
Abstract
Bacteriophages (phages) are viruses that infect bacteria. The “predator–prey” interactions are recognized as a potentially effective way to treat infections. Phages, as well as phage-derived proteins, especially enzymes, are intensively studied to become future alternative or supportive antibacterials used alone or in combination with standard antibiotic regimens treatment. There are many publications presenting phage therapy aspects, and some papers focused separately on the application of phage-derived enzymes. In this review, we discuss advantages and limitations of both agents concerning their specificity, mode of action, structural issues, resistance development, pharmacokinetics, product preparation, and interactions with the immune system. Finally, we describe the current regulations for phage-based product application.
Similar content being viewed by others

Introduction
Bacteriophages (bacterial viruses) are obligatory parasites propagating in bacterial hosts. The vast majority of discovered phages belong to dsDNA tailed viruses (Caudovirales) and can be distinguished into lytic and temperate phages. Each of these propagation strategies leads to the spread of viral DNA in a different way. Lytic phages are considered as professional host killers, whereas the temperate phages integrate within the host genome, what is often beneficial for the bacterial cell (lysogenic conversion) (Salmond and Fineran 2015). Phages are the most abundant biological particles in the world and playing a significant role in the environment being responsible for (1) dissolved and particulate organic matter circulation via host cell lysis, (2) regulation and biodiversity of populations by reducing the number of dominating bacteria, (3) horizontal gene transfer (HGT) via transduction, or indirectly via transformation of bacterial DNA released during cell lysis, and finally, (4) lysogenic conversion by temperate phages (Wommack and Colwell 2000; Brussaard et al. 2008). Therefore, phages greatly affect microbial diversification as an integral part of each ecological niche including the human body. The tremendous dynamics of the phage–host interactions results in the continuous flow of genetic material, which drives the co-evolution of both entities (Thierauf et al. 2009).
Phage life cycles—crucial differences
There are three types of life cycles in Caudovirales: lytic, lysogenic, and pseudolysogenic (Fig. 1). The typical lytic phage infection consists of six different stages and begins with the adhesion of viral particle to the surface of bacterial cell. Right after adhesion, phage activates various molecular mechanisms leading to the injection of viral DNA into the host cell. The host metabolism is hijacked to amplify viral DNA and to produce phage proteins. Consequently, phage capsids are assembled and packed with genetic material. After the host cell lysis, the phage progeny is released to the environment (Salmond and Fineran 2015).

The possible consequences of phage infection: a bacterial host lysis and release of phage progeny; b lack of virus propagation conditioned by bacterial resistance to phage infection; c lack of host lysis and phage DNA maintenance as an episome (pseudolysogeny, lytic and temperate phages); d lack of host lysis and phage DNA integration into bacterial genome (lysogeny, temperate phages)
Temperate phages can propagate in two different ways, either in the lytic strategy or by simultaneous propagation with the cell host as a prophage (lysogeny). The implementation of lysogenic or lytic cycle is governed by several phage-encoded repressors and regulators (e.g., λ phage CI protein), as well as specific phage enzymes such as integrases and excisionases. If environmental conditions stay favorable to the bacterial host, the repressor maintains the phage in lysogenic state. Under stress conditions, bacterial cells may mobilize the SOS response system (especially RecA co-protease) and inactivate the phage repressor, which triggers the expression of lytic cycle genes (Kim and Ryu 2013). A recent report proved the existence of a molecular phage quorum sensing based on the concentration of “arbitrium” molecule, which informs about the current state of phage population accumulation in a particular niche. At the low extracellular concentration of arbitrium, phages propagate intensively in the lytic cycle, whereas increasing number of arbitrium molecules switch lytic cycle to lysogenic (Erez et al. 2017).
The third type of phage existence is pseudolysogeny when the viral DNA is present within a host cell as an independent episome (plasmid-like form). The host is thus only a phage-carrier and the episome segregates asymmetrically during cell division. Formerly, pseudolysogeny was considered as a temporary suspension of phage developmental cycle preventing the release of phage progeny into environment deprived of the sensitive host cells. It should be emphasized that both lytic and temperate phages may undergo pseudolysogeny event and in some cases episomal genes can be expressed influencing host metabolism (Los and Wegrzyn 2012; Krylov et al. 2012; Latino et al. 2016; Argov et al. 2017).
Phage-based therapy—how did the story begin?
Phages were discovered in 1915 by Frederick William Twort and the term bacteriophages was coined by Felix d’Herelle, who in 1917 independently confirmed Twort’s discovery (Kutter et al. 2010). Phages were immediately recognized as potential antibacterials and used for the treatment of bacterial infections during the 1920s and 1930s. However, phage therapy was abandoned in favor of antibiotics exhibiting a broad activity against bacteria, and being easy to prepare, store, and distribute (Kutter et al. 2010). The benefits of antibiotics and chemotherapeutics were substantially lost in subsequent years following the emergence and dissemination of bacterial drug resistance. Today, multidrug-resistant (MDR) bacterial strains are a serious problem both in hospitals and community settings. Most frequent and especially difficult-to-treat MDR bacteria belong to the so-called “ESKAPE” group and include Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter spp. The pharmaceutical pipeline of antibiotics active against ESKAPE is extremely limited. This group spans methicillin-resistant S. aureus (MRSA), vancomycin-resistant enterococci (VRE), as well as carbapenemase (MBL, KPC, OXA-48) and extended spectrum beta-lactamase (ESBL) producers (http://www.eucast.org/resistance_mechanisms/). The emergence of infections caused by MDR pathogens generated a critical need to find alternatives to classical antibiotics (Barrow and Soothill 1997; Alisky et al. 1998; Carlton 1999; Sulakvelidze et al. 2001). For this reason, the phage therapy gets revitalized.
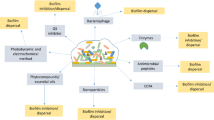
Considerable interest arose on phage-encoded proteins with antibacterial potential (Fig. 2). These include viral enzymes such as endolysins, virion-associated lysins (VALs), and polysaccharide depolymerases. Endolysins are the lytic enzymes used by phages at the end of the replication cycle to degrade bacterial peptidoglycan (PG) from within, resulting in a rapid host lysis and the release of phage progeny. VALs and depolymerases are linked to the virion particle and serve at the beginning of infection to overcome bacterial cell surface barriers. VALs are responsible for PG degradation required for phage genetic material injection to the infected host cell, whereas depolymerases degrade polysaccharide molecules such as capsule, lipopolysaccharide (LPS), or biofilm matrix (Nelson et al. 2012; Schmelcher et al. 2012a; Rodríguez-Rubio et al. 2013; Latka et al. 2017).

The main characteristics of phage-based products application. The application of lytic phage preparation may result in both bacterial host lysis (the effect observed even in minutes) or lack of lysis (transition into a pseudolysogeny state). The application of lysin-based product leads to the lysis of targeted host (the effect observed even in seconds). The application of depolymerase-based product leads to degradation of capsule (CPS), exopolysaccharides (EPS), or lipopolysaccharides (LPS) decreasing bacterial virulence followed by enhancement of immune system clearance (the effect observed even in minutes)
Phage therapy—issues to consider
Specificity and host range
Unlike wide spectrum antibiotics, phage therapy is characterized by selectivity (Table 1). The specificity of phages results from their relatively narrow host range limited usually to one bacterial species. The number of bacterial strains infected by particular phage varies depending on the type of surface receptor recognized and antiviral defense mechanisms by the targeted host. Wide host range phages that propagate on a large number of strains are generally more useful for therapy (Sulakvelidze et al. 2001).
Mode of action
One of the basic principles to select phages for therapy is excluding temperate phages because the bactericidal effect is only guaranteed for lytic phages. The most important factor ensuring the effectiveness of the treatment is the self-replicating nature of phages, which distinguishes them from conventional antibiotics. In addition to high burst size and propagation rate, the phage titer and MOI (multiplicity of infection—the number of phage particles per one bacterial cell) are critical factors. Since phages propagate only on actively growing host cells, the high MOI prevents the loss of antibacterial potential associated with phage adhesion to dormant and dead cells or cellular debris (Abedon 2016a).
Biofilm eradication
The most common cause of failure of antibiotic therapy in chronic infections is the ability of the bacteria to produce biofilms. Due to the impermeability of biofilm matrix and the clonal diversity of bacterial cells within this structure, the application of standard antibiotics usually fails. The activity of phage preparations against biofilm-forming bacteria is relatively high. Some phages are naturally equipped with virion-associated depolymerases that degrade the biofilm matrix (Lu and Collins 2007; Abedon 2015). Phages can also infect metabolically dormant bacteria if the surface receptor is still present. In this case, the lytic cycle stays suspended until bacteria switch from persistence to active growth (Pearl et al. 2008). However, the mature biofilm is a complex structure and its complete eradication by one phage is rather unlikely. Biofilm elimination takes time and requires the application of multi-phage cocktails or antibiotic supplementation (Abedon 2016b; Chaudhry et al. 2017). The ability of phages to biofilm degradation results from the existence of selective pressure in particular area, where depolymerase degrades matrix exopolysaccharides enabling the phage or other antimicrobials (combined therapy) to reach the bacterial cell (Abedon 2016b).
Development of resistance
Bacterial resistance to phage infection was documented by Felix d’Herelle at the very beginning of phage therapy. The interactions between phages and their hosts are commonly described as a parasite–host or predator–prey and both are subjected to the evolutionary mechanisms outlined in the “Red Queen” hypothesis (van Valen 1973). The bacterial resistance to phages can arise in several ways. The most common form is receptor modification due to point mutations of receptor-encoding genes or changes in their expression, which ultimately prevent phage adsorption. Discussing this common phage-resistance mechanism (loss/modification of phage receptor), it must be stressed out that most phages target bacterial surface molecules, especially those of carbohydrate nature. Surface glycans and glycoconjugates such as capsules and LPS serve as molecular patterns for recognition by the innate immune system, and also provide shields to antibiotic entry and host defense mechanisms (e.g., complement system and phagocytosis). Loss or alteration of these molecules could result in bacteria more susceptible to host clearance mechanisms by the immune system. Another resistance mechanism to phage infection is superinfection exclusion systems encoded by other prophages already present in the target bacterial cell, which protect bacteria against infection by other closely related phages. Bacteria may also activate restriction-modification systems, which are responsible for destruction of invading foreign DNA. The more complex mechanism of resistance, operating at the DNA/RNA level, is CRISPR/Cas system, also called the acquired immunity of bacteria (Labrie et al. 2010). Another multi-threaded mechanism protecting bacteria against lytic and temperate phage infection is the bacteriophage exclusion system (BREX), which inhibits foreign DNA replication (Goldfarb et al. 2015). The last resort for resistance mechanism, which operates in the context of the entire population, is the abortive infection system. This mechanism leads to the death of the invaded host cell, preventing phage multiplication and further infection of susceptible population (Labrie et al. 2010). The appearance of phage-resistant bacterial clones cannot be avoided since it is a natural mechanism of bacteria-virus co-evolution, which also occurs in phage therapy. To enhance the abundance reduction of pathogenic strain in treated patient, the polyvalent phage cocktails are composed (Ormälä and Jalasvuori 2013). Nevertheless, recent in vivo studies show that the emergence of phage-resistant mutants does not affect the effectiveness of therapy in immunocompetent patients, where both phage-sensitive and phage-resistant population were cleared out by innate immune mechanisms, especially neutrophils (Bull et al. 2002; Roach et al. 2017).
Product modification
Phage preparations used for a specific infection (e.g., wound infection) or against a particular bacterial group (e.g., anti-staphylococci) are usually composed of a multi-phage cocktail, which may be further modified by adding more phages to an existing cocktail or replacing one phage with another. These improvements can be made by selecting phage from an existing collection or by isolating new phage from the environment (Goodridge 2010; Chan and Abedon 2012). Although the molecular engineering tools are currently available to create genetically modified phages, their use is not permitted for human therapy by the Food and Drug Administration or the European Medicines Agency. Nevertheless, there is interest in creating genetically modified temperate phages or lytic phages equipped with specific dedicated biofilm-degrading enzymes, increasing the effectiveness of treatment for chronic, biofilm-related infections (Lu and Collins 2007; Edgar et al. 2012).
Influence of phages on normal flora
The narrow host range of phages ensures that phage therapy plays does not adversely affect the natural microbiota. Phage cocktails specific to different bacterial strains or species usually do not contain phages capable of infecting saprophytic bacteria. Nevertheless, the microbiota of each person differs and some people may be the carriers of potentially dangerous species (e.g., multidrug-resistant ESKAPE representatives). In this unique situation, the therapy directed against those potentially pathogenic bacteria may cause imbalance in the microbiota (Loc-Carrillo and Abedon 2011).
Impact on immune system
The success of phage therapy largely depends on the patient’s immune system. The interactions between phages and the immune system should be considered in various ways. First, the immune system may recognize and inactivate viral particles (Górski et al. 2012). In vertebrates, the effectiveness of phage clearance depends on the structure of viral capsid (Merril et al. 1996). Even minor changes in phage coat protein composition can affect their bloodstream circulation time and immunogenicity. Fast clearance of phage particles is carried out by the reticuloendothelial system, especially in the liver and spleen. Kupffer cells (macrophages located in the liver) engulf phages four times more efficiently than splenic macrophages. This phenomenon is probably related to the different function of these organs. Kupffer cells are meant to purify the blood of the most serious microorganisms, immune complexes, and cellular debris, whereas splenic macrophages are more involved in stimulating lymphocytes to antibody production (Dabrowska et al. 2005; Górski et al. 2012). Low level of anti-phage antibodies can naturally occur in patients, but their titer against particular phages may increase during phage therapy. Interestingly, the vertebrate immune system does not trigger a specific cellular response against bacteriophages (T cells do not participate in phage elimination) (Górski et al. 2012; Cisek et al. 2017). In addition, the activation of immune responses associated with phage proteins may also exhibit immunomodulating properties. Phages affect phagocytosis and the development of an inflammatory response, but depending on the phage preparation (species, dose, purity, and route of administration) they can either intensify or inhibit these processes (Górski et al. 2012).
Safety
The onset of phage therapy dates back to the early twentieth century. Due to the ease of administration and no side effects, phages were used as oral and topical preparations. Despite the primitive methods of purification, the first attempts at intravenous administration of phage preparations began in the mid-1920s (Smith 1924; D’Herelle 1931). The first clinical trial of intravenous therapy was effective despite the occasional adverse effects of a “specific therapeutic shock” (Hugh Young reaction). The elimination of peptone and other animal protein components from the propagation medium reduced the negative effects of phage injections. The introduction of routine phage purification in cesium chloride density gradient, ammonium sulfate precipitation, and filtration on anion-exchange diethylaminoethyl cellulose columns (DEAE) eliminated or significantly diminished potential hazards (Abedon et al. 2011; Speck and Smithyman 2016). Although phage therapy is generally considered safe, its use in immunocompromised patients may be riskier and less effective (Speck and Smithyman 2016; Roach et al. 2017). Another aspect of safety issues is the probability of HGT carried out by phages. Because phages multiply at the site of infection, there is always the risk of some form of HGT that might affect/increase the virulence of co-existing bacterial population or introduction of new antibiotic resistance genes into the population (Lin et al. 2017). The safety of phage therapy in the context of rapid release of bacterial toxins (especially LPS) might be also considered. During phage therapy of Gram-negative bacterial infections, especially using a high dose of phages, a simultaneous lysis of high numbers of bacteria may release endotoxins in such quantities that they might cause endotoxic shock. However, a similar outcome could be also be possible during bactericidal antibiotic (e.g., β-lactams) treatment (van Langevelde et al. 1998).
Product preparation
Each step of phage preparation must be strictly controlled to ensure safety. From the very beginning, at the stage of host selection, special attention must be paid to the exclusion of bacterial strains carrying phage-related entities (prophages, satellite phages, episomes containing viral DNA). This will prevent the contamination at the initial stage of production and will reduce the risk of HGT (Abedon 2017). While the multiplication of phages is not complicated, the lysate purification could be troublesome. For the safety issues, the lysate should be purified from toxic products of bacterial metabolism and any cell debris, especially endotoxins (LPS). The multi-stage purification procedure involves centrifugation, filtration (0.22 μm pore filters), organic solvents treatment (chloroform, n-butanol, 1-octanol), condensation (polyethylene glycol precipitation, ultracentrifugation in cesium chloride gradient), and dialysis (Bonilla et al. 2016). Due to the wide variety of phage particles, there is no universal protocol for their purification. Phage capsule morphology differences often affect the stability of preparations and the sensitivity of phages to various chemical and physical factors (Alper 1954). Some phage particles are rapidly inactivated by chloroform, others can be damaged during ultracentrifugation or dry freezing processes, and others have a very short shelf-life. On the other hand, there are also phages resistant to high salt concentrations, extreme temperatures or pH values, and long-lasting drying (Jończyk et al. 2011). All above factors make it difficult to obtain a pure phage preparation and to maintain viral particles infective.
Formulations and delivery route
As phages are easily propagated in bacteria cultivated in liquid media, these formulations are the most popular form of phage preparations. Moreover, liquid formulations prevent phages from drying and inactivation. Liquids can be administered by the oral, intravenous, or topical route. Phages can be used to prepare aerosols (inhalants), creams/ointments (for topical applications), moist dressings and tampons, and even powders and tablets (Abedon et al. 2011; Weber-Dabrowska et al. 2016). Phage preparations can also be administered intravenously, intramuscularly, vaginally, rectally, or by inhalation. Above methods allow for treatment of many types of infection including gastrointestinal, respiratory, urinary, and even sepsis. Phages easily penetrate from the intestine to the blood and urinary tract, but their delivery to peripheral tissues is usually not sufficient. The transfer of phages through the blood–brain barrier is sometimes problematic. The blood–brain barrier limits passive diffusion between the blood and the brain compartments even for large proteins (> 400 kDa), making it permeable for phages only in the case of blood–brain barrier dysfunction or inflammatory conditions (Weiss et al. 2009). For localized infections such as sinusitis, pharyngitis, or skin infections, the best efficacy is obtained by topical application of aerosols/suspensions or creams/ointments (Letkiewicz et al. 2010; Abedon et al. 2011; Weber-Dabrowska et al. 2016).
Pharmacokinetics
The pharmacokinetics of phage preparations depends on many factors. Important aspects are the size of the phages and the structure/composition of their capsids. Phage capsular proteins can interact in various ways with enterocytes and immune cells (especially dendritic cells). The number of phage particles that enter the body fluids depends on the initial phage titer, its resistance to gastrointestinal conditions (pH, digestive enzymes), and the rate of penetration trough the intestinal epithelium. In most of the cases, phages easily cross the barrier of gut epithelium and reach the bloodstream, but their persistence in the circulation varies depending on the efficacy of reticuloendothelial system clearance. In addition, phage particles are also removed by secretions, which in some way facilitates the possible treatment of urinary tract infections. Since the vertebrate immune system may produce Ab specific for phages, this may additionally cause phage inactivation and its elimination. An indirect solution for this problem is the application of phage cocktails composed of relatively distantly related phages, which prevent the cross-reactivity of emerging Ab (Dabrowska et al. 2005; Skurnik and Strauch 2006; Gorski et al. 2006).
Combined therapy
At the very beginning of phage therapy age, Felix d’Herelle noted that the effectiveness of single phage therapy rapidly decreased. To maintain a high bactericidal efficacy, it is necessary to use polyvalent phage preparations, composed of phages recognizing several different bacterial receptors. This reduces the risk of therapy failure due to inactivation of phages by the immune system as well as the emergence of phage-resistant strains (Chan et al. 2013). The combination of phages with antibiotics can also have positive effects. Synergism is especially seen in infections caused by biofilm-producing bacteria (Chaudhry et al. 2017).
Therapy using phage enzymes—issues to consider (Table 1)
Specificity and host range
Specificity and host range of PG degrading lysins (endolysins and VALs) vary and depend on protein characteristics as well as on phage species/genus from which the protein is derived (Paul et al. 2011; Rodríguez-Rubio et al. 2013; Latka et al. 2017). Lysins encoded by Gram-positive-specific phages have evolved along its target which is characterized by a strong variation in the peptide composition, crosslinks, and modification of glycan chain (Schleifer and Kandler 1972). Therefore, the activity of those enzymes is limited to certain bacterial species or even serotype (Table 1). This narrow specificity allows for selective killing of a given target pathogen, saving accompanying microflora and reducing the risk of resistance development (Borysowski et al. 2006). In contrast, PG of Gram-negatives has a highly conservative structure with significant similarities shared among different species. Therefore, endolysins and VALs are usually active against a wide host range (Briers et al. 2007; Latka et al. 2017). The third type of antibacterial enzymes (depolymerases) shows high substrate specificity as bacteria can produce a huge diversity of glycans such as capsule (CPS, K-serotype), O-polysaccharide chains (LPS, O-serotype), or extracellular polysaccharides (EPS). Therefore, glycan-degrading phage depolymerases can be useful even for targeting or detecting particular bacterial serotype (Latka et al. 2017).
Mode of action
Phage-encoded lysins fall into two major classes according to their mechanism of action: (1) hydrolases degrading PG bonds via hydrolysis and (2) lytic transglycosylases, cleaving glycoside bonds in glycan chain forming 1,6-anhydro ring at the N-acetylmuramic acid residue (Höltje et al. 1975). Depending on the type of chemical bond that is hydrolyzed in PG, we distinguish (1) amidases hydrolyzing amide bond, (2) endopeptidases cleaving bonds within peptide chains, and (3) glucosaminidases and lysozymes (muramidases) both hydrolyzing glycoside bonds in the glycan chain (Nelson et al. 2012). The effect of the degrading activity of lysins can be manifested in seconds as osmotic lysis of the targeted cell (Fig. 2). Numerous in vivo trials have been conducted proving lysin’s high effectiveness against Gram-positive pathogens, including Streptococcus pneumoniae, MRSA, or Bacillus anthracis (Table 2). In contrast, those enzymes applied exogenously have limited effect on Gram-negatives because of the outer membrane layer. To date, only a few endolysins (e.g., SPN9CC, PlyF307, and CfP1gp153) were shown to cross the outer membrane and degrade Gram-negative PG when used as external agents (Lim et al. 2014; Lood et al. 2015; Oliveira et al. 2016). Although phage lysins differ vastly in their lytic activities, ranging from 100 to 108 U/mg, they are still recognized as the strongest PG hydrolyzers. Nanogram amounts of PlyC endolysin derived from streptococcal C1 phage clear a bacterial culture within seconds, being several orders more active than any other described PG hydrolase of non-phage origin (Schmelcher et al. 2012a).
Phage depolymerases are responsible for degrading carbohydrate macromolecules in the bacterial cell envelope. Depolymerases are divided according to their mode of action into (1) hydrolases and (2) lyases cleaving a glycosidic bond by trans-β-elimination. The hydrolases comprise sialidases (hydrolyzing internal α-2,8-linkages in capsular polysialic acid), rhamnosidases (cleaving α-1,3 O-glycosidic bonds between l-rhamnose and d-galactose in the O-antigen of Salmonella LPS), levanases (hydrolyzing β-2,6-bonds between fructose monomers in levan), xylanases (cutting β-1,4 bonds within xylan), dextranases (cleaving α-1,6-linkages between glucose units in dextran), and LPS deacetylases which deacetylate the O-antigen rather than breaking the polysaccharide chain (Prokhorov et al. 2017; Latka et al. 2017). The lyases include hyaluronate lyase (cleaving β-1,4 bonds in hyaluronic acid), pectate lyase (cleaving α-1,4 bonds of polygalacturonic acid), alginate lyase (cutting α-1,4 bonds of alginate), and K5 lyase (cleaving α-1,4 bonds of E. coli K5 capsules). Depolymerases as antimicrobials can be successfully implemented as external agents to degrade bacterial capsules, LPS, and exopolysaccharides, acting indeed as anti-virulent agents and sensitizing bacteria to antimicrobials, the immune system, and desiccation (Pires et al. 2016; Latka et al. 2017). Like for endolysins, the therapeutic efficacy of recombinant depolymerases was confirmed in animal models (Table 2).
Biofilm eradication
Phage depolymerases have evolved as a response against the thick polysaccharide layer covering bacterial cell and hiding phage receptor required for successful attachment to the host. One of such layer is biofilm matrix composed mostly of exopolysaccharides. Moreover, LPS-degrading enzymes are also able to loosen biofilm structure as LPS-containing outer membrane vesicles are embedded within the matrix (Olszak et al. 2017). There are many reports confirming the efficacy of depolymerases in eradication of biofilms formed by both Gram-positive and Gram-negative bacteria (Mushtaq et al. 2004, 2005; Cornelissen et al. 2011; Gutiérrez et al. 2012, 2015); Bansal et al. 2014; Pan et al. 2015). Besides depolymerases, also phage lysins have been successfully used in the removal of bacterial biofilms. Most of the studies were dedicated to S. aureus (Sass and Bierbaum 2007; Son et al. 2010; Fenton et al. 2013; Schmelcher et al. 2014; Singh et al. 2014; Gutiérrez et al. 2014; Yang et al. 2015) and streptococcal biofilm treatment (Domenech et al. 2011; Meng et al. 2011; Shen et al. 2013; Rico-Lastres et al. 2015). Concerning the biofilms of Gram-negative bacteria, endolysins Lys68 (Oliveira et al. 2014), LysPA26 (Guo et al. 2017) and PlyF307 (Lood et al. 2015) were proved to be effective against Salmonella Typhimurium, P. aeruginosa, and A. baumannii, respectively.
Development of resistance
Phage lysins are in a certain sense unique relative to whole phages and antibiotics since resistance is an extremely rare event. This is due to lysin’s ability to bind and cleave highly conserved targets within the cell wall (Fischetti 2010). Moreover, high specificity of most endolysins reduces the probability of developing bacterial resistance (Fischetti 2005). Nevertheless, secondary modifications of bacterial cell walls, such as O-acetylation and N-deacetylation in PG or d-alanylation in teichoic acids, can be considered as potential resistance mechanisms against phage lysins, in analogy to what was reported for human lysozyme (Vollmer et al. 2008; Guariglia-Oropeza and Helmann 2011). There are some studies addressing the repeated exposure to low concentrations of the enzyme, which proved no resistance phenotypes to native or engineered phage lysins (Loeffler et al. 2001; Schuch et al. 2002; Fischetti 2005; Pastagia et al. 2011; Schmelcher et al. 2012a; Gilmer et al. 2013). Resistance against phage depolymerases develops quite often due to modifications or variations in polysaccharide composition of capsule, exopolysaccharides, or LPS. The application of depolymerase resulted in the rapid emergence of E. coli O9:K30 and Klebsiella O1:K20 resistant mutants (McCallum et al. 1989).
Product modification
Current synthetic biology techniques can be used to improve the efficacy of phage lysins. Random mutagenesis within enzymatic domain (EAD) or the exchange of cell wall binding domain (CBD) can increase lytic activity. The spectrum of enzymes was experimentally extended by (1) the fusion of two full-length endolysins, (2) the addition of a heterologous EAD to a full-length enzyme, (3) the addition of a heterologous CBD to a truncated endolysin, (4) the duplication of CBD, or (5) the combination of two heterologous CBDs (Cheng and Fischetti 2007; Schmelcher et al. 2012a). Recent studies propose the application of genetically modified endolysins (Artilysins®) or enzymes combined with membrane permeabilizers to efficiently destroy PG in Gram-negatives (Briers et al. 2011, 2014; Oliveira et al. 2014; Yang et al. 2015). These modifications involve the attachment of short (6–100 aa) membrane-penetrating or membrane-destabilizing peptides usually of polycationic, hydrophobic, or amphipathic nature causing membrane disruption or pore formation (Briers et al. 2014; Peng et al. 2017). Numerous endolysins (Ply511, PlyA, CD27L, OBPgpLYS) were recently modified this way and patented as Artilysins® (Briers et al. 2014; Schirmeier et al. 2017). In addition to structure-based modification, outer membrane destabilizing agents such as EDTA, weak organic acids (citric acid), and polycationic agents could be mixed with lysin preparation to enhance antibacterial activity against Gram-negatives (Oliveira et al. 2014). There is not much data on the modification of phage depolymerases. One of the main reasons for this is the relatively big size of these enzymes (usually ~ 1000 aa) forming a complicated spatial structure of trimers or sometimes tetramers. Depolymerases being an integral part of phage particle are still not well-studied proteins concerning enzymatic activity and specificity (Latka et al. 2017).
Influence on normal flora
A high specificity of lysins targeted to Gram-positives allows for the selective killing of given pathogens with little to no effect on normal human microbiota. Nevertheless, in some cases, phage enzymes may show a broad spectrum as recently reported for enterococcal phage lysin active against enterococci, S. aureus, Streptococcus pyogenes, and S. agalactiae (Yoong et al. 2004). Another example is streptococcal lysin PlySs2, able to eradicate staphylococci, several species of Streptococcus (S. agalactiae, S. pyogenes, and S. pneumoniae), and Listeria sp. as well (Gilmer et al. 2013). Lysins derived from Gram-negatives infecting phages show theoretically a broad spectrum when combined with permeabilizing agents. In this regard, such preparation might influence the accompanying microflora with the same efficiency as for the targeted pathogen.
Effect on the immune system
Due to their proteinaceous nature, phage enzymes stimulate a rapid immune response and generation of neutralizing antibodies (Fischetti 2010). Antibodies against Cpl-1, Pal, MV-L, ClyS, and SAL-1 endolysins were confirmed in several animal studies (Table 2) (Jado et al. 2003; Loeffler et al. 2003; Rashel et al. 2007; Daniel et al. 2010; Jun et al. 2014). The first clinical trial on SAL200 preparation (endolysin SAL-1) also revealed anti-endolysin antibodies in collected serum samples (Jun et al. 2017). Although antibodies were poorly effective in lysin inactivation, their presence sufficiently reduced the systemic half-life of enzymes to approximately 20 min (Loeffler et al. 2003). In vitro and in vivo studies on different endolysins and pathogens confirmed that antibodies slow down the antimicrobial efficacy of lysins but do not abolish their activity completely (Loeffler et al. 2003; Fischetti 2005; Rashel et al. 2007; Jun et al. 2014). The modification of lysins to extend their half-life is possible. Attempted dimerization of Cpl-1 endolysin through the introduction of C-terminal cysteine residues and subsequent formation of disulfide bonds resulted in a twofold increase of anti-pneumococcal activity and tenfold reduction of plasma clearance (Resch et al. 2011). Interestingly, a recently described chimeric endolysin ClyS turned out to be completely insensitive to generated antibodies (Daniel et al. 2010; Pastagia et al. 2011).
Safety
Phages are an integral part of the natural human microbiota and the constant release of lysins and depolymerases has no adverse effects on human health (Navarro and Muniesa 2017). For this reason, phage enzymes are considered to have a good safety profile, which was confirmed in many trials using animal models (Table 2). The clinical trials on intravenous administration of SAL200, conducted accordingly to good laboratory practice, demonstrated good tolerance in healthy male volunteers (Jun et al. 2017). Phage enzymes, like other proteins, can theoretically induce an allergic response or some adverse side effects, but these have not been reported in animal models (Jado et al. 2003; Gu et al. 2011a; Gupta and Prasad 2011a; Jun et al. 2014; Pan et al. 2015). Both lysins and depolymerases are specific for unique and highly conserved bacterial structures (polysaccharides or PG) that are absent in mammalian cells, and therefore are non-toxic agents. The good safety profile also includes relatively fast biodegradability (Nelson et al. 2012). The side effects of lysin applications are similar to those of lytic phages and bactericidal drugs, and are associated to the release of endotoxin, as well as bacterial cellular contents and debris during rapid cell lysis, especially in the case of massive infections. This release may induce strong immune responses leading to endothelial and tissue damage, and severe hemodynamic and metabolic derangements, namely, toxic shock (Prins et al. 1994; Nau and Eiffert 2002; Ramachandran 2014). The in vivo administration of phage depolymerases (endosialidase, endorhamnosidase, lyase) against E. coli, S. Typhimurium, or K. pneumoniae was effective in killing bacteria and safe (Table 2). There is a lack of controlled clinical trials dealing with the systemic application of phage-based enzymes in the treatment of infected patients. Detailed evaluation including clinical trials of multiple increasing doses and assessing the effects of therapy on vital functions, such as in the respiratory, central nervous, and cardiovascular systems, are necessary to improve our understanding of the safety profile of phage enzymes.
Product preparation
Preparation of lysin/depolymerase formulations should not pose any major problems. Methodologies and strategies for recombinant protein production and purification are systematically improved, and well-established conditions allow for rapid preparation of ultrapure protein in a large scale (Wingfield 2015). The phage-based products characterized so far possess the desired formulation parameters; they remain stable at fridge storage temperature (4 °C) for weeks or even months, and can be kept frozen or lyophilized (Cheng and Fischetti 2007; Pastagia et al. 2011; Gilmer et al. 2013; Jun et al. 2013). Protein stability can be further increased by the selection of optimal conditions (protein concentration, buffer, pH, temperature, additional stabilizers) (Jun et al. 2013) or by molecular engineering (Heselpoth et al. 2015). The vast majority of currently described phage-enzymes was relatively stable at wide pH range (Yoong et al. 2006; Maciejewska et al. 2017; Olszak et al. 2017), suggesting that they might remain functional even after oral administration. Several phage-based products showed to be highly thermostable (above 80 °C), a property that extends their application to industry as for instance food preservatives (Matsushita and Yanase 2008; Plotka et al. 2014; Oliveira et al. 2014; Rodríguez-Rubio et al. 2016; Majkowska-Skrobek et al. 2016; Maciejewska et al. 2017; Olszak et al. 2017).
Formulations and delivery route
Enzyme-based formulations applied to date are prepared as injections, aerosols for inhalations, and formulas for local application (liquids, ointments, and gels) (Table 2). Numerous commercially available formulations (emollients ointments, petrolatum for topical application, surfactants, or injection buffers like Dulbecco’s phosphate-buffered saline) were applied for phage-based products preparation (Pastagia et al. 2011; Jun et al. 2013). Like phages, the enzyme-based products must be preceded by thorough and multistep protein purification with the removal of bacterial endotoxins. The reduction of endotoxins to a maximum level of 5 U/kg of body weight per hour for intravenous applications is a challenging procedure but crucial for safe therapy (Pan et al. 2015). The aerosolized and topical enzyme delivery ensures a drug direct accumulation at the site of infection with relatively low systemic exposure (Ryan et al. 2011). To date, the in vivo tests covered the following delivery routes of phage enzymes: injections (intravenous, intraperitoneal, and intravitreal), trans-nasal, and vaginal administration, oral delivery, inhalations, topical application, and injection directly under the skin (Table 2). Each of the listed routes provided effective treatment. The enteral delivery of phage proteins poses a challenge to maintain enzyme activity at low pH and in the presence of proteolytic enzymes in the stomach. There is one example of successful oral application of P22sTsp depolymerase insusceptible to trypsin and partially to chymotrypsin activity (Waseh et al. 2010). To avoid this obstacle, phage enzymes could be encapsulated in polymeric nanoparticles and thus protected from the harsh gastric environment (Chan et al. 2010). An inventive strategy proposed to preserve lysins in the gastrointestinal tract involved the administration of engineered lactic acid bacteria excreting the endolysin while colonizing intestines (Mayer et al. 2008; Gervasi et al. 2014).
Pharmacokinetics
A successful treatment depends on well-characterized pharmacokinetic/pharmacodynamic properties of the individual medical product. Despite a wealth of knowledge about antibacterial potential and biochemical parameters of phage enzymes, little is known about their capacity to penetrate mammalian tissues and cells, which influences their effective concentration and dose, timing of administration, or optimal treatment duration. Currently, we can only rely on assumptions and scarce reports in animal models (Table 2). Due to the much smaller size comparing to phages itself, phage enzymes should penetrate more efficiently to human tissues. Indeed, the PlyC endolysin can cross the epithelial cell membrane to reach and lyse intracellular S. pyogenes (Shen et al. 2016). The effective intravenous, intraperitoneal, or oral administration in animal bacteremia indicated rapid distribution of lysins and depolymerases within the body and a good penetration to adjoining tissues (Table 2). In the clinical trial of SAL200 preparation, the doses provided a maximum concentration of 10 mg/kg of body weight (Jun et al. 2017). The majority of reports described the effectiveness of a single dose of recombinant phage enzyme for infection eradication (Nelson et al. 2001; Jado et al. 2003; Cheng et al. 2005; Mushtaq et al. 2005; Grandgirard et al. 2008; Daniel et al. 2010; Gu et al. 2011a; Doehn et al. 2013; Lood et al. 2015; Majkowska-Skrobek et al. 2016). Nevertheless, the multiple lysin doses increased the systematic drug concentration followed by a significant improvement in animal survival rate (Oechslin et al. 2013; Jun et al. 2016).
Combined therapy
Antimicrobial synergy was demonstrated for several lysins and depolymerases in combination with other PG hydrolases, as well as with numerous classes of antimicrobials (antimicrobial peptides, antibiotics). The in vivo synergy of glycopeptides and β-lactams with MV-L (Rashel et al. 2007) and ClyS (Daniel et al. 2010) endolysins was confirmed in the treatment of systemic MRSA infections. Chimeric endolysin λSA2-E-LysK-SH3b acts synergistically with lysostaphin in a mice model (Schmelcher et al. 2012b), similar to the combination of Cpl-1 with Pal endolysins (Jado et al. 2003).
Summary: applications of bacteriophages versus phage enzymes to combat and cure bacterial infections: an ambitious and also a realistic application?
The rapid development of phage therapy that took place in the 1920s and 1930s significantly slowed down by the invention of antibiotics. Discovery of penicillin led to almost complete abandonment of phage therapy in the Western countries. However, several research centers (in Georgia, Russia, and Poland) continued research on bacteriophages and their cumulative experience is crucial in the present era of rapid antibiotic resistance development. Currently, the use of phages and phage-borne enzymes in the EU and USA is considered as experimental therapy, which can only be applied under the umbrella of the Article 37 of Helsinki Declaration (World Medical Association 2013; Debarbieux et al. 2016).
“In the treatment of an individual patient, where proven interventions do not exist or other known interventions have been ineffective, the physician, after seeking expert advice, with informed consent from the patient or a legally authorised representative, may use an unproven intervention if in the physician’s judgement it offers hope of saving life, re-establishing health or alleviating suffering. This intervention should subsequently be made the object of research, designed to evaluate its safety and efficacy. In all cases, new information must be recorded and, where appropriate, made publicly available.”
There are two different approaches to phage therapy. One focuses on the “sur-mesure” therapy individually matching of phages to the bacterial strain isolated from a particular patient. The second one is called the “prêt-à-porter” model, which is based on the application of already-made, polyvalent phage cocktail dedicated to the treatment of a particular type of infection or targeted to the selected pathogen. It is difficult to decide which model is better, but legal constraints make the “prêt-à-porter” model a little easier to implement today.
In the USA and EU, the phages and phage-based products (enzymes) classified as human therapeutic products are subjected to the same implementation rigors as conventional drugs. That regulation raises some controversy because of the biological nature of phage preparations (especially based on infective phages). The Food and Drug Administration in the USA and the European Medicines Agency do not allow any modifications to finished medicinal products. Thus, the potentially registered phage preparations cannot be improved in any way after approval. In practice, a long and extremely expensive registration procedure results in a product with a very restrictive scope of activity and suitable only for a “prêt-à-porter” model. Further, because phage products are classified as Biological Medicinal Products (BMPs), their use is not allowed under the “hospital exemption,” as in the case of Advanced Therapy Medicinal Products (ATMPs). This regulation limits the use of targeted phage therapy designed for a particular patient. In conclusion, the legislative gaps listed above make the large pharmaceutical companies uninterested in developing phage preparations (Verbeken et al. 2016). Despite many institutional and legislative shortcomings, phage therapy is successfully used in EU in the Ludwik Hirszfeld Institute of Immunology and Experimental Therapy of Wroclaw, Poland (Miedzybrodzki et al. 2012), as well as in Queen Astrid Military Hospital in Brussels, Belgium (Jennes et al. 2017). Nonetheless, European law must undergo serious modifications to the status of phage therapy and to the registration of phages and phage derivatives. Otherwise, in the age of an increasing drug resistance, it may not be possible to draw from the advantages of phages as an effective alternative to antimicrobial therapy (Pirnay et al. 2011).
Concerning the application of phage-based enzymes, the preliminary studies involving animal models and clinical trials are demonstrating promising antibacterial efficacy and confirming their safety (Table 2). However, the current regulations also hamper the use of recombinantly produced phage proteins for therapeutic purposes, especially for long-term systemic treatment (Chan and Abedon 2012; Schmelcher et al. 2012a). The main reason is the limited data of phage enzyme interactions with the human body, which will require to perform further detailed studies concerning pharmacokinetic/pharmacodynamic properties. Nevertheless, the first phage-lysin-based preparations for topical applications, i.e., Staphefekt™ (developed by Micreos), is already registered and commercially available (Totté et al. 2017). Moreover, the first clinical trial on anti-staphylococci endolysin (SAL200 preparation) has also started (Jun et al. 2017).
References
Abedon ST (2015) Ecology of anti-biofilm agents II: bacteriophage exploitation and biocontrol of biofilm bacteria. Pharmaceuticals 8:559–589. https://doi.org/10.3390/ph8030559
Abedon ST (2016a) Phage therapy dosing: the problem(s) with multiplicity of infection (MOI). Bacteriophage 6:e1220348. https://doi.org/10.1080/21597081.2016.1220348
Abedon ST (2016b) Bacteriophage exploitation of bacterial biofilms: phage preference for less mature targets? FEMS Microbiol Lett 363:fnv246. https://doi.org/10.1093/femsle/fnv246
Abedon ST (2017) Information phage therapy research should report. Pharmaceuticals 10:43. https://doi.org/10.3390/ph10020043
Abedon ST, Kuhl SJ, Blasdel BG, Kutter EM (2011) Phage treatment of human infections. Bacteriophage 1:66–85. https://doi.org/10.4161/bact.1.2.15845
Alisky J, Iczkowski K, Rapoport A, Troitsky N (1998) Bacteriophages show promise as antimicrobial agents. J Inf Secur 36:5–15
Alper T (1954) The inactivation of free bacteriophage by irradiation and by chemical agents. J Gen Microbiol 11:313–324. https://doi.org/10.1099/00221287-11-2-313
Argov T, Azulay G, Pasechnek A, Stadnyuk O, Ran-Sapir S, Borovok I, Sigal N, Herskovits AA (2017) Temperate bacteriophages as regulators of host behavior. Curr Opin Microbiol 38:81–87. https://doi.org/10.1016/j.mib.2017.05.002
Bansal S, Soni SK, Harjai K, Chhibber S (2014) Aeromonas punctata derived depolymerase that disrupts the integrity of Klebsiella pneumoniae capsule: optimization of depolymerase production. J Basic Microbiol 54:711–720. https://doi.org/10.1002/jobm.201300356
Barrow PA, Soothill JS (1997) Bacteriophage therapy and prophylaxis: rediscovery and renewed assessment of potential. Trends Microbiol 5:268–271. https://doi.org/10.1016/S0966-842X(97)01054-8
Bonilla N, Rojas MI, Netto Flores Cruz G, Hung S-H, Rohwer F, Barr JJ (2016) Phage on tap—a quick and efficient protocol for the preparation of bacteriophage laboratory stocks. PeerJ 4:e2261. https://doi.org/10.7717/peerj.2261
Borysowski J, Weber-Dabrowska B, Górski A (2006) Bacteriophage endolysins as a novel class of antibacterial agents. Exp Biol Med (Maywood) 231:366–377. https://doi.org/10.1177/153537020623100402
Briers Y, Volckaert G, Cornelissen A, Lagaert S, Michiels CW, Hertveldt K, Lavigne R (2007) Muralytic activity and modular structure of the endolysins of Pseudomonas aeruginosa bacteriophages φKZ and EL. Mol Microbiol 65:1334–1344. https://doi.org/10.1111/j.1365-2958.2007.05870.x
Briers Y, Walmagh M, Lavigne R (2011) Use of bacteriophage endolysin EL188 and outer membrane permeabilizers against Pseudomonas aeruginosa. J Appl Microbiol 110:778–785. https://doi.org/10.1111/j.1365-2672.2010.04931.x
Briers Y, Walmagh M, Van Puyenbroeck V, Cornelissen A, Cenens W, Aertsen A, Oliveira H, Azeredo J, Verween G, Pirnay J-P, Miller S, Volckaert G, Lavigne R (2014) Engineered endolysin-based “Artilysins” to combat multidrug-resistant gram-negative pathogens. MBio 5:e01379–e01314. https://doi.org/10.1128/mBio.01379-14
Brussaard CPD, Wilhelm SW, Thingstad F, Weinbauer MG, Bratbak G, Heldal M, Kimmance SA, Middelboe M, Nagasaki K, Paul JH, Schroeder DC, Suttle CA, Vaqué D, Wommack KE (2008) Global-scale processes with a nanoscale drive: the role of marine viruses. ISME J 2:575–578. https://doi.org/10.1038/ismej.2008.31
Bull JJ, Levin BR, DeRouin T, Walker N, Bloch CA (2002) Dynamics of success and failure in phage and antibiotic therapy in experimental infections. BMC Microbiol 2:35. https://doi.org/10.1186/1471-2180-2-35
Carlton RM (1999) Phage therapy: past history and future prospects. Arch Immunol Ther Exp 47:267–274
Chan BK, Abedon ST (2012) Phage therapy pharmacology. Phage cocktails. Adv Appl Microbiol 78:1–23. https://doi.org/10.1016/B978-0-12-394805-2.00001-4
Chan BK, Abedon ST, Loc-Carrillo C (2013) Phage cocktails and the future of phage therapy. Future Microbiol 8:769–783. https://doi.org/10.2217/fmb.13.47
Chan JM, Valencia PM, Zhang L, Langer R, Farokhzad OC (2010) Polymeric nanoparticles for drug delivery. Methods Mol Biol 624:163–175. https://doi.org/10.1007/978-1-60761-609-2_11
Chaudhry WN, Concepción-Acevedo J, Park T, Andleeb S, Bull JJ, Levin BR (2017) Synergy and order effects of antibiotics and phages in killing Pseudomonas aeruginosa biofilms. PLoS One 12:e0168615. https://doi.org/10.1371/journal.pone.0168615
Cheng Q, Fischetti VA (2007) Mutagenesis of a bacteriophage lytic enzyme PlyGBS significantly increases its antibacterial activity against group B streptococci. Appl Microbiol Biotechnol 74:1284–1291. https://doi.org/10.1007/s00253-006-0771-1
Cheng Q, Nelson D, Zhu S, Fischetti VA (2005) Removal of group B streptococci colonizing the vagina and oropharynx of mice with a bacteriophage lytic enzyme. Antimicrob Agents Chemother 49:111–117. https://doi.org/10.1128/AAC.49.1.111-117.2005
Cisek AA, Dąbrowska I, Gregorczyk KP, Wyżewski Z (2017) Phage therapy in bacterial infections treatment: one hundred years after the discovery of bacteriophages. Curr Microbiol 74:277–283. https://doi.org/10.1007/s00284-016-1166-x
Cornelissen A, Ceyssens P-J, T’Syen J, Van Praet H, Noben J-P, Shaburova OV, Krylov VN, Volckaert G, Lavigne R (2011) The T7-related Pseudomonas putida phage φ15 displays virion-associated biofilm degradation properties. PLoS One 6:e18597. https://doi.org/10.1371/journal.pone.0018597
D’Herelle F (1931) An address on bacteriophagy and recovery from infectious diseases. Can Med Assoc J 24:619
Dabrowska K, Switala-Jelen K, Opolski A, Weber-Dabrowska B, Gorski A (2005) Bacteriophage penetration in vertebrates. J Appl Microbiol 98:7–13. https://doi.org/10.1111/j.1365-2672.2004.02422.x
Daniel A, Euler C, Collin M, Chahales P, Gorelick KJ, Fischetti VA (2010) Synergism between a novel chimeric lysin and oxacillin protects against infection by methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother 54:1603–1612. https://doi.org/10.1128/AAC.01625-09
Debarbieux L, Pirnay JP, Verbeken G, De Vos D, Merabishvili M, Huys I, Patey O, Schoonjans D, Vaneechoutte M, Zizi M, Rohde C (2016) A bacteriophage journey at the European Medicines Agency. FEMS Microbiol Lett 363:fnv225. https://doi.org/10.1093/femsle/fnv225
Díez-Martínez R, De Paz HD, García-Fernández E, Bustamante N, Euler CW, Fischetti VA, Menendez M, García P (2014) A novel chimeric phage lysin with high in vitro and in vivo bactericidal activity against Streptococcus pneumoniae. J Antimicrob Chemother 70:1763–1773. https://doi.org/10.1093/jac/dkv038
Doehn JM, Fischer K, Reppe K, Gutbier B, Tschernig T, Hocke AC, Fischetti V a, Löffler J, Suttorp N, Hippenstiel S, Witzenrath M (2013) Delivery of the endolysin Cpl-1 by inhalation rescues mice with fatal pneumococcal pneumonia. J Antimicrob Chemother 68:2111–2117. doi: https://doi.org/10.1093/jac/dkt131
Domenech M, Garciá E, Moscoso M (2011) In vitro destruction of Streptococcus pneumoniae biofilms with bacterial and phage peptidoglycan hydrolases. Antimicrob Agents Chemother 55:4144–4148. https://doi.org/10.1128/AAC.00492-11
Edgar R, Friedman N, Shahar MM, Qimron U (2012) Reversing bacterial resistance to antibiotics by phage-mediated delivery of dominant sensitive genes. Appl Environ Microbiol 78:744–751. https://doi.org/10.1128/AEM.05741-11
Entenza JM, Loeffler JM, Grandgirard D, Fischetti VA, Moreillon P (2005) Therapeutic effects of bacteriophage Cpl-1 lysin against Streptococcus pneumoniae endocarditis in rats. Antimicrob Agents Chemother 49:4789–4792. https://doi.org/10.1128/AAC.49.11.4789-4792.2005
Erez Z, Steinberger-Levy I, Shamir M, Doron S, Stokar-Avihail A, Peleg Y, Melamed S, Leavitt A, Savidor A, Albeck S, Amitai G, Sorek R (2017) Communication between viruses guides lysis-lysogeny decisions. Nature:1–18. https://doi.org/10.1038/nature21049
Fenton M, Keary R, McAuliffe O, Ross RP, O’Mahony J, Coffey A (2013) Bacteriophage-derived peptidase CHAPK eliminates and prevents staphylococcal biofilms. Int J Microbiol 2013:625341. https://doi.org/10.1155/2013/625341
Fischetti VA (2010) Bacteriophage endolysins: a novel anti-infective to control gram-positive pathogens. Int J Med Microbiol 300:357–362. https://doi.org/10.1016/j.ijmm.2010.04.002
Fischetti VA (2005) Bacteriophage lytic enzymes: novel anti-infectives. Trends Microbiol 13:491–496. https://doi.org/10.1016/j.tim.2005.08.007
Gervasi T, Horn N, Wegmann U, Dugo G, Narbad A, Mayer MJ (2014) Expression and delivery of an endolysin to combat Clostridium perfringens. Appl Microbiol Biotechnol 98:2495–2505. https://doi.org/10.1007/s00253-013-5128-y
Gilmer DB, Schmitz JE, Euler CW, Fischetti VA (2013) Novel bacteriophage lysin with broad lytic activity protects against mixed infection by Streptococcus pyogenes and methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother 57:2743–2750. https://doi.org/10.1128/AAC.02526-12
Gilmer DB, Schmitz JE, Thandar M, Euler CW, Fischetti VA (2017) The phage lysin PlySs2 decolonizes Streptococcus suis from murine intranasal mucosa. PLoS One 12:e0169180. https://doi.org/10.1371/journal.pone.0169180
Goldfarb T, Sberro H, Weinstock E, Cohen O, Doron S, Charpak-Amikam Y, Afik S, Ofir G, Sorek R (2015) BREX is a novel phage resistance system widespread in microbial genomes. EMBO J 34:169–183. https://doi.org/10.15252/embj.201489455
Goodridge LD (2010) Designing phage therapeutics. Curr Pharm Biotechnol 11:15–27. https://doi.org/10.2174/138920110790725348
Gorski A, Wazna E, Dabrowska B-W, Dabrowska K, Switała-Jelen K, Miedzybrodzki R (2006) Bacteriophage translocation. FEMS Immunol Med Microbiol 46:313–319. https://doi.org/10.1111/j.1574-695X.2006.00044.x
Górski A, Międzybrodzki R, Borysowski J, Dąbrowska K, Wierzbicki P, Ohams M, Korczak-Kowalska G, Olszowska-Zaremba N, Łusiak-Szelachowska M, Kłak M, Jończyk E, Kaniuga E, Gołaś A, Purchla S, Weber-Dąbrowska B, Letkiewicz S, Fortuna W, Szufnarowski K, Pawełczyk Z, Rogóz P, Kłosowska D (2012) Phage as a modulator of immune responses. Practical implications for phage therapy. Adv Virus Res 83:41–71. https://doi.org/10.1016/B978-0-12-394438-2.00002-5
Grandgirard D, Loeffler JM, Fischetti VA, Leib SL (2008) Phage lytic enzyme Cpl-1 for antibacterial therapy in experimental pneumococcal meningitis. J Infect Dis 197:1519–1522. https://doi.org/10.1086/587942
Gu J, Xu W, Lei L, Huang J, Feng X, Sun C, Du C, Zuo J, Li Y, Du T, Li L, Han W (2011a) LysGH15, a novel bacteriophage lysin, protects a murine bacteremia model efficiently against lethal methicillin-resistant Staphylococcus aureus infection. J Clin Microbiol 49:111–117. https://doi.org/10.1128/JCM.01144-10
Gu J, Zuo J, Lei L, Zhao H, Sun C, Feng X, Du C, Li X, Yang Y, Han W (2011b) LysGH15 reduces the inflammation caused by lethal methicillin-resistant Staphylococcus aureus infection in mice. Bioeng Bugs 2:96–99. https://doi.org/10.4161/bbug.2.2.14883
Guariglia-Oropeza V, Helmann JD (2011) Bacillus subtilis σ(V) confers lysozyme resistance by activation of two cell wall modification pathways, peptidoglycan O-acetylation and D-alanylation of teichoic acids. J Bacteriol 193:6223–6232. https://doi.org/10.1128/JB.06023-11
Guo M, Feng C, Ren J, Zhuang X, Zhang Y, Zhu Y, Dong K, He P, Guo X, Qin J (2017) A novel antimicrobial endolysin, LysPA26, against Pseudomonas aeruginosa. Front Microbiol 8. https://doi.org/10.3389/fmicb.2017.00293
Gupta R, Prasad Y (2011a) P-27/HP Endolysin as antibacterial agent for antibiotic resistant Staphylococcus aureus of human infections. Curr Microbiol 63:39–45. https://doi.org/10.1007/s00284-011-9939-8
Gupta R, Prasad Y (2011b) Efficacy of polyvalent bacteriophage P-27/HP to control multidrug resistant Staphylococcus aureus associated with human infections. Curr Microbiol 62:255–260. https://doi.org/10.1007/s00284-010-9699-x
Gutiérrez D, Briers Y, Rodríguez-Rubio L, Martínez B, Rodríguez A, Lavigne R, García P (2015) Role of the pre-neck appendage protein (Dpo7) from phage vB_SepiS-phiIPLA7 as an anti-biofilm agent in staphylococcal species. Front Microbiol 6:1315. https://doi.org/10.3389/fmicb.2015.01315
Gutiérrez D, Martínez B, Rodríguez A, García P (2012) Genomic characterization of two Staphylococcus epidermidis bacteriophages with anti-biofilm potential. BMC Genomics 13:228. https://doi.org/10.1186/1471-2164-13-228
Gutiérrez D, Ruas-Madiedo P, Martínez B, Rodríguez A, García P (2014) Effective removal of staphylococcal biofilms by the endolysin LysH5. PLoS One 9:e107307. https://doi.org/10.1371/journal.pone.0107307
Heselpoth RD, Yin Y, Moult J, Nelson DC (2015) Increasing the stability of the bacteriophage endolysin PlyC using rationale-based FoldX computational modeling. Protein Eng Des Sel 28:85–92. https://doi.org/10.1093/protein/gzv004
Höltje JV, Mirelman D, Sharon N, Schwarz U (1975) Novel type of murein transglycosylase in Escherichia coli. J Bacteriol 124:1067–1076
Jado I, López R, García E, Fenoll A, Casal J, García P, Pallares R, de la Campa AG, Bouza E, Baquero F, Soriano F, Prieto J, Pallarés R, Liñares J, Garau J, Martínez Lacasa J, Latorre C, Pérez-Trallero E, García de Lomas J, Fleites A (2003) Phage lytic enzymes as therapy for antibiotic-resistant Streptococcus pneumoniae infection in a murine sepsis model. J Antimicrob Chemother 52:967–973. https://doi.org/10.1093/jac/dkg485
Jennes S, Merabishvili M, Soentjens P, Pang KW, Rose T, Keersebilck E, Soete O, François P-M, Teodorescu S, Verween G, Verbeken G, De Vos D, Pirnay J-P (2017) Use of bacteriophages in the treatment of colistin-only-sensitive Pseudomonas aeruginosa septicaemia in a patient with acute kidney injury—a case report. Crit Care 21:129. https://doi.org/10.1186/s13054-017-1709-y
Jończyk E, Kłak M, Międzybrodzki R, Górski A (2011) The influence of external factors on bacteriophages—review. Folia Microbiol 56:191–200. https://doi.org/10.1007/s12223-011-0039-8
Jun SY, Jang IJ, Yoon S, Jang K, Yu K-S, Cho JY, Seong M-W, Jung GM, Yoon SJ, Kang SH (2017) Pharmacokinetics and tolerance of the phage endolysin-based candidate drug SAL200 after a single intravenous administration among healthy volunteers. Antimicrob Agents Chemother 61:e02629–e02616. https://doi.org/10.1128/AAC.02629-16
Jun SY, Jung GM, Yoon SJ, Choi YJ, Koh WS, Moon KS, Kang SH (2014) Preclinical safety evaluation of intravenously administered SAL200 containing the recombinant phage endolysin SAL-1 as a pharmaceutical ingredient. Antimicrob Agents Chemother 58:2084–2088. https://doi.org/10.1128/AAC.02232-13
Jun SY, Jung GM, Yoon SJ, Oh MD, Choi YJ, Lee WJ, Kong JC, Seol JG, Kang SH (2013) Antibacterial properties of a pre-formulated recombinant phage endolysin, SAL-1. Int J Antimicrob Agents 41:156–161. https://doi.org/10.1016/j.ijantimicag.2012.10.011
Jun SY, Jung GM, Yoon SJ, Youm SY, Han H-Y, Lee J-H, Kang SH (2016) Pharmacokinetics of the phage endolysin-based candidate drug SAL200 in monkeys and its appropriate intravenous dosing period. Clin Exp Pharmacol Physiol 43:1013–1016. https://doi.org/10.1111/1440-1681.12613
Kim M, Ryu S (2013) Antirepression system associated with the life cycle switch in the temperate Podoviridae phage SPC32H. J Virol 87:11775–11786. https://doi.org/10.1128/JVI.02173-13
Krylov V, Shaburova O, Krylov S, Pleteneva E (2012) A genetic approach to the development of new therapeutic phages to fight Pseudomonas aeruginosa in wound infections. Viruses 5:15–53. https://doi.org/10.3390/v5010015
Kutter E, De Vos D, Gvasalia G, Alavidze Z, Gogokhia L, Kuhl S, Abedon ST (2010) Phage therapy in clinical practice: Treatment of human infections. Curr Pharm Biotechnol 11:69–86. https://doi.org/10.2174/138920110790725401
Labrie SJ, Samson JE, Moineau S (2010) Bacteriophage resistance mechanisms. Nat Rev Microbiol 8:317–327. https://doi.org/10.1038/nrmicro2315
Latino L, Midoux C, Hauck Y, Vergnaud G, Pourcel C (2016) Pseudolysogeny and sequential mutations build multiresistance to virulent bacteriophages in Pseudomonas aeruginosa. Microbiol (United Kingdom) 162:748–763. https://doi.org/10.1099/mic.0.000263
Latka A, Maciejewska B, Majkowska-Skrobek G, Briers Y, Drulis-Kawa Z (2017) Bacteriophage-encoded virion-associated enzymes to overcome the carbohydrate barriers during the infection process. Appl Microbiol Biotechnol 101:3103–3119. https://doi.org/10.1007/s00253-017-8224-6
Letkiewicz S, Miedzybrodzki R, Klak M, Jonczyk E, Weber-Dabrowska B, Gorski A (2010) The perspectives of the application of phage therapy in chronic bacterial prostatitis. FEMS Immunol Med Microbiol 60:99–112. https://doi.org/10.1111/j.1574-695X.2010.00723.x
Lim JA, Shin H, Heu S, Ryu S (2014) Exogenous lytic activity of SPN9CC endolysin against gram-negative bacteria. J Microbiol Biotechnol 24:803–811. https://doi.org/10.4014/jmb.1403.03035
Lin DM, Koskella B, Lin HC (2017) Phage therapy: an alternative to antibiotics in the age of multi-drug resistance. World J Gastrointest Pharmacol Ther 8:162. https://doi.org/10.4292/wjgpt.v8.i3.162
Loc-Carrillo C, Abedon ST (2011) Pros and cons of phage therapy. Bacteriophage 1:111–114. https://doi.org/10.4161/bact.1.2.14590
Loeffler JM, Djurkovic S, Fischetti VA (2003) Phage lytic enzyme Cpl-1 as a novel antimicrobial for pneumococcal bacteremia. Infect Immun 71:6199–6204. https://doi.org/10.1128/IAI.71.11.6199-6204.2003
Loeffler JM, Nelson D, Fischetti VA (2001) Rapid killing of Streptococcus pneumoniae with a bacteriophage cell wall hydrolase. Science 294:2170–2172. https://doi.org/10.1126/science.1066869
Lood R, Raz A, Molina H, Euler CW, Fischetti VA (2014) A highly active and negatively charged Streptococcus pyogenes lysin with a rare d-alanyl-l-alanine endopeptidase activity protects mice against streptococcal bacteremia. Antimicrob Agents Chemother 58:3073–3084. https://doi.org/10.1128/AAC.00115-14
Lood R, Winer BY, Pelzek AJ, Diez-Martinez R, Thandar M, Euler CW, Schuch R, Fischetti VA (2015) Novel phage lysin capable of killing the multidrug-resistant gram-negative bacterium Acinetobacter baumannii in a mouse bacteremia model. Antimicrob Agents Chemother 59:1983–1991. https://doi.org/10.1128/AAC.04641-14
Łoś M, Węgrzyn G (2012) Pseudolysogeny. Adv Virus Res 82:339–49. https://doi.org/10.1016/B978-0-12-394621-8.00019-4
Lu TK, Collins JJ (2007) Dispersing biofilms with engineered enzymatic bacteriophage. Proc Natl Acad Sci U S A 104:11197–11202. https://doi.org/10.1073/pnas.0704624104
Maciejewska B, Roszniowski B, Espaillat A, Kesik-Szeloch A, Majkowska-Skrobek G, Kropinski AM, Briers Y, Cava F, Lavigne R, Drulis-Kawa Z (2017) Klebsiella phages representing a novel clade of viruses with an unknown DNA modification and biotechnologically interesting enzymes. Appl Microbiol Biotechnol 101:673–684. https://doi.org/10.1007/s00253-016-7928-3
Majkowska-Skrobek G, Latka A, Berisio R, Maciejewska B, Squeglia F, Romano M, Lavigne R, Struve C, Drulis-Kawa Z (2016) Capsule-targeting depolymerase, derived from Klebsiella KP36 phage, as a tool for the development of anti-virulent strategy. Viruses 8:324. https://doi.org/10.3390/v8120324
Matsushita I, Yanase H (2008) A novel thermophilic lysozyme from bacteriophage phiIN93. Biochem Biophys Res Commun 377:89–92. https://doi.org/10.1016/j.bbrc.2008.09.101
Mayer MJ, Narbad A, Gasson MJ (2008) Molecular characterization of a Clostridium difficile bacteriophage and its cloned biologically active endolysin. J Bacteriol 190:6734–6740. https://doi.org/10.1128/JB.00686-08
McCallum KL, Laakso DH, Whitfield C (1989) Use of a bacteriophage-encoded glycanase enzyme in the generation of lipopolysaccharide O-side chain deficient mutants of Escherichia coli O9:K30 and Klebsiella O1:K20: Role of O and K antigens in resistance to complement-mediated serum killing. Can J Microbiol 35:994–999. https://doi.org/10.1139/m89-166
McCullers JA, Karlström Å, Iverson AR, Loeffler JM, Fischetti VA (2007) Novel strategy to prevent otitis media caused by colonizing Streptococcus pneumoniae. PLoS Pathog 3:e28. https://doi.org/10.1371/journal.ppat.0030028
Meng X, Shi Y, Ji W, Meng X, Zhang J, Wang H, Lu C, Sun J, Yan Y (2011) Application of a bacteriophage lysin to disrupt biofilms formed by the animal pathogen Streptococcus suis. Appl Environ Microbiol 77:8272–8279. https://doi.org/10.1128/AEM.05151-11
Merril CR, Biswas B, Carlton R, Jensen NC, Creed GJ, Zullo S, Adhya S (1996) Long-circulating bacteriophage as antibacterial agents. Proc Natl Acad Sci U S A 93:3188–3192
Miedzybrodzki R, Borysowski J, Weber-Dabrowska B, Fortuna W, Letkiewicz S, Szufnarowski K, Pawelczyk Z, Rogoz P, Klak M, Wojtasik E, Gorski A (2012) Clinical aspects of phage therapy. Adv Virus Res 83:73–121. https://doi.org/10.1016/B978-0-12-394438-2.00003-7
Mushtaq N, Redpath MB, Luzio JP, Taylor PW (2004) Prevention and cure of systemic Escherichia coli K1 infection by modification of the bacterial phenotype. Antimicrob Agents Chemother 48:1503–1508. https://doi.org/10.1128/AAC.48.5.1503-1508.2004
Mushtaq N, Redpath MB, Luzio JP, Taylor PW (2005) Treatment of experimental Escherichia coli infection with recombinant bacteriophage-derived capsule depolymerase. J Antimicrob Chemother 56:160–165. https://doi.org/10.1093/jac/dki177
Nau R, Eiffert H (2002) Modulation of release of proinflammatory bacterial compounds by antibacterials: potential impact on course of inflammation and outcome in sepsis and meningitis. Clin Microbiol Rev 15:95–110. https://doi.org/10.1128/CMR.15.1.95-110.2002
Navarro F, Muniesa M (2017) Phages in the human body. Front Microbiol 8:566. https://doi.org/10.3389/fmicb.2017.00566
Nelson D, Loomis L, Fischetti VA (2001) Prevention and elimination of upper respiratory colonization of mice by group a streptococci by using a bacteriophage lytic enzyme. Proc Natl Acad Sci U S A 98:4107–4112. https://doi.org/10.1073/pnas.061038398
Nelson DC, Schmelcher M, Rodriguez-Rubio L, Klumpp J, Pritchard DG, Dong S, Donovan DM (2012) Endolysins as antimicrobials. In: Advances in Virus Research. 83:299-365. https://doi.org/10.1016/B978-0-12-394438-2.00007-4
Oechslin F, Daraspe J, Giddey M, Moreillon P, Resch G (2013) In vitro characterization of PlySK1249, a novel phage lysin, and assessment of its antibacterial activity in a mouse model of Streptococcus agalactiae bacteremia. Antimicrob Agents Chemother 57:6276–6283. https://doi.org/10.1128/AAC.01701-13
Oliveira H, Thiagarajan V, Walmagh M, Sillankorva S, Lavigne R, Neves-Petersen MT, Kluskens LD, Azeredo J (2014) A thermostable Salmonella phage endolysin, Lys68, with broad bactericidal properties against gram-negative pathogens in presence of weak acids. PLoS One 9:e108376. https://doi.org/10.1371/journal.pone.0108376
Oliveira H, Vilas Boas D, Mesnage S, Kluskens LD, Lavigne R, Sillankorva S, Secundo F, Azeredo J (2016) Structural and enzymatic characterization of ABgp46, a novel phage endolysin with broad anti-gram-negative bacterial activity. Front Microbiol 7:208. https://doi.org/10.3389/fmicb.2016.00208
Olszak T, Shneider MM, Latka A, Maciejewska B, Browning C, Sycheva LV, Cornelissen A, Danis-Wlodarczyk K, Senchenkova SN, Shashkov AS, Gula G, Arabski M, Wasik S, Miroshnikov KA, Lavigne R, Leiman PG, Knirel YA, Drulis-Kawa Z (2017) The O-specific polysaccharide lyase from the phage LKA1 tailspike reduces Pseudomonas virulence. Sci Rep 7:16302. https://doi.org/10.1038/s41598-017-16411-4
Ormälä A-M, Jalasvuori M (2013) Phage therapy: should bacterial resistance to phages be a concern, even in the long run? Bacteriophage 3:e24219. https://doi.org/10.4161/bact.24219
Pan Y-J, Lin T-L, Lin Y-T, Su P-A, Chen C-T, Hsieh P-F, Hsu C-R, Chen C-C, Hsieh Y-C, Wang J-T (2015) Identification of capsular types in carbapenem-resistant Klebsiella pneumoniae strains by wzc sequencing and implications for capsule depolymerase treatment. Antimicrob Agents Chemother 59:1038–1047. https://doi.org/10.1128/AAC.03560-14
Pastagia M, Euler C, Chahales P, Fuentes-Duculan J, Krueger JG, Fischetti VA (2011) A novel chimeric lysin shows superiority to mupirocin for skin decolonization of methicillin-resistant and sensitive Staphylococcus aureus strains. Antimicrob Agents Chemother 55:738–744. https://doi.org/10.1128/AAC.00890-10
Paul V, Rajagopalan S, Sundarrajan S, George SE, Asrani JY, Pillai R, Chikkamadaiah R, Durgaiah M, Sriram B, Padmanabhan S (2011) A novel bacteriophage tail-associated muralytic enzyme (TAME) from phage K and its development into a potent antistaphylococcal protein. BMC Microbiol 11:226. https://doi.org/10.1186/1471-2180-11-226
Pearl S, Gabay C, Kishony R, Oppenheim A, Balaban NQ (2008) Nongenetic individuality in the host–phage interaction. PLoS Biol 6:957–964. https://doi.org/10.1371/journal.pbio.0060120
Peng S-Y, You R-I, Lai M-J, Lin N-T, Chen L-K, Chang K-C (2017) Highly potent antimicrobial modified peptides derived from the Acinetobacter baumannii phage endolysin LysAB2. Sci Rep 7:11477. https://doi.org/10.1038/s41598-017-11832-7
Pires DP, Oliveira H, Melo LDR, Sillankorva S, Azeredo J (2016) Bacteriophage-encoded depolymerases: their diversity and biotechnological applications. Appl Microbiol Biotechnol 100:2141–2151. https://doi.org/10.1007/s00253-015-7247-0
Pirnay J-P, De Vos D, Verbeken G, Merabishvili M, Chanishvili N, Vaneechoutte M, Zizi M, Laire G, Lavigne R, Huys I, Van den Mooter G, Buckling A, Debarbieux L, Pouillot F, Azeredo J, Kutter E, Dublanchet A, Gorski A, Adamia R (2011) The phage therapy paradigm: prêt-à-porter or sur-mesure? Pharm Res 28:934–937. https://doi.org/10.1007/s11095-010-0313-5
Plotka M, Kaczorowska AK, Stefanska A, Morzywolek A, Fridjonsson OH, Dunin-Horkawicz S, Kozlowski L, Hreggvidsson GO, Kristjansson JK, Dabrowski S, Bujnicki JM, Kaczorowski T (2014) Novel highly thermostable endolysin from Thermus scotoductus MAT2119 bacteriophage Ph2119 with amino acid sequence similarity to eukaryotic peptidoglycan recognition proteins. Appl Environ Microbiol 80:886–895. https://doi.org/10.1128/AEM.03074-13
Prins JM, van Deventer SJ, Kuijper EJ, Speelman P (1994) Clinical relevance of antibiotic-induced endotoxin release. Antimicrob Agents Chemother 38:1211–1218. https://doi.org/10.1128/AAC.38.6.1211
Prokhorov NS, Riccio C, Zdorovenko EL, Shneider MM, Browning C, Knirel YA, Leiman PG, Letarov AV (2017) Function of bacteriophage G7C esterase tailspike in host cell adsorption. Mol Microbiol 105:385–398. https://doi.org/10.1111/mmi.13710
Ramachandran G (2014) Gram-positive and gram-negative bacterial toxins in sepsis. Virulence 5:213–218. https://doi.org/10.4161/viru.27024
Rashel M, Uchiyama J, Ujihara T, Uehara Y, Kuramoto S, Sugihara S, Yagyu K-I, Muraoka A, Sugai M, Hiramatsu K, Honke K, Matsuzaki S (2007) Efficient elimination of multidrug-resistant Staphylococcus aureus by cloned lysin derived from bacteriophage phi MR11. J Infect Dis 196:1237–1247. https://doi.org/10.1086/521305
Resch G, Moreillon P, Fischetti VA (2011) A stable phage lysin (Cpl-1) dimer with increased antipneumococcal activity and decreased plasma clearance. Int J Antimicrob Agents 38:516–521. https://doi.org/10.1016/j.ijantimicag.2011.08.009
Rico-Lastres P, Díez-Martínez R, Iglesias-Bexiga M, Bustamante N, Aldridge C, Hesek D, Lee M, Mobashery S, Gray J, Vollmer W, García P, Menéndez M (2015) Substrate recognition and catalysis by LytB, a pneumococcal peptidoglycan hydrolase involved in virulence. Sci Rep 5:16198. https://doi.org/10.1038/srep16198
Roach DR, Leung CY, Henry M, Morello E, Singh D, Di Santo JP, Weitz JS, Debarbieux L (2017) Synergy between the host immune system and bacteriophage is essential for successful phage therapy against an acute respiratory pathogen. Cell Host Microbe 22:38–47.e4. https://doi.org/10.1016/j.chom.2017.06.018
Rodríguez-Rubio L, Gerstmans H, Thorpe S, Mesnage S, Lavigne R, Briers Y (2016) DUF3380 domain from a Salmonella phage endolysin shows potent N-acetylmuramidase activity. Appl Environ Microbiol 82:4975–4981. https://doi.org/10.1128/AEM.00446-16
Rodríguez-Rubio L, Martínez B, Donovan DM, García P, Rodríguez A (2013) Potential of the virion-associated peptidoglycan hydrolase HydH5 and its derivative fusion proteins in milk biopreservation. PLoS One 8:e54828. https://doi.org/10.1371/journal.pone.0054828
Ryan EM, Gorman SP, Donnelly RF, Gilmore BF (2011) Recent advances in bacteriophage therapy: how delivery routes, formulation, concentration and timing influence the success of phage therapy. J Pharm Pharmacol 63:1253–1264. https://doi.org/10.1111/j.2042-7158.2011.01324.x
Salmond GPC, Fineran PC (2015) A century of the phage: past, present and future. Nat Rev Microbiol 13:777–786. https://doi.org/10.1038/nrmicro3564
Sass P, Bierbaum G (2007) Lytic activity of recombinant bacteriophage phi11 and phi12 endolysins on whole cells and biofilms of Staphylococcus aureus. Appl Environ Microbiol 73:347–352. https://doi.org/10.1128/AEM.01616-06
Schirmeier E, Zimmermann P, Hofmann V, Biebl M, Gerstmans H, Maervoet VE, Briers Y (2017) Inhibitory and bactericidal effect of Artilysin® Art-175 against colistin-resistant mcr-1-positive Escherichia coli isolates. Int J Antimicrob Agents. https://doi.org/10.1016/j.ijantimicag.2017.08.027
Schleifer KH, Kandler O (1972) Peptidoglycan types of bacterial cell walls and their taxonomic implications. Bacteriol Rev 36:407–477
Schmelcher M, Donovan DM, Loessner MJ (2012a) Bacteriophage endolysins as novel antimicrobials. Future Microbiol 7:1147–1171. https://doi.org/10.2217/fmb.12.97
Schmelcher M, Powell AM, Becker SC, Camp MJ, Donovan DM (2012b) Chimeric phage lysins act synergistically with lysostaphin to kill mastitis-causing Staphylococcus aureus in murine mammary glands. Appl Environ Microbiol 78:2297–2305. https://doi.org/10.1128/AEM.07050-11
Schmelcher M, Shen Y, Nelson DC, Eugster MR, Eichenseher F, Hanke DC, Loessner MJ, Dong S, Pritchard DG, Lee JC, Becker SC, Foster-Frey J, Donovan DM (2014) Evolutionarily distinct bacteriophage endolysins featuring conserved peptidoglycan cleavage sites protect mice from MRSA infection. J Antimicrob Chemother 70:1453–1465. https://doi.org/10.1093/jac/dku552
Schuch R, Nelson D, Fischetti VA (2002) A bacteriolytic agent that detects and kills Bacillus anthracis. Nature 418:884–889. https://doi.org/10.1038/nature01026
Shen Y, Barros M, Vennemann T, Gallagher DT, Yin Y, Linden SB, Heselpoth RD, Spencer DJ, Donovan DM, Moult J, Fischetti VA, Heinrich F, Lösche M, Nelson DC (2016) A bacteriophage endolysin that eliminates intracellular streptococci. elife 5:e13152. https://doi.org/10.7554/eLife.13152
Shen Y, Köller T, Kreikemeyer B, Nelson DC (2013) Rapid degradation of Streptococcus pyogenes biofilms by PlyC, a bacteriophage-encoded endolysin. J Antimicrob Chemother 68:1818–1824. https://doi.org/10.1093/jac/dkt104
Singh PK, Donovan DM, Kumar A (2014) Intravitreal injection of the chimeric phage endolysin Ply187 protects mice from Staphylococcus aureus endophthalmitis. Antimicrob Agents Chemother 58:4621–4629. https://doi.org/10.1128/AAC.00126-14
Skurnik M, Strauch E (2006) Phage therapy: Facts and fiction. Int. J. Med. Microbiol. 296:5–14. https://doi.org/10.1016/j.ijmm.2005.09.002
Smith J (1924) The bacteriophage in the treatment of typhoid fever. Br Med J 2:47–49
Son J-S, Lee S-J, Jun SY, Yoon SJ, Kang SH, Paik HR, Kang JO, Choi Y-J (2010) Antibacterial and biofilm removal activity of a podoviridae Staphylococcus aureus bacteriophage SAP-2 and a derived recombinant cell-wall-degrading enzyme. Appl Microbiol Biotechnol 86:1439–1449. https://doi.org/10.1007/s00253-009-2386-9
Speck P, Smithyman A (2016) Safety and efficacy of phage therapy via the intravenous route. FEMS Microbiol Lett 363:fnv242. https://doi.org/10.1093/femsle/fnv242
Sulakvelidze A, Alavidze Z, Morris JG Jr (2001) Bacteriophage therapy minireview. Antimicrob Agents Chemother 45:649–659. https://doi.org/10.1128/AAC.45.3.649
Thierauf A, Perez G, Maloy S (2009) Generalized transduction. In: Clokie MRJ, Kropinski A (eds) Bacteriophages: methods and protocols. Volume 1: isolation, characterization, and interactions. Humana press, pp 267–286
Totté JEE, van Doorn MB, Pasmans SGMA (2017) Successful treatment of chronic Staphylococcus aureus-related dermatoses with the topical endolysin staphefekt SA.100: a report of 3 cases. Case Rep Dermatol 9:19–25. https://doi.org/10.1159/000473872
van Langevelde P, Kwappenberg KM, Groeneveld PH, Mattie H, van Dissel JT (1998) Antibiotic-induced lipopolysaccharide (LPS) release from Salmonella Typhi: delay between killing by ceftazidime and imipenem and release of LPS. Antimicrob Agents Chemother 42:739–743
van Valen L (1973) A new evolutionary law. Evol Theory 1:1–30. https://doi.org/10.1017/CBO9781107415324.004
Verbeken G, Huys I, De Vos D, De Coninck A, Roseeuw D, Kets E, Vanderkelen A, Draye JP, Rose T, Jennes S, Ceulemans C, Pirnay JP (2016) Access to bacteriophage therapy: discouraging experiences from the human cell and tissue legal framework. FEMS Microbiol Lett 363:fnv241. https://doi.org/10.1093/femsle/fnv241
Vollmer W, Joris B, Charlier P, Foster S (2008) Bacterial peptidoglycan (murein) hydrolases. FEMS Microbiol Rev 32:259–286. https://doi.org/10.1111/j.1574-6976.2007.00099.x
Waseh S, Hanifi-Moghaddam P, Coleman R, Masotti M, Ryan S, Foss M, MacKenzie R, Henry M, Szymanski CM, Tanha J (2010) Orally administered P22 phage tailspike protein reduces Salmonella colonization in chickens: prospects of a novel therapy against bacterial infections. PLoS One 5:e13904. https://doi.org/10.1371/journal.pone.0013904
Weber-Dabrowska B, Jonczyk-Matysiak E, Zaczek M, Lobocka M, Lusiak-Szelachowska M, Gorski A (2016) Bacteriophage procurement for therapeutic purposes. Front Microbiol 7:1177. https://doi.org/10.3389/fmicb.2016.01177
Weiss N, Miller F, Cazaubon S, Couraud P-O (2009) The blood–brain barrier in brain homeostasis and neurological diseases. Biochim Biophys Acta Biomembr 1788:842–857. https://doi.org/10.1016/j.bbamem.2008.10.022
Wingfield PT (2015) Overview of the purification of recombinant proteins. Curr Protoc protein Sci 80:6.1.1–6.135. https://doi.org/10.1002/0471140864.ps0601s80
Witzenrath M, Schmeck B, Doehn JM, Tschernig T, Zahlten J, Loeffler JM, Zemlin M, Müller H, Gutbier B, Schütte H, Hippenstiel S, Fischetti VA, Suttorp N, Rosseau S (2009) Systemic use of the endolysin Cpl-1 rescues mice with fatal pneumococcal pneumonia. Crit Care Med 37:642–649. https://doi.org/10.1097/CCM.0b013e31819586a6
Wommack KE, Colwell RR (2000) Virioplankton: viruses in aquatic ecosystems. Microbiol Mol Biol Rev 64:69–114. https://doi.org/10.1128/MMBR.64.1.69-114.2000
World Medical Association (2013) World medical association declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA 310:2191–2194. https://doi.org/10.1001/jama.2013.281053
Yang H, Wang M, Yu J, Wei H (2015) Antibacterial activity of a novel peptide-modified lysin against Acinetobacter baumannii and Pseudomonas aeruginosa. Front Microbiol 6:1471. https://doi.org/10.3389/fmicb.2015.01471
Yoong P, Schuch R, Nelson D, Fischetti VA (2006) PlyPH, a bacteriolytic enzyme with a broad pH range of activity and lytic action against Bacillus anthracis. J Bacteriol 188:2711–2714. https://doi.org/10.1128/JB.188.7.2711-2714.2006
Yoong P, Schuch R, Nelson D, Fischetti VA (2004) Identification of a broadly active phage lytic enzyme with lethal activity against antibiotic-resistant Enterococcus faecalis and Enterococcus faecium. J Bacteriol 186:4808–4812. https://doi.org/10.1128/JB.186.14.4808-4812.2004
Zhang L, Li D, Li X, Hu L, Cheng M, Xia F, Gong P, Wang B, Ge J, Zhang H, Cai R, Wang Y, Sun C, Feng X, Lei L, Han W, Gu J (2016) LysGH15 kills Staphylococcus aureus without being affected by the humoral immune response or inducing inflammation. Sci Rep 6:29344. https://doi.org/10.1038/srep29344
Acknowledgements
This project was financed by National Science Centre, Poland, granted based on decisions No. UMO-2016/21/B/NZ6/01157.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Maciejewska, B., Olszak, T. & Drulis-Kawa, Z. Applications of bacteriophages versus phage enzymes to combat and cure bacterial infections: an ambitious and also a realistic application?. Appl Microbiol Biotechnol 102, 2563–2581 (2018). https://doi.org/10.1007/s00253-018-8811-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-018-8811-1