate qualitatively different behaviors in mice with wild-derived alleles at the Tpnr6 locus, and are consistent with a study by Chesler et al. (2013) and Philip et al. (2011) that examined hot-plate response in 62 strains of mice, including the three wild-derived DO progenitors. We interpret our results to indicate that the historical inbreeding in commonly used mapping populations has inadvertently reduced the genetic and phenotypic variation available for studies of hot-plate behavior. A clear advantage of DO mice is that their heterozygous nature allows for the segregation of “lost” alleles back into the mapping population, thereby providing a broader quantitative distribution of behavior.
The suggestive QTL peak on chromosome 12 overlaps the previously identified chemical/inflammatory pain QTL Nociq3. Nociq3 is 28 Mbp wide and its underlying candidate gene has not yet been elucidated. Positionally overlaying our suggestive chromosome 12 QTL onto the Nociq3 locus provides a means for narrowing the Nociq3 interval; the overlapping region can be taken to have the greatest probability of containing a pain-related gene, thus reducing the number of positional candidates. Overlaying our chromosome 12 locus onto Nociq3 reduces the number of Nociq3 candidate genes from 492 to 72 (85 %). If a mutual candidate gene was found to underlie both loci, functional characterization of the gene and its related molecular genetic pathways could shed light on nociceptive networks common to both thermal and inflammatory pain. Allelic information for the Nociq3 locus is limited, as the study was conducted using three (C57BL/6 × MOLF/Ei) congenic strains. The genetic diversity afforded by the DO could contribute valuable allelic information to the genetic characterization of a shared candidate gene.
Hydin is a structural protein critical for motility in secondary cilia. Cilia are highly conserved hair-like structures generally comprised of an axoneme consisting of nine outer doublet microtubules. Motile cilia typically possess radial spokes, inner and outer dynein arms on the doublet microtubules, and a central pair of two singlet microtubules. Hydin is an axonemal protein, helping to position the central pair microtubules in motile cilia (Dawe et al. 2007). The synchronous beating of cilia on the ependymal surface of brain ventricles is responsible for the circulation of CSF throughout the central nervous system (CNS) (Breunig et al. 2010; Yamadori and Nara 1979). The CSF and the tissue that secrete it, the CP, have traditionally been thought to provide three primary functions within the CNS: (1) providing the brain with essential nutrients, (2) removing neuronal activity waste products from the CNS, and (3) providing mechanical support for the brain itself (Skipor and Thiery 2008). Recent evidence, however, indicates that the CP–CSF system plays a major functional role in CNS signaling. Numerous studies have demonstrated the importance of the CP–CSF system in neuroendocrine signaling within the brain (see (Skipor and Thiery 2008) for a review), suggesting that the CP–CSF system may also play a role in neuroendocrine mediated processes such as pain sensitivity and response. At the time of this writing, the CP–CSF system has only been loosely associated with pain through its known involvement in inflammatory response (Marques et al. 2009). It is known that heat sensitivity is fundamental to the experience of inflammatory pain, and recent studies have revealed genetic correlations between baseline thermal nociception and the hypersensitivity states associated with inflammatory and neuropathic pain (Mogil et al. 2005b).
The results of our plausibility analysis implicate Hydin in biological processes relating to pain and inflammation, suggesting the relevance of Hydin not only to acute thermal pain but also to inflammatory and central sensitization-mediated chronic pain as well, perhaps through involvement of the CP–CSF system. To this end, we investigated the expression levels of Hydin in the strains of mice reported to contribute pain-sensitive (C57BL/6J) and pain-resistant (A/J) alleles at the Tpnr6 locus using real-time quantitative PCR. After normalizing to brain weight, no Hydin expression differences were observed between the two strains. The murine Hydin gene contains 1100 SNPs: 33 coding-synonymous, 8 coding-nonsynonymous, 1059 intronic (dbSNP Build 128, http://www.ncbi.nlm.nih.gov/projects/SNP/; NCBI Build 37). Only seven Hydin SNPs differ between A/J and C57BL/6J, all of which are located in intronic sequence (rs32704439, rs13479984, rs33400842, rs33170521, rs32685085, rs33110413, rs32952051). There are three SNPs located in Hydin regulatory regions: two proximal upstream SNPs located in the predicted gene Gm17466 (rs50054724 and rs49903084) and one downstream SNP located 1,111 bp downstream of Hydin (rs31253280), but there are no upstream regulatory polymorphisms between A/J and C57BL/6J. The Wellcome Trust Sanger Institute (REL-1211; NCBI build 37) reports four CAST/EiJ-exclusive SNPs present in Hydin splice regions (rs214233349, rs234089050, rs253098678, no rsID), suggesting a Hydin functional difference private to CAST/EiJ. These observations suggest that Hydin may influence pain sensitivity through mechanisms other than functional polymorphisms regulating gene expression, such as differences in mRNA splicing.
The precise mechanism(s) by which Hydin influences hot-plate sensitivity are not known. It is most likely that CSF circulation of endogenous peptides plays a role, thereby establishing pain as a ciliopathic condition. However, indirect mechanisms are also possible; for example, Hydin may influence hot-plate behavior through biological mechanisms related to stress, locomotion, or other behavioral phenotypes. Future studies to perform deeper functional characterization of the role of Hydin and the particular SNPs discussed in this report will be needed to confirm Hydin’s functional role in conditioning pain sensitivity. Through the use of QTL mapping in the Diversity Outcross population we were able to rapidly identify multiple genetic variants that may modify the activity of Hydin and its role in pain sensitivity in an intact mammalian system.
References
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G (2000) Gene Ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25:25–29
Baker EJ, Jay JJ, Bubier JA, Langston MA, Chesler EJ (2012) GeneWeaver: a web-based system for integrative functional genomics. Nucleic Acids Res 40:D1067–D1076
Bannon AW, Malmberg AB (2007) Models of nociception: hot-plate, tail-flick, and formalin tests in rodents. Curr Protoc Neurosci Chapter 8:Unit 8 9
Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Holko M, Yefanov A, Lee H, Zhang N, Robertson CL, Serova N, Davis S, Soboleva A (2013) NCBI GEO: archive for functional genomics data sets—update. Nucleic Acids Res 41:D991–D995
Breunig JJ, Arellano JI, Rakic P (2010) Cilia in the brain: going with the flow. Nat Neurosci 13:654–655
Carbon S, Ireland A, Mungall CJ, Shu S, Marshall B, Lewis S (2009) AmiGO: online access to ontology and annotation data. Bioinformatics 25:288–289
Cheng R (2011) Package ‘QTLRel’
Chesler EJ, Miller DR, Branstetter LR, Galloway LD, Jackson BL, Philip VM, Voy BH, Culiat CT, Threadgill DW, Williams RW, Churchill GA, Johnson DK, Manly KF (2008) The Collaborative Cross at Oak Ridge National Laboratory: developing a powerful resource for systems genetics. Mamm Genome 19:382–389
Chesler EJ, Philip VM, Voy BH (2013) Multi-system assessment of 8 inbred founder strains and 54 F1 hybrids of the Collaborative Cross. MPD:43474
Chou WY, Yang LC, Lu HF, Ko JY, Wang CH, Lin SH, Lee TH, Concejero A, Hsu CJ (2006) Association of mu-opioid receptor gene polymorphism (A118G) with variations in morphine consumption for analgesia after total knee arthroplasty. Acta Anaesthesiol Scand 50:787–792
Churchill GA, Gatti DM, Munger SC, Svenson KL (2012) The Diversity Outbred mouse population. Mamm Genome 23:713–718
Compton P, Geschwind DH, Alarcon M (2003) Association between human mu-opioid receptor gene polymorphism, pain tolerance, and opioid addiction. Am J Med Genet B Neuropsychiatr Genet 121B:76–82
Davy B (2004) Identification and characterization of hydin, a large novel gene disrupted in a murine model of congenital hydrocephalus. In: Graduate Program in Molecular, Cellular, and Developmental Biology. Ohio State University, Columbus, p 172
Dawe HR, Shaw MK, Farr H, Gull K (2007) The hydrocephalus inducing gene product, Hydin, positions axonemal central pair microtubules. BMC Biol 5:33
Devor M, Gilad A, Arbilly M, Yakir B, Raber P, Pisante′ A, Darvasi A (2005) Pain1: a neuropathic pain QTL on mouse chromosome 15 in a C3H×C58 backcross. Pain 116:289–293
Devor M, Gilad A, Arbilly M, Nissenbaum J, Yakir B, Raber P, Minert A, Pisanté A, Darvasi A (2007) Sex-specific variability and a ‘cage effect’ independently mask a neuropathic pain quantitative trait locus detected in a whole genome scan. Eur J Neurosci 26:681–688
Diatchenko L, Slade GD, Nackley AG, Bhalang K, Sigurdsson A, Belfer I, Goldman D, Xu K, Shabalina SA, Shagin D, Max MB, Makarov SS, Maixner W (2005) Genetic basis for individual variations in pain perception and the development of a chronic pain condition. Hum Mol Genet 14:135–143
Diatchenko L, Nackley AG, Slade GD, Bhalang K, Belfer I, Max MB, Goldman D, Maixner W (2006) Catechol-O-methyltransferase gene polymorphisms are associated with multiple pain-evoking stimuli. Pain 125:216–224
Doerr A (2010) A global map of gene function. Nat Methods 7:176
Eddy NB, Leimbach D (1953) Synthetic analgesics. II. Dithienylbutenyl- and dithienylbutylamines. J Pharmacol Exp Ther 107:385–393
Eppig JT, Blake JA, Bult CJ, Kadin JA, Richardson JE (2012) The Mouse Genome Database (MGD): comprehensive resource for genetics and genomics of the laboratory mouse. Nucleic Acids Res 40:D881–D886
Espejo EF, Mir D (1993) Structure of the rat’s behaviour in the hot plate test. Behav Brain Res 56:171–176
Fillingim RB, Kaplan L, Staud R, Ness TJ, Glover TL, Campbell CM, Mogil JS, Wallace MR (2005) The A118G single nucleotide polymorphism of the mu-opioid receptor gene (OPRM1) is associated with pressure pain sensitivity in humans. J Pain 6:159–167
Flint J, Eskin E (2012) Genome-wide association studies in mice. Nat Rev Genet 13:807–817
Furuse T, Miura Y, Yagasaki K, Shiroishi T, Koide T (2003) Identification of QTLs for differential capsaicin sensitivity between mouse strains KJR and C57BL/6. Pain 105:169–175
Goodson M, Rust MB, Witke W, Bannerman D, Mott R, Ponting CP, Flint J (2012) Cofilin-1: a modulator of anxiety in mice. PLoS Genet 8:e1002970
Guan Y, Myers CL, Lu R, Lemischka IR, Bult CJ, Troyanskaya OG (2008) A genomewide functional network for the laboratory mouse. PLoS Comput Biol 4:e1000165
Guan Y, Gorenshteyn D, Burmeister M, Wong AK, Schimenti JC, Handel MA, Bult CJ, Hibbs MA, Troyanskaya OG (2012) Tissue-specific functional networks for prioritizing phenotype and disease genes. PLoS Comput Biol 8:e1002694
Hammond DL (1989) Inference of pain and its modulation from simple behaviors. In: Chapman CR, Loeser JD (eds) Advances in pain research and therapy: issues in pain management. Raven Press, New York, pp 69–91
Hibbs MA, Hess DC, Myers CL, Huttenhower C, Li K, Troyanskaya OG (2007) Exploring the functional landscape of gene expression: directed search of large microarray compendia. Bioinformatics 23:2692–2699
Honda K, Takano Y (2009) New topics in vasopressin receptors and approach to novel drugs: involvement of vasopressin V1a and V1b receptors in nociceptive responses and morphine-induced effects. J Pharmacol Sci 109:38–43
Hunter S, Jones P, Mitchell A, Apweiler R, Attwood TK, Bateman A, Bernard T, Binns D, Bork P, Burge S, de Castro E, Coggill P, Corbett M, Das U, Daugherty L, Duquenne L, Finn RD, Fraser M, Gough J, Haft D, Hulo N, Kahn D, Kelly E, Letunic I, Lonsdale D, Lopez R, Madera M, Maslen J, McAnulla C, McDowall J, McMenamin C, Mi H, Mutowo-Muellenet P, Mulder N, Natale D, Orengo C, Pesseat S, Punta M, Quinn AF, Rivoire C, Sangrador-Vegas A, Selengut JD, Sigrist CJ, Scheremetjew M, Tate J, Thimmajanarthanan M, Thomas PD, Wu CH, Yeats C, Yong SY (2012) InterPro in 2011: new developments in the family and domain prediction database. Nucleic Acids Res 40:D306–D312
Ibanez-Tallon I, Heintz N, Omran H (2003) To beat or not to beat: roles of cilia in development and disease. Hum Mol Genet 12(Spec No. 1):R27–R35
Indo Y, Tsuruta M, Hayashida Y, Karim MA, Ohta K, Kawano T, Mitsubuchi H, Tonoki H, Awaya Y, Matsuda I (1996) Mutations in the TRKA/NGF receptor gene in patients with congenital insensitivity to pain with anhidrosis. Nat Genet 13:485–488
Janicki PK, Schuler G, Francis D, Bohr A, Gordin V, Jarzembowski T, Ruiz-Velasco V, Mets B (2006) A genetic association study of the functional A118G polymorphism of the human mu-opioid receptor gene in patients with acute and chronic pain. Anesth Analg 103:1011–1017
Jaxwest (2013) Multi-system analysis of physiology on 7 inbred strains of mice. MPD:15123
Klepstad P, Rakvag TT, Kaasa S, Holthe M, Dale O, Borchgrevink PC, Baar C, Vikan T, Krokan HE, Skorpen F (2004) The 118 A>G polymorphism in the human mu-opioid receptor gene may increase morphine requirements in patients with pain caused by malignant disease. Acta Anaesthesiol Scand 48:1232–1239
Koide T, Ikeda K, Ogasawara M, Shiroishi T, Moriwaki K, Takahashi A (2011) A new twist on behavioral genetics by incorporating wild-derived mouse strains. Exp Anim 60:347–354
Koide T, Ikeda K, Moriwaki K, Niki H, Shiroishi T (2013) Behavior in females of 11 inbred strains of mice, mostly wild-derived (Mishima) strains. MPD:1809
LaCroix-Fralish ML, Mo G, Smith SB, Sotocinal SG, Ritchie J, Austin JS, Melmed K, Schorscher-Petcu A, Laferriere AC, Lee TH, Romanovsky D, Liao G, Behlke MA, Clark DJ, Peltz G, Seguela P, Dobretsov M, Mogil JS (2009) The beta3 subunit of the Na+, K+-ATPase mediates variable nociceptive sensitivity in the formalin test. Pain 144:294–302
Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, Chen L, Chen TM, Chin MC, Chong J, Crook BE, Czaplinska A, Dang CN, Datta S, Dee NR, Desaki AL, Desta T, Diep E, Dolbeare TA, Donelan MJ, Dong HW, Dougherty JG, Duncan BJ, Ebbert AJ, Eichele G, Estin LK, Faber C, Facer BA, Fields R, Fischer SR, Fliss TP, Frensley C, Gates SN, Glattfelder KJ, Halverson KR, Hart MR, Hohmann JG, Howell MP, Jeung DP, Johnson RA, Karr PT, Kawal R, Kidney JM, Knapik RH, Kuan CL, Lake JH, Laramee AR, Larsen KD, Lau C, Lemon TA, Liang AJ, Liu Y, Luong LT, Michaels J, Morgan JJ, Morgan RJ, Mortrud MT, Mosqueda NF, Ng LL, Ng R, Orta GJ, Overly CC, Pak TH, Parry SE, Pathak SD, Pearson OC, Puchalski RB, Riley ZL, Rockett HR, Rowland SA, Royall JJ, Ruiz MJ, Sarno NR, Schaffnit K, Shapovalova NV, Sivisay T, Slaughterbeck CR, Smith SC, Smith KA, Smith BI, Sodt AJ, Stewart NN, Stumpf KR, Sunkin SM, Sutram M, Tam A, Teemer CD, Thaller C, Thompson CL, Varnam LR, Visel A, Whitlock RM, Wohnoutka PE, Wolkey CK, Wong VY, Wood M, Yaylaoglu MB, Young RC, Youngstrom BL, Yuan XF, Zhang B, Zwingman TA, Jones AR (2007) Genome-wide atlas of gene expression in the adult mouse brain. Nature 445:168–176
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25:402–408
Logan RW, Robledo RF, Recla JM, Philip VM, Bubier JA, Jay JJ, Harwood C, Wilcox T, Gatti DM, Bult CJ, Churchill GA, Chesler EJ (2013) High-precision genetic mapping of behavioral traits in the Diversity Outbred mouse population. Genes Brain Behav 12:424–437
Marques F, Falcao AM, Sousa JC, Coppola G, Geschwind D, Sousa N, Correia-Neves M, Palha JA (2009) Altered iron metabolism is part of the choroid plexus response to peripheral inflammation. Endocrinology 150:2822–2828
Mogil JS, Grisel JE (1998) Transgenic studies of pain. Pain 77:107–128
Mogil JS, Richards SP, O’Toole LA, Helms ML, Mitchell SR, Belknap JK (1997) Genetic sensitivity to hot-plate nociception in DBA/2 J and C57BL/6J inbred mouse strains: possible sex-specific mediation by d2-opioid receptors. Pain 70:267–277
Mogil JS, Wilson SG, Bon K, Lee SE, Chung K, Raber P, Pieper JO, Hain HS, Belknap JK, Hubert L, Elmer GI, Chung JM, Devor M (1999a) Heritability of nociception I: responses of 11 inbred mouse strains on 12 measures of nociception. Pain 80:67–82
Mogil JS, Wilson SG, Bon K, Lee SE, Chung K, Raber P, Pieper JO, Hain HS, Belknap JK, Hubert L, Elmer GI, Chung JM, Devor M (1999b) Heritability of nociception II. ‘Types’ of nociception revealed by genetic correlation analysis. Pain 80:83–93
Mogil JS, Wilson SG, Chesler EJ, Rankin AL, Nemmani KV, Lariviere WR, Groce MK, Wallace MR, Kaplan L, Staud R, Ness TJ, Glover TL, Stankova M, Mayorov A, Hruby VJ, Grisel JE, Fillingim RB (2003) The melanocortin-1 receptor gene mediates female-specific mechanisms of analgesia in mice and humans. Proc Natl Acad Sci USA 100:4867–4872
Mogil JS, Miermeister F, Seifert F, Strasburg K, Zimmermann K, Reinold H, Austin J-S, Bernardini N, Chesler EJ, Hofmann HA, Hordo C, Messlinger K, Nemmani KVS, Rankin AL, Ritchie J, Siegling A, Smith SB, Sotocinal S, Vater A, Lehto SG, Klussmann S, Quirion R, Michaelis M, Devor M, Reeh PW (2005a) Variable sensitivity to noxious heat is mediated by differential expression of the CGRP gene. Proc Natl Acad Sci USA 102:12938–12943
Mogil JS, Ritchie J, Smith SB, Strasburg K, Kaplan L, Wallace MR, Romberg RR, Bijl H, Sarton EY, Fillingim RB, Dahan A (2005b) Melanocortin-1 receptor gene variants affect pain and mu-opioid analgesia in mice and humans. J Med Genet 42:583–587
Mogil JS, Ritchie J, Sotocinal SG, Smith SB, Croteau S, Levitin DJ, Naumova AK (2006) Screening for pain phenotypes: analysis of three congenic mouse strains on a battery of nine nociceptive assays. Pain 126:24–34
Mogil JS, Devor M, Lariviere WR (2013) Investigation of basal nociception in males of 12 inbred strains of mice. MPD:2201
Nackley AG, Shabalina SA, Tchivileva IE, Satterfield K, Korchynskyi O, Makarov SS, Maixner W, Diatchenko L (2006) Human catechol-O-methyltransferase haplotypes modulate protein expression by altering mRNA secondary structure. Science 314:1930–1933
Nair HK, Hain H, Quock RM, Philip VM, Chesler EJ, Belknap JK, Lariviere WR (2011) Genomic loci and candidate genes underlying inflammatory nociception. Pain 152:599–606
Neogen (2010) Mouse Genotyping—Universal Genotyping Array Service. Neogen Corporation, Lansing
Nissenbaum J, Devor M, Seltzer Z, Gebauer M, Michaelis M, Tal M, Dorfman R, Abitbul-Yarkoni M, Lu Y, Elahipanah T, delCanho S, Minert A, Fried K, Persson AK, Shpigler H, Shabo E, Yakir B, Pisante A, Darvasi A (2010) Susceptibility to chronic pain following nerve injury is genetically affected by CACNG2. Genome Res 20:1180–1190
Philip VM, Sokoloff G, Ackert-Bicknell CL, Striz M, Branstetter L, Beckmann MA, Spence JS, Jackson BL, Galloway LD, Barker P, Wymore AM, Hunsicker PR, Durtschi DC, Shaw GS, Shinpock S, Manly KF, Miller DR, Donohue KD, Culiat CT, Churchill GA, Lariviere WR, Palmer AA, O’Hara BF, Voy BH, Chesler EJ (2011) Genetic analysis in the Collaborative Cross breeding population. Genome Res 21:1223–1238
Poulsen L, Brosen K, Arendt-Nielsen L, Gram LF, Elbaek K, Sindrup SH (1996) Codeine and morphine in extensive and poor metabolizers of sparteine: pharmacokinetics, analgesic effect and side effects. Eur J Clin Pharmacol 51:289–295
Rakvag TT, Klepstad P, Baar C, Kvam TM, Dale O, Kaasa S, Krokan HE, Skorpen F (2005) The Val158Met polymorphism of the human catechol-O-methyltransferase (COMT) gene may influence morphine requirements in cancer pain patients. Pain 116:73–78
Reyes-Gibby CC, Shete S, Rakvag T, Bhat SV, Skorpen F, Bruera E, Kaasa S, Klepstad P (2007) Exploring joint effects of genes and the clinical efficacy of morphine for cancer pain: OPRM1 and COMT gene. Pain 130:25–30
Seltzer Z, Wu T, Max MB, Diehl SR (2001) Mapping a gene for neuropathic pain-related behavior following peripheral neurectomy in the mouse. Pain 93:101–106
Sindrup SH, Brosen K, Bjerring P, Arendt-Nielsen L, Larsen U, Angelo HR, Gram LF (1990) Codeine increases pain thresholds to copper vapor laser stimuli in extensive but not poor metabolizers of sparteine. Clin Pharmacol Ther 48:686–693
Skipor J, Thiery JC (2008) The choroid plexus–cerebrospinal fluid system: undervaluated pathway of neuroendocrine signaling into the brain. Acta Neurobiol Exp (Wars) 68:414–428
Smith CL, Eppig JT (2009) The mammalian phenotype ontology: enabling robust annotation and comparative analysis. Wiley Interdiscip Rev Syst Biol Med 1:390–399
Stranger BE, Stahl EA, Raj T (2011) Progress and promise of genome-wide association studies for human complex trait genetics. Genetics 187:367–383
Svenson KL, Gatti DM, Valdar W, Welsh CE, Cheng R, Chesler EJ, Palmer AA, McMillan L, Churchill GA (2012) High-resolution genetic mapping using the Mouse Diversity Outbred population. Genetics 190:437–447
Tsao D, Shabalina SA, Gauthier J, Dokholyan NV, Diatchenko L (2011) Disruptive mRNA folding increases translational efficiency of catechol-O-methyltransferase variant. Nucleic Acids Res 39(14):6201–6212
Wahlsten D, Crabbe JC (2013) Survey of motor activity, behavior, and forebrain morphometry in 21 inbred strains of mice across two laboratories. MPD:10804
Wahlsten D, Bachmanov A, Finn DA, Crabbe JC (2006) Stability of inbred mouse strain differences in behavior and brain size between laboratories and across decades. Proc Natl Acad Sci USA 103:16364–16369
Wilson SG, Mogil JS (2001) Measuring pain in the (knockout) mouse: big challenges in a small mammal. Behav Brain Res 125:65–73
Wilson SG, Chesler EJ, Hain H, Rankin AJ, Schwarz JZ, Call SB, Murray MR, West EE, Teuscher C, Rodriguez-Zas S, Belknap JK, Mogil JS (2002) Identification of quantitative trait loci for chemical/inflammatory nociception in mice. Pain 96:385–391
Woolfe G, MacDonald A (1944) The evaluation of the analgesic action of pethidine hydrochloride (Demerol). J Pharmacol Exp Ther 80:300–307
Yamadori T, Nara K (1979) The directions of ciliary beat on the wall of the lateral ventricle and the currents of the cerebrospinal fluid in the brain ventricles. Scanning Electron Microsc (3):335-340
Zubieta JK, Heitzeg MM, Smith YR, Bueller JA, Xu K, Xu Y, Koeppe RA, Stohler CS, Goldman D (2003) COMT val158met genotype affects mu-opioid neurotransmitter responses to a pain stressor. Science 299:1240–1243
Acknowledgments
This work was supported financially by the National Science Foundation IGERT Training Grant, NIH GM076468, and start-up funding from The Jackson Laboratory to EJC. This project was completed by JMR as part of her graduate dissertation research in the Graduate School of Biomedical Sciences and Engineering (GSBSE) at the University of Maine, Orono. The authors would like to thank Drs. Matthew Hibbs (Trinity University) and Yuanfang Guan (University of Michigan) for their knowledge and contributions to the MouseMap and SPELL analyses, as well as the reviewers of the manuscript for their insights and timely suggestions.
Conflict of interest
The authors declare no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Recla, J.M., Robledo, R.F., Gatti, D.M. et al. Precise genetic mapping and integrative bioinformatics in Diversity Outbred mice reveals Hydin as a novel pain gene. Mamm Genome 25, 211–222 (2014). https://doi.org/10.1007/s00335-014-9508-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00335-014-9508-0