
Abstract
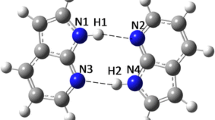
We analyzed the monohydration effect on the hydrogen-bonded structure between the adenine–thymine base pair using path integral molecular dynamics simulations including the nuclear quantum and thermal effects. We focused on two monohydration models for an adenine–thymine base pair with a water molecule bound to each adenine and thymine site. The adenine–thymine base pair without a water molecule was also discussed to reveal the role of a water molecule in monohydrated models. We found that the monohydration effect varies depending on the location of the water molecule. The monohydration effect on the inter-molecular motions is also investigated using the principle component analysis. The monohydration alters the inter-molecular motions of adenine–thymine base pair. We found that the nuclear quantum effect on the motion depends on the positions of the bound water molecule. The nuclear quantum effect on the hydrogen-bonded structure of adenine, thymine and water molecules is rather small, but we found significantly large nuclear quantum effect on the inter-molecular motions of the monohydrated base pair systems.



Similar content being viewed by others


References
Watson JD, Crick FHC (1953) Nature 171:737–738
Plützer C, Hünig I, Kleinermanns K, Nir E, Vries DSM (2003) Chem Phy Chem 4:838–842
Krishnan GM, Kühn O (2007) Chem Phys Lett 435:132–135
Plützer C, Hünig I, Kleinermanns K (2003) Phys Chem Chem Phys 5:1158–1163
Urashima S, Asami H, Ohba M, Saigusa H (2010) J Phys Cham A 114:11231–11237
Abo-Riziq A, Crews B, Grace L, Vries SM (2005) J Am Chem Soc 127:2374–2375
Bakker JM, Compagnon I, Meijer G, Helden G, Kabelác M, Hobza P, Vries MS (2004) Phys Chem Chem Phys 6:2810–2815
Guerra CF, Bickelhaupt FM, Sniders JG, Baerends EJ (2000) J Am Chem Soc 122:4117–4128
Cerón-Carrasco JP, Requena A, Michaux C, Perpete EA, Jacquemin D (2009) J Phys Chem A 113:7892–7898
Belau L, Wilson KR, Leone AR, Ahmed M (2007) J Phys Chem A 111:7562–7568
Rueda M, Kalko SG, Luque FJ, Orozco M (2003) J Am Chem Soc 125:8007–8014
Daido M, Koizumi A, Shiga M, Tachikawa M (2011) Theor Chem Acc 130:385–391
Daido M, Kawashima Y, Tachikawa M (2013) J Comp Chem 34:2403–2411
Jiří H, Jan R, Pavel H (2013) Chem Phys Lett 568–569:161–166
Antone M, Gerald M, Manuel FR (2014) J Chem Phys 141:034106
Tachikawa M, Shiga M (2005) J Am Chem Soc 127:11908–11909
Suzuki K, Shiga M, Tachikawa M (2008) J Chem Phys 129:144310
Suzuki K, Tachikawa M, Shiga M (2010) J Chem Phys 132:144108
Koizumi A, Suzuki K, Shiga M, Tachikawa M (2011) J Chem Phys 134:031101
Kawashima Y, Tachikawa M (2013) Chem Phys Lett 571:23–27
Ogata Y, Daido M, Kawashima Y, Tachikawa M (2013) RSC Adv 3:25252–25257
Sugioka Y, Yoshikawa T, Takayanagi T (2014) J Phys Chem A 117:11403–11410
Villani G (2005) Chem Phys 316:1–8
Villani G (2006) Chem Phys 324:438–446
Villani G (2008) J Chem Phys 128:114306
Kitao A, Go N (1999) Curr Opin Struct Biol 9:164
Potoyan DA, Papoian GA (2011) J Am Chem Soc 133:7405
Martyna GJ, Klein ML, Tuckerman M (1992) J Chem Phys 97:2635
Korth M (2010) J Chem Theory Comput 6:3808–3816
MOPAC2009, James JPS (2008) Stewart computational chemistry, Colorado Springs, CO, USA. http://OpenMOPAC.net
Srinivasan AR, Sauers RR, Fenley MO, Boschitsch AH, Matsumoto A, Colasanti AV, Olson WK (2009) Biophys Rev 1:13–20
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam MJ, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2009) Gaussian 09, revision D.01. Gaussian, Inc., Wallingford CT
Brooks B, Karplus M (1983) Proc Natl Acad Sci USA 80:6571–6575
Head-Gordon M, Pople JA, Frisch MJ (1988) Chem Phys Lett 153:503–506
Saebø S, Almlöf J (1989) Chem Phys Lett 154:83–89
Frisch MJ, Head-Gordon M, Pople JA (1990) Chem Phys Lett 166:275–280
Frisch MJ, Head-Gordon M, Pople JA (1990) Chem Phys Lett 166:281–289
Head-Gordon M, Head-Gordon T (1994) Chem Phys Lett 166:122–128
Dewar MJS, Zoebisch EG, Healy EF, Stewart JJP (1985) J Am Chem Soc 107:3902–3909
Stewart JJP (1989) J Comp Chem 10:209–220
Stewart JJP (1989) J Comp Chem 10:221–264
Stewart JJP (1991) J Comp Chem 12:320–341
Anders E, Koch R, Freunscht P (1993) J Comp Chem 14:1301–1312
Stewart JJP (2007) J Mol Model 13:1173–1213
Acknowledgments
This study was partly supported by a JSPS/MEXT KAKENHI Grant-in-Aid for Scientific Research.
Author information
Authors and Affiliations
Corresponding author
Additional information
Published as part of the special collection of articles derived from the 9th Congress on Electronic Structure: Principles and Applications (ESPA 2014).
Electronic supplementary material
Below is the link to the electronic supplementary material.
Appendix
Appendix
We analyzed the inter-molecular motions of buckle and propeller modes of adenine–thymine pair introducing a simple geometric model as shown in Fig. 4. The model is constructed with two plates \(h_{1} h_{2} r_{1} r_{2}\) and \(h_{1} h_{2} r_{3} r_{4}\) connected by two hinges \(h_{1}\) and \(h_{2}\). We assign 2a to both hydrogen-bond lengths and \(z_{0}\) to the distance between two hydrogen bonds in this model. We fix the plate \(h_{1} h_{2} r_{3} r_{4}\) on the z–x plane. The angles \(\theta_{1}\) and \(\theta_{2}\) are defined as angles between vector \(\overrightarrow {{r_{1} h_{1} }}\) and the z–x plane and vector \(\overrightarrow {{r_{2} h_{2} }}\) and the z–x plane, respectively. The angle between \(\overrightarrow {{{\text{OM}}_{1} }}\) and \(\overrightarrow {{{\text{M}}_{2} {\text{O}}}}\) is \(\phi_{1}\), and the angle between \(r_{1} - r_{2}\) and \(r_{3} - r_{4}\) is \(\phi_{2}\). The displacement of \(\phi_{1}\) and \(\phi_{2}\) corresponds to the buckle and propeller modes, respectively. These angles \(\phi_{1}\) and \(\phi_{2}\) are expressed as follows using \(\theta_{1}\) and \(\theta_{2}\).
and
When \(\theta_{1}\) and \(\theta_{2}\) are small, Eqs. (3) and (4) can be approximated as
and
The displacement ratio of buckle and propeller modes corresponds to the ratio of \(\phi_{1}\) and \(\phi_{2}\). The relation can be written as
Figure 5 shows the relation between \(\phi_{2} /\phi_{1}\) and \(\theta_{2} /\theta_{1}\) in Eq. (7). We set the parameter \(a\) to be \(a/z_{0} = 1\). Only the buckle mode exists when \(\theta_{1} = \theta_{2}\) and \(\phi_{2} /\phi_{1} = 0\), while only the propeller mode exists when \(\theta_{1} = - \theta_{2}\) and \(\phi_{2} /\phi_{1} = \infty\). In other areas, both buckle and propeller modes exist. The figure shows that the ratio of buckle and propeller modes may alter if the vibrational frequencies of the hydrogen-bond angle shift. The ratio depends on the difference between \(\theta_{1}\) and \(\theta_{2}\) and can rapidly change from buckle dominant motion (region around \(\theta_{1} = \theta_{2}\)) to propeller dominant motion (region around \(\theta_{1} = - \theta_{2}\)). Our finding indicates that the small difference in the hydrogen-bond structure may lead to large difference in molecular motion.

A simple geometric model for the buckle and propeller modes of the adenine–thymine pair

The relation between \(\phi_{2} /\phi_{1}\) and \(\theta_{2} /\theta_{1}\) in Eq. (7)
Rights and permissions
About this article

Cite this article
Watanabe, S., Ogata, Y., Kawatsu, T. et al. Effects of monohydration on an adenine–thymine base pair. Theor Chem Acc 134, 84 (2015). https://doi.org/10.1007/s00214-015-1686-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00214-015-1686-7