
Abstract
Ebstein’s anomaly is frequently detected before birth, with prenatal detection accounting for the majority of cases in the current population. This study aimed to identify the outcome variables among these infants. The medical records of 59 patients with neonatal Ebstein’s anomaly managed at the Asan Medical Center between January, 2001 and June, 2012 were investigated retrospectively. In 46 cases, the diagnosis was made prenatally. Surgical/interventional procedures were performed for 27 of the analyzed patients. Biventricular repair was successful for 12 patients but not for 9 patients with pulmonary atresia. The median follow-up period was 1.96 years (range 0.0–10.4 years). The overall mortality rate was 23.7 % (14/59). Of the 14 deaths, 5 occurred within several hours after birth. The 1- and 5-year survival rates were 78.6 and 76.3 %, respectively. Univariate analysis identified several variables related to the time to death: fetal distress (p = 0.002), prematurity (p = 0.036), low birth weight (p = 0.003), diameter of the atrial septal defect (p = 0.022), and pulmonary stenosis/atresia (p = 0.001). Neither the Carpentier classification (p = 0.175) nor the Celermajer index (p = 0.958) was a significant variable. According to the multivariate analysis, fetal distress (p = 0.004) and pulmonary atresia/stenosis (p < 0.001) were significant determinants of outcome. In conclusion, fetal distress and pulmonary atresia/stenosis are significant predictors of mortality in the current population of patients with neonatal Ebstein’s anomaly. A close cooperation of associated clinicians is required for an improvement in outcome. To establish a better surgical strategy for patients with Ebstein’s anomaly and pulmonary atresia, studies of larger populations are required.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
It is well known that young age is a significant risk factor for patients with Ebstein’s anomaly [10, 13]. In studies reported before 2000, the mortality rate of affected neonates ranged from 26 to 81 % [4, 5, 16]. This broad range reflects gradual improvements in the treatment of these patients, such that whereas the mortality rate was as high as 81 % from 1954 to 1985, it decreased to 47 % between 1986 and 1996 [16]. The survival rate for neonates with Ebstein’s anomaly continues to increase [7, 15] with improved conservative management and surgical strategies [1, 3, 8, 14, 15, 17].
Many cases of Ebstein’s anomaly currently are detected before birth. These patients account for the majority of cases in the current population with neonatal Ebstein’s anomaly. Consequently, the diagnosis can be determined for a considerable number of patients before significant illness develops. Furthermore, cases with fatal presentations also can be detected before death. Therefore, it was necessary for a study to reinvestigate the clinical course of neonates with Ebstein’s anomaly in the recently changed diagnostic and therapeutic situation. This study aimed to determine the outcome variables in a series of recent neonatal Ebstein’s anomaly cases.
Methods
Subjects
Between January 2001 and June 2012, 80 children with Ebstein’s anomaly were managed in the Division of Pediatric Cardiology at the Asan Medical Center. The study enrolled 59 of these children who were neonates at their diagnosis. Patients with a corrected transposition of the great arteries or complex conotruncal anomalies were excluded from the study. Of the 59 subjects, 46 were born at our center. The remaining 13 had been transferred from other institutions. Ebstein’s anomaly was diagnosed prenatally in 46 subjects, 45 of whom were born at our center.
The medical records of the patients and their mothers, chest radiographs, and electrocardiograms were reviewed. Echocardiographic data from a review of stored images were obtained whenever possible. Otherwise, we referred to the examination reports. The Institutional Review Board of the Medical Center approved this retrospective study (2013–0008) and waived the need for patient consent.
Echocardiography
Morphologic classification of the tricuspid valve (types A to D) was based on a review of the echocardiographic images or the surgical/autopsy records according to the guidelines published by Carpentier et al. [3] and Chauvaud [6]. The echocardiographic grade of severity was calculated according to the guidelines of Celermajer et al. [4]. The ratio of the combined area of the right atrium and atrialized right ventricle to that of the functional right ventricle, left atrium, and left ventricle, as determined on a four-chamber view at end diastole, was used to define four grades of increasing severity: grade 1 (ratio, <0.5), grade 2 (ratio, 0.5–0.99), grade 3 (ratio, 1–1.49), and grade 4 (ratio, ≥1.5) [4].
The maximum diameter of the atrial septal defect was measured on any available echocardiographic view. Tricuspid regurgitation was assessed as more than moderate if the jet extended more than halfway from the valve to the posterior wall. The size of the ductus arteriosus was based on the ratio of its smallest diameter to the diameter of the left pulmonary artery, according to the guidelines of Wald et al. [15], with a ratio of 1 or higher defined as large, a ratio of 0.5–1 defined as moderate, and a ratio less than 0.5 defined as small. Left ventricular function was not analyzed as part of the study due to invariable paradoxic interventricular septal motion.
Statistical Analysis
The data are presented as frequencies or as medians with ranges. All statistical analyses were performed using SPSS 13.0 (SPSS Korea Data Solution, Seoul, South Korea). Statistical significance was defined as a p value lower than 0.05. Survival analysis was performed using the Kaplan–Meier method. Univariate analyses of the relationship between all presented independent variables and time to death were conducted using the Cox proportional-hazards model. Any variable with a p value lower than 0.10, as determined in the univariate analysis, was used as input in the multivariable model.
Results
Clinical Characteristics
Table 1 lists the clinical characteristics of the 59 patients at their initial presentation. The initial presentation types, except for the prenatal diagnosis, were cyanosis (n = 7, 12.3 %), cardiac murmur (n = 5, 8.5 %), and an abnormal heart shape on chest radiography (n = 1, 1.7 %). An associated pulmonary atresia was suspected in 21 of the 46 patients with a prenatal diagnosis. After birth, anatomic pulmonary atresia was confirmed in eight and functional pulmonary atresia in eight of these patients.
Fetal distress, defined by reviewing the results of a nonstress test, a contraction stress test, and a biophysical profile of the mother, was observed in nine patients. The birth of 17 patients occurred before the full gestation period of 37 weeks. The body weight of 17 patients was less than 2.5 kg.
Signs of low cardiac output (sinus tachycardia, metabolic acidosis, elevated serum lactate, low diastolic arterial pressure, and the need for inotropics) before any palliative or corrective procedure were present in 12 patients. Two patients had Wolff-Parkinson-White syndrome.
Echocardiographic Results
For two patients, postnatal echocardiographic examination was not possible because they died immediately after birth. The echocardiographic characteristics of the remaining 57 subjects at initial presentation are shown in Table 2. Other associated cardiac anomalies, except for atrial septal defect and patent ductus arteriosus, were pulmonary atresia in nine patients (including 1 case postnatally detected, number 10 in Table 3), pulmonary stenosis in four patients, ventricular septal defect in six patients, partial atrioventricular septal defect in one patient, and juxtaposition of the right atrial appendage in one patient. Functional pulmonary atresia was found in 16 patients, including 8 postnatally detected cases.
Procedures and Courses
A total of 25 patients survived without undergoing any procedure. Palliative or corrective surgical/interventional procedures were performed for 27 patients (Fig. 1). Procedures for biventricular repair were successful for 12 patients. Five patients underwent a one-and-a-half ventricular repair, but the Fontan operation was performed late for one of them. For four of the five patients who underwent a right ventricular exclusion procedure, a bidirectional Glenn shunt was performed. Additional surgical procedures such as reduction of the right atrium, patch closure of a ventricular septal defect, ductus arteriosus ligation, and pulmonary angioplasty were performed as needed.

Palliative or corrective surgical/interventional procedures performed for patients with neonatal Ebstein’s anomaly. All numbers indicate the number of patients. ASD atrial septal defect, BAS balloon atrial septostomy, PDA patent ductus arteriosus, RVOT right ventricular outflow tract, TVP/TAP tricuspid valvuloplasty/annuloplasty, BCPS bidirectional cavopulmonary shunt, RV right ventricular
Seven patients died before undergoing any surgical/interventional procedures, including five patients who died within several hours after birth, three of whom (including no. 5 in Table 3) experienced fetal distress. One patient died of severe heart failure aggravated by a refractory supraventricular tachycardia on day 10 after birth. One patient (no. 1 in Table 3) with an associated ventricular septal defect, pulmonary stenosis, and left ventricular failure died of severe biventricular failure at the age of 3 months while on a waiting list for heart transplantation.
Seven patients died after surgical/interventional procedures. Three of these patients (nos. 6, 7, and 9 in Table 3) had an associated pulmonary atresia and experienced hemodynamic deterioration after tricuspid annuloplasty/valvuloplasty. One patient (no. 8 in Table 3) died of a thrombosis involving a Blalock-Taussig shunt after right ventricular exclusion. Three unexpected sudden deaths occurred. The first patient, with cat-eye syndrome (no. 3 in Table 3), died at 4 months after widening of a right ventricular outflow tract. The second patient (no. 2 in Table 3) died at 7 months despite a successful interventional pulmonary valvotomy, and the third patient, with Down syndrome, died at the age of 7 years after tricuspid valvuloplasty.
Only one of the patients with a functional pulmonary atresia died. The characteristics of the patients with pulmonary stenosis/atresia are presented in Table 3. Biventricular repair was not possible for all the patients with pulmonary atresia. For two patients (nos. 10 and 11 in Table 3) classified as Carpentier type A, a one-and-a-half ventricular repair was possible.
Follow-up and Survival Analyses
The median follow-up period was 1.96 years (range, 0.0–10.4 years). Seven patients were lost to follow-up evaluation at a median age of 0.79 years (range, 0.32–2.77 years) when they were transferred to other hospitals after undergoing the required procedures. Their survival was confirmed, but only the observation period at our hospital was used in the survival analysis.
Supraventricular tachycardia was found in 11 patients (including nos. 4, 6, 7, and 12 in Table 3) and idioventricular rhythm in 1 patient (no. 9 in Table 3). A permanent pacemaker was inserted in two patients for the control of a postoperative Mobitz type 2 atrioventricular block and tachycardia-bradycardia syndrome (no. 12 in Table 3), respectively.
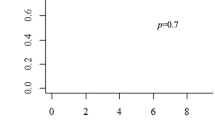
The overall mortality rate was 23.7 % (14/59). Of the 14 patients, 10 (including patients 5 and 9 in Table 3) died during their hospital stay, either after a procedure or after the diagnosis of Ebstein’s anomaly. The 1- and 5-year survival rates were 78.6 and 76.3 %, respectively (Fig. 2). Univariate analysis identified fetal distress (p = 0.002), prematurity (p = 0.036), low birth weight (p = 0.003), diameter of the atrial septal defect (p = 0.022), and pulmonary stenosis/atresia (p = 0.001) as the variables related to the time to death, whereas neither the Carpentier classification (p = 0.175) nor the Celermajer index (p = 0.958) was significant.

Survival after the diagnosis of Ebstein’s anomaly for 59 patients (Kaplan–Meier method)
In the multivariate analysis, fetal distress (p = 0.004) and pulmonary stenosis/atresia (p < 0.001) remained significant. Of the patients who survived, 45 met the criteria for New York Heart Association (NYHA) functional class 1 or 2 at the latest follow-up evaluation.
Discussion
This study showed that two extreme groups of patients (a group with no significant clinical presentation of illness during the neonatal period and a group with severe presentation) were included in the current population with neonatal Ebstein’s anomaly. Fetal distress and pulmonary atresia/stenosis were the significant risk factors for mortality among the patients with neonatal Ebstein’s anomaly in this study. Previously suggested risk factors of mortality were Celermajer index [4, 5], fetal presentation [5], right ventricular outflow obstruction [5, 16], functional pulmonary atresia [16], large atrial septal defect [16], and left ventricular dysfunction [16]. However, in a recent study by Barre et al. [2] of fetuses with Ebstein’s anomaly, retrograde flow through the pulmonary valve was a significant prognostic factor, whereas Celermajer index was not. In pulmonary atresia, which was also identified in our study as a significant prognostic factor, retrograde flow through the pulmonary valve cannot be present. Together, these findings suggest that the prognostic factors of Ebstein’s anomaly differ between fetuses and neonates.
According to our findings, fetal distress has an effect on mortality within several hours after birth and might be a risk factor during the immediate postnatal period. Because a preterm delivery aimed at preventing fetal distress would cause problems related to prematurity or low birth weight, the appropriate therapeutic strategy for these patients still is lacking. However, it is clear that adequate management of fetuses with Ebstein’s anomaly requires strict cooperation between obstetricians, neonatologists, pediatric cardiologists, and pediatric cardiac surgeons.
This study identified the size of the atrial septal defect as a risk factor for mortality by univariate but not multivariate analysis. A large atrial septal defect might be a formative result of the small size of a functional right ventricle or right ventricular outflow obstruction.
Left ventricular function, reported to be a prognostic factor of mortality [16], was not investigated in this study. However, according to Jaquiss and Imamura [7], left ventricular function may subsequently prove to be quite adequate after a right-sided pathology has been addressed surgically. Moreover, in one report, left ventricular noncompaction was not related to signs of left heart failure [10].
Despite some evidence that functional pulmonary atresia is a prognostic factor of mortality [16], it was not significant in this study. For patients with neonatal Ebstein’s anomaly and functional pulmonary atresia, we have applied a waiting approach without infusing prostaglandin E1 until spontaneous closure of the ductus arteriosus was achieved. By contrast, Wald et al. [15] proposed the relatively aggressive closure of a ductus arteriosus in patients who had Ebstein’s anomaly without anatomic obstruction. Although clinical status was stabilized in one of our patients with functional pulmonary atresia after surgical closure of a ductus arteriosus (Fig. 1), we have our reservations concerning the strategy advocated by Wald et al., given that other authors recommend a preservation approach to ductus arteriosus because it allows the stabilization of patients with neonatal Ebstein’s anomaly and functional pulmonary atresia [11, 12]. In the case reported by Santoro et al. [12], the right ventricular cavity was tiny, and the Celermajer index was grade 3.
The question concerning the best surgical strategy for neonates with Ebstein’s anomaly and pulmonary atresia cannot be answered clearly. According to the report by Knott-Craig et al. [9], biventricular repair was possible for three patients with anatomic pulmonary atresia (37.5 % of their subjects). On the other hand, Jaquiss and Imamura [7] recommended a univentricular approach in cases with an atretic pulmonary valve.
Biventricular repair failed in all nine of our patients with pulmonary atresia, including two for whom one-and-a-half ventricular repair was performed. We therefore do not recommend biventricular repair for patients with neonatal Ebstein’s anomaly and anatomic pulmonary atresia. However, in these cases, the initial procedure should not be a right ventricular exclusion because a one-and-a-half ventricular repair may be possible for patients with good right ventricular morphology. To establish an adequate surgical strategy for neonatal Ebstein’s anomaly with pulmonary atresia, additional studies based on a large number of patients are required.
We suggest a surgical decision algorithm (Fig. 3) in which Carpentier classification is adopted. We think it still is a valuable parameter in a surgical decision, although it was not a significant risk factor of mortality in this study.

Recommendation of surgical decision. The perinatal risk factors are prematurity, low birth weight, and fetal distress. If findings of right ventricular dysfunction are observed through close monitoring after any procedure, a change of surgical plan should be considered (dashed arrows). TVP/TAP tricuspid valvuloplasty/annuloplasty, BVR biventricular repair, RV right ventricular, BCPS bidirectional cavo-pulmonary shunt
The limitations of this study were those inherent to its retrospective design. For example, several variables could not be completely investigated or were not measured in some of the infants.
In conclusion, fetal distress and pulmonary atresia/stenosis are significant predictors of mortality in the current population with neonatal Ebstein’s anomaly. A close cooperation of associated clinicians is required for an improvement in outcome. To establish a better surgical strategy for patients with Ebstein’s anomaly and pulmonary atresia, studies of larger populations are required.
References
Atz AM, Nunoz RA, Adatia I, Wessel DL (2003) Diagnostic and therapeutic uses of inhaled nitric oxide in neonatal Ebstein’s anomaly. Am J Cardiol 91:906–908
Barre E, Durand I, Hazelzet T (2012) Ebstein’s anomaly and tricuspid valve dysplasia: prognosis after diagnosis in utero. Pediatr Cardiol 33:1391–1396
Carpentier A, Chauvaud S, Mace L, Relland J, Mihaileanu S, Marino JP, Abry B, Guibourt P (1988) A new reconstructive operation for Ebstein’s anomaly of the tricuspid valve. J Thorac Cardiovasc Surg 96:92–101
Celermajer DS, Cullen S, Sullivan ID, Spiegelhalter DJ, Wyse RKH, Deanfield JE (1992) Outcome in neonates with Ebstein’s anomaly. J Am Coll Cardiol 19:1041–1045
Celermajer DS, Bull C, Till JA, Cullen S, Vassillikos VP, Sullivan ID, Allan L, Nihoyannopoulos P, Soerville J, Deanfield JE (1994) Ebstein’s anomaly: presentation and outcome from fetus to adult. J Am Coll Cardiol 23:170–176
Chauvaud S (2000) Ebstein’s malformation: surgical treatment and results. Thorac Cardiovasc Surg 48:220–223
Jaquiss RDB, Imamura M (2007) Management of Ebstein’s anomaly and pure tricuspid insufficiency in the neonate. Semin Thorac Cardiovasc Surg 19:258–263
Knott-Craig CJ, Overholt ED, Ward KE, Razook JD (2000) Neonatal repair of Ebstein’s anomaly: indications, surgical technique, and medium-term follow-up. Ann Thorac Surg 69:1505–1510
Knott-Craig CJ, Overholt ED, Ward KE, Ringewald JM, Baker SS, Razook JD (2002) Repair of Ebstein’s anomaly in the symptomatic neonate: an evolution of technique with 7-year follow-up. Ann Thorac Surg 73:1786–1793
Oxenius A, Attenhofer Jost CH, Prêtre R, Dave H, Bauersfeld U, Kretschmar O, Seifert B, Balmer C, Valsangiacomo Buechel ER (2012) Management and outcome of Ebstein’s anomaly in children. Cardiol Young 15:1–8. doi:10.1017/S1047951112000224
Pflaumer A, Eicken A, Augustin N, Hess J (2004) Symptomatic neonates with Ebstein anomaly. J Thorac Cardiovasc Surg 127:1208–1209
Santoro G, Palladino MT, Russo MG, Calabrò R (2008) Neonatal patent ductus arteriosus recanalization and stenting in critical Ebstein’s anomaly. Pediatr Cardiol 29:176–179
Sarris EG, Giannopoulos NM, Tsoutsinos AJ, Chatzis AK, Kirvassilis G, Brawn WJ, Comas JV, Corno AF, Di Carlo D, Fragata J, Hraska V, Jacobs JP, Krupianko S, Sairanen H, Stellin G, Urban A, Ziemer G (2006) Results of surgery for Ebstein anomaly: a multicenter study from the European Congenital Heart Surgeons Association. J Thorac Cardiovasc Surg 132:50–57
Starnes VA, Pitlick PT, Bernstein D, Griffin ML, Choy M, Shumway NE (1991) Ebstein’s anomaly appearing in the neonate. A new surgical approach. J Thorac Cardiovasc Surg 101:1082–1087
Wald RM, Adatia I, Van Arsdell GS, Hornberger LK (2005) Relation of limiting ductal patency to survival in neonatal Ebstein’s anomaly. Am J Cardiol 96:851–856
Yetman AT, Freedom RM, McCrindle BW (1998) Outcome in cyanotic neonates with Ebstein’s anomaly. Am J Cardiol 81:749–754
Yun TJ, Lee SH, Ko JK (2006) Neonatal stenotic Ebstein’s anomaly: a novel technique of right ventricular exclusion. J Thorac Cardiovasc Surg 131:469–471
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Yu, J.J., Yun, TJ., Won, HS. et al. Outcome of Neonates with Ebstein’s Anomaly in the Current Era. Pediatr Cardiol 34, 1590–1596 (2013). https://doi.org/10.1007/s00246-013-0680-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00246-013-0680-x