
Abstract
While in vitro testing is used to identify hazards of chemicals, nominal in vitro assay concentrations may misrepresent potential in vivo effects and do not provide dose–response data which can be used for a risk assessment. We used reverse dosimetry to compare in vitro effect concentrations-to-in vivo doses causing toxic effects related to endocrine disruption. Ten compounds (acetaminophen, bisphenol A, caffeine, 17α-ethinylestradiol, fenarimol, flutamide, genistein, ketoconazole, methyltestosterone, and trenbolone) have been tested in the yeast estrogen screening (YES) or yeast androgen-screening (YAS) assays for estrogen and androgen receptor binding, as well as the H295R assay (OECD test guideline no. 456) for potential interaction with steroidogenesis. With the assumption of comparable concentration–response ratios of these effects in the applied in vitro systems and the in vivo environment, the lowest observed effect concentrations from these assays were extrapolated to oral doses (LOELs) by reverse dosimetry. For extrapolation, an eight-compartment Physiologically Based Toxicokinetic (PBTK) rat model based on in vitro and in silico input data was used. The predicted LOEL was then compared to the LOEL actually observed in corresponding in vivo studies (YES/YAS assay versus uterotrophic or Hershberger assay and steroidogenesis assay versus pubertal assay or generation studies). This evaluation resulted in 6 out of 10 compounds for which the predicted LOELs were in the same order of magnitude as the actual in vivo LOELs. For four compounds, the predicted LOELs differed by more than tenfold from the actual in vivo LOELs. In conclusion, these data demonstrate the applicability of reverse dosimetry using a simple PBTK model to serve in vitro–in silico-based risk assessment, but also identified cases and test substance were the applied methods are insufficient.
Similar content being viewed by others

Introduction
The introduction of the “3Rs” principle, reduction, refinement and replacement, by Russell and Burch (1959), was instrumental in the development of alternative methods to animal experimentation. Since then, alternative methods have been developed and validated. One of the goals is to replace animal testing for toxicological hazard assessment and ultimate for risk assessment. The so-called Tox21 strategy is shifting the toxicological assessments away from traditional animal studies to target-specific, mechanism-based, biological observations largely obtained using in vitro assays (Tice et al. 2013). The overall toxicity of a compound in an in vivo organism is unlikely to be accurately reflected in a single stand-alone replacement model; rather, a battery of tests is required, such as those in the recently regulatory adopted strategy for skin sensitization (Bauch et al. 2011). Knowledge of physiological and toxicological pathways has allowed the development of adverse outcome pathways (AOPs), which describe the biological key events leading to an adverse outcome in vivo (Vinken 2013).
One adverse outcome of concern is interference of chemicals with sex hormone synthesis, regulation, and function, potentially disturbing reproduction and fetal development (WHO 2012). In 2012, the OECD has issued a guidance document on evaluating chemicals for endocrine disruption (OECD 2012). Since 1998, in the United States, the Environmental Protection Agency (EPA) requires a battery of in vitro and in vivo tests [developed from the Endocrine Disruptor Screening Program (EDSP)] for potential endocrine-disrupting chemicals (EDCs) (EPA 1998). The in vitro screening tests recommended by the EPA include an estrogen [yeast estrogen screening (YES)] or androgen [yeast androgen screening (YAS)] transcriptional activation assay (EPA 2009a; Kolle et al. 2010; OECD 2009a) and the steroidogenesis assay in the human derived cell line H295R (Kolle et al. 2010; OECD 2011). The first assays evaluate the effect of the compound on human steroid hormonal receptors, while the latter assesses any interference of a compound in the steroidogenesis pathway by measuring the steroid hormonal concentrations. Based on the known similarity of the steroid receptors in rats and humans (Chang et al. 1988; Sun et al. 2014), as well as on the common biochemical pathway of steroidogenesis in mammals, these in vitro data should also be predictive for the rat (Ankley and Gray 2013; Sun et al. 2008, 2014). The in vivo studies include one-generation [OECD test guideline (TG) 415 (OECD 1983)] and two-generation [OECD TG 416 (OECD 2001)] studies; the Hershberger [OECD 441 (OECD 2009b)]; and the uterotrophic [OECD 440, OECD 2007)] assays and also the in vivo pubertal assay. In addition, the in vitro assays provide information on the possible mechanisms of action of the endocrine activity observed in the in vivo tests (OECD 2009a). In this study, literature data for the YES/YAS and steroidogenesis in vitro assays from Kolle et al. (2012) were used to detect potential endocrine disruption.
While in vitro testing can provide an efficient way to identify potential hazards of chemicals, nominal in vitro assay concentrations may misrepresent potential in vivo effects (Wetmore et al. 2012) and do not provide dose–response data which can be used for a risk assessment. Therefore, an in vitro-to-in vivo extrapolation (IVIVE) that translates in vitro concentration–effect curves into in vivo dose–response curves, the so-called “reverse dosimetry approach”, is needed (Wetmore et al. 2012; Louisse et al. 2017; Paini et al. 2017). To investigate whether and how in vitro toxicity data can be used and extrapolated using reverse dosimetry to in vivo toxicity, we used data from the endocrine disruption assays in an in vitro–in silico-based concept. Accordingly, the lowest concentration that caused an effect in the toxicity assays in the absence of cytotoxicity or cross reactivity was defined as the in vitro point of departure (PoD). With the assumption of comparable concentration–response ratios of the addressed endocrine effects in the applied in vitro systems and the in vivo environment, this “lowest observed effect concentration (LOEC)” was then extrapolated to an in vivo oral dose using an eight-compartment PBTK model for the rat (including the potential target organs, namely, the adrenals and ovaries/testes). Thus, it is possible to use in vitro LOEC values to determine in vivo PoDs, which are required for risk assessment. This strategy has been applied to a number of toxicological endpoints (Punt et al. 2011) including developmental toxicity (Li et al. 2017; Louisse et al. 2010, 2015; Strikwold et al. 2013, 2017; Verwej et al. 2006); genotoxicity (Paini et al. 2010), acute (and repeated dose) toxicity and hepatotoxicity (Gubbels-van Hal et al. 2005); nephrotoxicity (Abdullah et al. 2016), neurotoxicity (DeJongh et al. 1999a, b; Forsby and Blaauboer 2007); and, more recently, endocrine disruption, which focused on (anti)estrogenicity (Zhang et al. 2018).
PBTK models can be used to acquire more kinetic information across species and for IVIVE (Paini et al. 2017). However, they are often focused on the modelling of a single chemical and adapted as specifically as possible for that compound. Therefore, we evaluated and applied a simple, transparent, and non-commercial PBTK model that could also be easily adapted to other toxicological endpoints. A set of 10 compounds were used in the evaluation, which were selected from a published dataset on YES/YAS and steroidogenesis studies (Kolle et al. 2010, 2012). In addition to in vitro hepatic metabolism data, in vivo rat endocrine disruption toxicity data were available for all compounds to compare in vitro–in silico-derived LOEALs with experimental data in rats.
Materials and methods
Test compounds
Test compounds were selected from a panel of compounds previously tested in steroidogenesis and YES/YAS in vitro assays to detect the potential for endocrine disruption (Kolle et al. 2012). From all compounds tested by Kolle et al. (2012), 10 compounds (Table 1) were selected based on the (1) internal in vitro database, (2) availability of input parameters for PBTK modeling, and (3) on available in vivo data (lowest observed effect levels, LOELs) in the rat for the evaluation of the in vitro–in silico-based dose–response description to facilitate risk assessments.
In vitro: LOEC
In the YES/YAS assays, each compound was analyzed for estrogen or androgen receptor-dependent reporter enzyme activity (agonistic and/or antagonistic). In the steroidogenesis assay, the effects of each compound on hormone synthesis of H295R human adrenocortical carcinoma cells were determined by measuring estradiol and testosterone levels (Kolle et al. 2012). In a conservative approach, the LOEC was set as the lowest concentration from the available in vitro data, in which an effect was observed without cytotoxicity or cross reactivity. If the lowest effect concentration in vitro was similar for different test principles as for BPA or GEN, the most sensitive lowest effect level was taken from available in vivo tests following a worst-case assumption. If the highest concentration tested was without effect in the absence of cytotoxicity, the LOEC was defined as above the highest concentration tested.
PBTK modeling
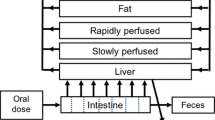
An eight-compartment model set up for male and female rats was applied to describe kinetics and distribution of the test compounds. The developed model included the target tissues/organs for endocrine disruption evaluated in vitro: adrenals and ovaries/testes. The principle of the model is shown in Fig. 1. Differential equations were used to describe a time-dependent mass balance for each defined compartment of the organism. The equations of the model are shown in Table 2.

Principle of the applied eight-compartmental physiologically based toxicokinetic (PBTK) model
In the model, the chemicals enter the bloodstream as a first-order process directly via the liver after uptake in the gastrointestinal tract. The distribution was based on diffusion. Hepatic metabolic clearance was integrated into the applied PBTK model and reflected the overall clearance of the test compound from the organism. The unbound fraction of the test compound was taken into account for the quantification of metabolic clearance. For parent compound, biliary and renal clearance are not considered in the applied PBTK model, based on the assumption that clearance via these pathways is mainly based on excretion of metabolites and is therewith addressed by hepatic metabolism as its prerequisite principle. The model is focused on the kinetics of the test compound, which reflects a postulated, parent compound-linked effect. Since in the applied in vitro systems, the identity of the metabolites may be unknown and the amounts negligible (OECD 2011; Routledge and Sumpter 1996), this approach is assessed to be appropriate within the applied concept of in vitro-to-in vivo extrapolation.
For the calculations, the oral dose was set to 50 mg/kg bw. This was an arbitrarily set dose to calculate corresponding Cmax values in plasma. Since all input parameters in the PBTK model are linear with dose, for each compound, a calculated constant Cmax/dose ratio can be used for reverse dosimetry by back-calculating the oral doses from the lowest effect levels in vitro (set to Cmax values) by the rule of proportion. The differential equations were solved using the software Berkeley Madonna™, version 8.3.18 (developed by Macey et al. 2009). All the compounds were analyzed in one task using the built ‘batch-run’. Microsoft Excel 2013 (Microsoft®) was used to import and analyze the data.
The target output parameters were the maximal concentrations (Cmax) in plasma (mean value for males and females) and in the target tissues (ovaries/testes and adrenal glands). To evaluate the PBTK predictions, the results obtained for Cmax in plasma were compared with measured Cmax from rats, extracted from the literature. As described above, in the applied PBTK model, the Cmax/dose ratio is constant and Cmax values for a given dose, from rat studies, can be calculated in a linear approach from the results obtained at the modeled dose of 50 mg/kg bw by the rule of proportion.
Physiological input parameters
The physiological parameters, including body weight, organ volumes, cardiac output, and blood flows, were taken from the literature (Brown et al. 1997; Davies and Morris 1993) and from in house data for male and female Wistar rats (Crl:Han, Charles River, Sulzfeld, Germany). Details are listed in Table 3.
Physicochemical input parameters
The physicochemical input parameters of the test compounds consist of the octanol/water partition coefficient log Kow and molecular weight (Table 1). Log Pow data were predicted using the ALOPGS 2.1 software. The tissue partition coefficients, used to describe and model the distribution between blood and defined tissues, were calculated based on the physicochemical parameters by the following equations as given by DeJongh et al. (1997):
Equation (1) was used for liver, kidneys, adrenals, ovaries/testes, and richly perfused tissues, Eq. (2) for fat and Eq. (3) for poorly perfused tissue.
Kinetic and metabolic input parameters
Intestinal absorption was predicted using a QSAR model based on Caco-2 data described by the following equation given by Hou et al. (2004):
where Papp is the apparent permeability coefficient and PSA is the polar surface area. PSA values were obtained in ChemAxon public database (2016). The results of this calculation in cm/s were thereafter converted to Papp in (cm/s)10−6.
Metabolic clearance was based on hepatic clearance reported in the literature or determined in S9 subcellular fractions of livers from Wistar rats at Cyprotex, Alderley Park, UK (see Table 4). Hence, the intrinsic clearance (CLint) in this model was based on hepatocytes, microsomal, or S9 subcellular hepatic fractions. Since some of the chemicals were expected to be directly conjugated, the microsomal and liver S9 incubations were used that contained cofactors for glucuronidation (UDPGA) and sulfation (PAPS), as well as NADPH for oxidation reactions. CLint data were normalized for hepatocytes as clearance per 106 cells, and for S9 and microsomal fraction as clearance per mg of protein. To estimate the in vivo hepatic clearance, the CLint values were scaled up using the factors of 135*106 cells/g liver for hepatocytes (Houston 1994); 91.3 and 50 mg protein/g liver for liver S9-fraction and microsomes, respectively (BASF internal data).
The fraction unbound to protein in the plasma (fup) was experimentally determined by performing rapid equilibrium dialysis (RED). Briefly, each test compound was incubated with rat plasma to a final concentration of 5 µM (1% DMSO) in a volume of 300 µL into a donor well of a RED plate in duplicates. 500 µL of DPBS was added to the receiver well of the plate. For the dialysis, this plate was sealed and incubated under shaking (300 rpm) at 37 °C with 5% CO2 for 6 h (Thermo Scientific 2012). A sample of 200 µL of each well was collected and frozen at − 40 °C until analysis. Warfarin (WAR) was used as a reference compound, which is known for its high protein binding (Waters et al. 2008; van Liempd et al. 2011; Zhang et al. 2012). The samples were analyzed using HPLC-MS/MS at Pharmacelsus, Saarbrücken, Germany. Applied methods are described in supplementary material 1.
The fup data were used to calculate the hepatic clearance in the model using the following equation described by Houston (1994):
where CLH is the hepatic clearance, CL is the intrinsic clearance scaled up, QL is the liver blood flow, AMint is the arterial blood metabolic intrinsic rate, and CVL is the concentration in the venous blood leaving the liver.
In vivo LOEL dose
The LOEL doses were extracted from literature data for the respective endpoints of endocrine disruption evaluated in the in vitro tests. The lowest dose levels were extracted based on (1) data availability and confirmed adequate studies which followed recommended guidelines; (2) oral route of administration, by gavage rather than feeding; and (3) the appropriate assay for determined in vitro LOEC.
The uterotrophic [OECD guideline 440 (OECD 2007)] or Hershberger [OECD guideline 441 (OECD 2009b)] tests were used to assess interference with estrogen or androgen receptors, respectively. The in vivo pubertal assay OECD guidance document 150 (OECD 2018a), EPA guidelines (EPA 2009b, c), or one- and two-generation studies OECD guidelines 416 and 443 (OECD 2001, 2018b) were chosen to evaluate the interference with steroid hormone synthesis, for which the main endpoints are hormone levels, vaginal opening (for females) and preputial separation (for males) observed in juvenile animals or in the offspring exposed in utero (Hayes et al. 2010). When the LOEC corresponded to a concentration from more than one assessed in vitro system, the LOEL was taken from any of the available in vivo tests described above. This approach is based on the assumption that LOECs in vitro take into consideration data assessments derived from the defined respective endpoints, such as steroid–receptor interaction and/or interaction with steroidogenesis.
Reverse dosimetry: IVIVE
Results from PBTK modelling and LOECs from in vitro experiments were used for IVIVE. In a linear manner, dose levels were calculated for respective LOECs based on the estimated plasma Cmax versus dose plot results from PBTK. The calculations are summarized in Fig. 2. For this analysis, the non-protein bound, free fraction of a test compound at the time point of the Cmax in plasma was considered as correlated tor the endocrine induced effects. In addition, extrapolated dose levels in plasma were compared with in vivo LOELs to evaluate the accuracy of the applied PBTK model.

In vitro-to-in vivo extrapolation (IVIVE) based on the in vitro LOEC concentration and Cmax in plasma
Sensitivity analysis
The sensitivity analysis of the model was performed for all compounds based on the description of Evans and Andersen (2000). In this approach, the sensitivity coefficient (SC) is defined by the initial maximum concentration (C) after prediction for plasma or tissue, the initial parameter of the model (P), the maximum concentration after increasing the parameter value by 5% (C′), and the changed parameter (P′) as shown below:
The resulting sensitivity coefficients were analyzed using Microsoft Excel 2013 (Microsoft®). The input parameter was considered to significantly affect the model output when the SC absolute value was higher than 0.5 (Rietjens et al. 2011). The output parameter assessed was the Cmax in plasma.
Results
Determination of in vitro LOEC and in vivo LOEL
For the selected compounds, the results of each in vitro assay including the thereof derived LOECs are shown in Table 5. APAP and CAF did not show any effects in these assays, and consequently, their LOECs for IVIVE were set to the highest concentrations tested without cytotoxicity (> 100 µM for both compounds). The LOECs were taken from YES/YAS for four compounds (EE, FLU, MTT, and TRE) and from steroidogenesis assay for two compounds (FEN and KET). The lowest concentrations in which an effect was observed were equal among the assay systems for two compounds (BPA and GEN).
In vivo LOELs for the defined endpoints with the respective assay are shown in Table 4. For APAP and CAF, no endocrine effects were described in literature and it was not possible to attribute any LOELs for the chosen endpoints. The LOELs were taken from uterotrophic/Hershberger assays for six compounds (BPA, EE, FLU, GEN, MTT, and TRE). Data from pubertal assay were taken for two compounds (FEN and KET).
PBPK model
Input parameters
Data for permeability and clearance for each compound are summarized in Table 5, together with their respective sources. Experimental data for fup, which was used for PBTK modelling, and the corresponding available literature data are also presented in Table 5. For BPA, FEN, GEN, KET, and TRE, literature values are slightly higher than experimentally determined fup. The highest difference was noted for KET, which is known for its high binding capacity. However, the experimentally determined fup for the reference compound WAR (0.4 ± 0.2%, mean ± standard deviation between 3 experiments) corroborates the literature data (< 1%, Waters et al. 2008; van Liempd et al. 2011; Zhang et al. 2012) and shows the reproducibility of the performed experiments.
Output parameters: predicted plasma C max
The predicted plasma Cmax using PBTK modelling were compared to in vivo data for six compounds for which data were available (Table 6). The PBTK model predicted the plasma Cmax for 67% of the test compounds (4/6—APAP, CAF, EE, and KET) in the same order of magnitude as that in vivo. BPA and GEN modeled concentrations were one order of magnitude higher than literature in vivo values. Further details are summarized in Table 6.
In vitro-to-in vivo extrapolation (IVIVE)
For 8 compounds, the IVIVE approach predicted doses in plasma that were correlated to the in vivo LOEL from literature (Table 4). For the evaluation, compounds with values that were within tenfold of the in vivo LOEL were considered to be correctly predicted. The correctly predicted compounds were BPA, FEN, GEN, and KET. Two compounds were over-predicted (MTT and TRE) and two compounds were under-predicted (EE and FLU). There was no trend between compounds under or over-predicted in terms of physicochemical properties or clearance pathways. Using this decision criterion, 50% of the test compounds (4/8) were correctly predicted. Assuming no literature evidence in vivo for the assessed endpoints for correct negative compounds in the in vitro assays (APAP and CAF), the current IVIVE model correctly predicted 60% (6/10) using this in vitro in silico-based risk assessment approach.
Sensitivity analysis
A sensitivity analysis of the model was performed for all compounds (supplementary data, Table S1). Sensitivity analysis demonstrated that, generally, the intestinal permeability and the lipophilicity input parameters highly influenced the output parameter, Cmax in plasma. SC absolute values were higher than 0.5 for Log Kow for BPA, EE, FEN, FLU, GEN, KET, MTT, and TRE for intestinal absorption (quantified via Papp) for APAP, BPA, EE, FEN, GEN, KET, MTT, and TRE.
Discussion
In the current case study, we followed basic principles of the concept of animal-free risk assessment and used, as starting point, available in vitro data for endpoints of endocrine disruption. Although the species of interest for animal- free risk assessment is human, especially when taking tests in human in vitro systems into consideration, we applied the principle of in vitro-to-in vivo extrapolation for rats. We did this to be able to assess obtained in vitro–in silico-based results versus available literature data from corresponding standardized in vivo tests in the rat. For this purpose, we applied PBTK modeling for an in vitro–in silico-based risk assessment for a set of 10 compounds. A first assumption was that the concentration–response ratios of the addressed endocrine effects in the applied in vitro systems were comparable to the in vivo environment. This assumption is a general issue in in vitro toxicology and drug discovery (Smith et al. 2010; Lu et al. 2011) and is a prerequisite for IVIVE, since the concentration–effect ratio is given for the test substance concentration in the buffer/medium and the readout for in vitro testing and is related directly to plasma concentration–effect ratios in vivo. As shown for endocrine effects of 17β-estradiol (E2) and BPA based on YES assays, results of these in vitro tests yielded better in vitro in vivo correlation than the MCF-7/BOS proliferation assay or the U2OS ER-CALUX assay (Zhang et al. 2018). The LOEC in vitro was extrapolated to an oral dose by reverse dosimetry applying a PBTK model for the rat. This correlates to a predicted LOEL in vivo, which was compared with the measured LOELs in in vivo studies for endocrine disruption to gain experience on the predictivity of such a concept and therewith knowledge on its potential future applicability.
As a simple approach, it was assumed that for each compound, the LOEC in the in vitro tests indicates the most sensitive endpoint and is a relevant parameter for risk assessment. The lowest LOEC values were from YES or YAS binding assays for EE, KET, MTT, and TRE; therefore, the corresponding in vivo endpoints used for comparison were the uterotrophic or Hershberger assay, respectively. For FEN and FLU, the LOEC values were from the steroid mapping assays. Consequently, predicted LOEL values were compared with the LOELs from the in vivo pubertal assay or one- and two-generation studies. APAP and CAF did not show clear effects in the in vitro assays up to the highest concentrations tested. Therefore, the highest concentration was translated into a corresponding in vivo dose that should be considered not to induce any endocrine effect.
For the applied IVIVE approach, it was assumed that a concentration (not area under the curve)-related endocrine effect was due to the parent compound. This is justified by in vitro tests that take receptor binding and/or enzyme interactions, both established as typical concentration-driven processes, as relevant modes of action into account. In addition, the nominal compound concentration is assumed to be responsible for effects in tests which have limited metabolic functions (Coecke et al. 2006). However, the understanding of exposure in in vitro testing becomes critical in the applied concept, since the nominal dose may not be the effective-free concentration if it binds to the plastic culture vessel or medium components, or evaporates (Groothuis et al. 2013). The assumption that the parent compound causes the toxic effect also implies that its metabolites do not contribute to the endocrine effects. The relevance of metabolic activation (Dekant 2009) is widely recognized and ways to implement this in vitro systems are described generally (Landsiedel et al. 2011), but these principles are not yet universally applied.
For the IVIVE, the maximum plasma concentration (Cmax) after a simulated single oral dose was adjusted to equal to the LOEC in vitro. This is a simplified and straightforward approach for compounds with short half-lives. However, this approach has a number of caveats. First, it may fail for compounds that accumulate, resulting in significantly higher steady-state concentrations after multiple dosing than after single dosing. Second, it is assumed that the test compounds do not induce or inhibit metabolizing enzymes in the liver, which would also significantly change compound kinetics after multiple dosing compared to single dosing. In addition, inter-individual differences in kinetics that may be addressed in Monte Carlo models are not taken into consideration in the presented basic IVIVE approach.
For PBTK modeling, Cmax in plasma was defined as the dose metric related to the lowest nominal effect concentrations in the respective in vitro assay. With the assumption that medium in the in vitro test system mirrors blood in a living organism, both providing nutrients to the cells and distributing the test substances within the system to the cells, this compartment was chosen to bridge the nominal in vitro concentrations to in vivo. The targets of the endocrine effects (ovaries, testes and/or adrenals) were added to the PBTK model as a future option, but should then be linked to in vitro effect concentrations of test substances in the tested cells or directly at the site of action. Based on available literature data that describe the observed effects as a function of nominal concentrations, this approach may be followed by the application of in vitro dosimetry concepts as described by (Groothuis et al. 2013), but was not addressed within the current case study. Since for the investigated potential endocrine disruptors, the investigated key events of receptor binding and enzyme interactions are concentration, not AUC-mediated effects, the maximum-free concentration of the test compound in plasma was applied for extrapolation. This approach reflects a worst-case scenario, since Cmax as dose metric results in the lowest possible estimated LOEL.
For in vitro-to-in vivo extrapolation, plasma protein binding was taken into consideration and the free concentration of the test compound in plasma was calculated. It was assumed that the free fraction of compound affects the toxicological activity (Smith et al. 2010). This implies that the compound: (1) does not induce irreversible inactivation of the target, e.g., by covalent binding; (2) does not act via multiple mechanisms and by activation of target-mediated events; and (3) has an equal action/potency in the in vitro assays, as it does in vivo. Rapid equilibrium dialysis (RED) was chosen for the determination of the unbound fraction of the compound in plasma, because in our experiments, this method was robust in respect of recovery, replicability, and obtained results of the positive control Warfarin (Zhang et al. 2012).
Further assumptions for PBTK modeling were that hepatic clearance drives the overall clearance of the test compound in the organism and other clearance mechanisms were not taken into consideration. This is true for many compounds, such that extrahepatic metabolism is minor compared to hepatic metabolism (Gundert-Remy et al. 2014; Oesch et al. 2018), and the metabolites, not the parent compound, are excreted via urine and/or bile. Consequently, in the overall clearance process, metabolism is the time critical process of elimination. This assumption is likely to be valid for most of the modeled compounds in our analysis, since the major metabolites of most of the test compounds are either oxidized and/or direct glucuronic acid and/or sulfate conjugates, which are excreted in the feces or urine (see Table 1 for metabolic and excretion pathways). The exception to this was EE and GEN, whereby intestinal first-pass metabolism contributes extensively to their overall metabolism in the rat, such that conjugation in the intestine reduces significantly their bioavailability (Hirai et al. 1981; Schwenk et al. 1982; Sfakianos et al. 1997). Since the current IVIVE concept did not take extrahepatic metabolism of EE or GEN into account and assumes direct uptake of the compounds from the GI tract, the model could consequently underestimate the extrapolated LOEL, although this was not the case for EE and GEN (Table 4). Therefore, the total clearance of the modeled compounds in this evaluation may be well reflected by their hepatic clearance. In this strategy, intrinsic clearance was linearly correlated to substrate concentrations and for higher plasma concentrations, this approach may overestimate hepatic clearance. Therefore, it may be considered for a potential higher tier modeling to apply Michaelis Menten parameters for the description of the kinetics of the metabolism of the test compound if available, or to determine them in appropriate experiments.
Another assumption of the PBTK modeling is that distribution is based on diffusion and the resulting steady-state concentrations are described by blood/tissue partition coefficients that are derived from physicochemical parameters of the test compounds, as described by (Jones and Rowland-Yeo 2013). This means that active transport was not addressed for modeling uptake and distribution of the test compound between defined compartments. Hence, this diffusion-based modeling is correct only if active transport processes are negligible. Although for the current modeled test compounds, APAP, EE, KET, BPA, and GEN are described in the literature to be substrates or inhibitors of active transporters (Manov et al. 2006; Zamek-Gliszczynski et al. 2011; Englund et al. 2014; Mazur et al. 2012; Ge et al. 2017), the quantitative input in the overall kinetics of these compounds is difficult to judge and should be addressed in future works.
Sensitivity analysis demonstrated that, generally, permeability (based on Papp), and log Kow input parameters highly influenced the output parameter cmax in plasma. First, the SC absolute value was higher than 0.5 for Log Kow for 8 of 10 substances (BPA, EE, FEN, FLU, GEN, KET, MTT, and TRE). Likewise, the SC absolute value was higher than 0.5 for Papp for also 8 of 10 substances (APAP, BPA, EE, FEN, GEN, KET, MTT, and TRE). SC absolute values were lower than 0.5 for all other investigated input parameters. In general, hepatic clearance of the compounds had minor influence on Cmax. This means that with respect to the kinetics of the test compounds, absorption and partitioning of the compounds are the main drivers of Cmax. Therefore, special attention should be given to these input parameters when the current dose metric is applied. It should be mentioned here that changing the dose metric will also change the sensitivity of input parameters and AUC or average plasma concentration, which are more dependent on clearance than Cmax.
Plasma kinetics data from in vivo experiments in rats were available for 6 of the 10 test compounds and were used to compare with predicted Cmax values (see Table 6). The modeled Cmax values for APAP, CAF, EE, and KET are in general accordance with the measured in vivo data and differ by less than threefold. By contrast, predicted Cmax values for BPA and GEN clearly overestimate the in vivo Cmax by more than tenfold. Thus, modeled plasma concentrations could be considered valid (within threefold of the in vivo value) for 4 out of 6 compounds, i.e., 66% were correctly predicted. These results are similar to those of a study in which a six-compartment rat PBTK model was used for a set of active ingredients of plant protection products (50% were correctly predicted) (Fabian et al. 2015).
Using reverse dosimetry to predict the in vivo LOEL for endocrine disruption, 6 of 10 LOELs were predicted within the correct order of magnitude. The predicted LOELs differed by more than tenfold from the described in vivo value for 4 compounds, 2 of which were over and 2 were under-predicted. Interestingly, for EE, although the LOEL estimation was more than tenfold higher than the in vivo value, the calculation of its Cmax was well predicted. This implies that the in vitro result may not reflect a relevant value. In contrast to EE, the LOEL estimations for BPA and GEN were within tenfold of the in vivo value, whereas the predictions of their Cmax values were more than tenfold higher than in vivo values. This observation is interesting, since a correct prediction of an LOEL based on PBTK modelling is critical in respect of the defined assumptions of the applied strategy. The LOELs for FLU, MTT, and TRE were not well predicted, with more than one order of magnitude difference between the modeled and observed in vivo values (in vivo Cmax values were unavailable for these three compounds). Reasons for these lacks of correlation could be limitations of the applied models which do not consider, e.g., deviation from the assumed linear kinetics of hepatic clearance, extrahepatic metabolism, enterohepatic recirculation, renal clearance, and active transport of the compound (as described above).
Endocrine disruption served as an example to correlate concentrations causing in vitro effects-to-in vivo effect doses using IVIVE reverse dosimetry. This IVIVE concept is also applicable to other toxicological effects. To make risk assessments based on in vitro and in silico methods widely applicable and acceptable, those cases or test substances, for which the IVIVE predictions (or in vitro models) are not correlating with in vivo data, need to be reliably identified and the inadequacies of the models need to be clarified. This can then be used to amend and improve the methods accordingly.
References
Abdelwahab NS, Elshemy HAH, Farid NF (2018) Determination of flutamide and two major metabolites using HPLC-DAD and HPTLC methods. Chem Cent J 12(1):4
Abdullah R, Alhusainy W, Woutersen J, Rietjens IMCM, Punt A (2016) Predicting points of departure for risk assessment based on in vitro cytotoxicity data and physiologically based kinetic (PBK) modeling: the case of kidney toxicity induced by aristolochic acid I. Food Chem Toxicol 92:104–116
Abu-Qare AW, Abou-Donia MB (2001) A validated HPLC method for the determination of pyridostigmine bromide, acetaminophen, acetylsalicylic acid and caffeine in rat plasma and urine. J Pharm Biomed Anal 26(5–6):939–947. https://doi.org/10.1016/S0731-7085(01)00448-4
An JH, Lee HJ, Jung BH (2012) Quantitative analysis of acetaminophen and its six metabolites in rat plasma using liquid chromatography/tandem mass spectrometry. Biomed Chromatogr 26(12):1596–1604. https://doi.org/10.1002/bmc.2737
Ankley GT, Gray LE (2013) Cross-species conservation of endocrine pathways: a critical analysis of tier 1 fish and rat screening assays with 12 model chemicals. Environ Toxicol Chem 32(5):1084–1087
Arms AD, Travis CC (1988) Reference physiological parameters in pharmacokinetic modeling. Office of Health and Environmental Assessment, Office of Research and Development U.S. Environmental Protection Agency, Washington (EPA/600/6-88/004)
Bauch C, Kolle SN, Fabian E, Pachel C, Ramirez T, Wiench B, Wruck CJ, van Ravenzwaay B, Landsiedel R (2011) Intralaboratory validation of four in vitro assays for the prediction of the skin sensitizing potential of chemicals. Toxicol In Vitro 25(6):1162–1168
Berry LM, Roberts J, Be X, Zhao Z, Lin MH (2010) Prediction of Vss from in vitro tissue-binding studies. Drug Metab Dispos 38(1):115–121. https://doi.org/10.1124/dmd.109.029629
Brown RP, Delp MD, Lindstedt SL, Rhomberg LR, Beliles RP (1997) Physiological parameter values for physiologically based pharmacokinetic models. Toxicol Ind Health 13(4):407–484
Bruce NW (1976) The distribution of blood flow to the reproductive organs of rats near term. J Reprod Fertil 46(2):359–362
Chang CS, Kokontis J, Liao ST (1988) Molecular cloning of human and rat complementary DNA encoding androgen receptors. Science 240(4850):324–326
ChemAxon public database (2016) https://chemaxon.com/. Accessed 01 Feb 2018
Chen C-Y, Bakhiet RM (2006) Age decreased steady-state concentrations of genistein in plasma, liver, and skeletal muscle in Sprague-Dawley rats. Mech Ageing Dev 127(4):344–348. https://doi.org/10.1016/j.mad.2005.12.003
Coecke S, Ahr H, Blaauboer BJ, Bremer S et al (2006) Metabolism: a bottleneck in in vitro toxicological test development. The report and recommendations of ECVAM workshop 54. Altern Lab Anim 34:49–84
Coldham NG, Sauer MJ (2000) Pharmacokinetics of [(14)C]Genistein in the rat: gender-related differences, potential mechanisms of biological action, and implications for human health. Toxicol Appl Pharmacol 164(2):206–215. https://doi.org/10.1006/taap.2000.8902
Cyprotex (2014) Cyprotex metabolic stability measurements in S9 subcellular fraction (contract measurement). Macclesfield
Davies B, Morris T (1993) Physiological parameters in laboratory animals and humans. Pharm Res 10(7):1093–1095
DeJongh J, Verhaar HJ, Hermens JL (1997) A quantitative property–property relationship (QPPR) approach to estimate in vitro tissue-blood partition coefficients of organic chemicals in rats and humans. Arch Toxicol 72(1):17–25
DeJongh J, Forsby A, Houston JB, Beckman M, Combes R, Blaauboer BJ (1999a) An integrated approach to the prediction of systemic toxicity using computer-based biokinetic models and biological in vitro test methods: overview of a prevalidation study based on the ECITTS project. Toxicol In Vitro 13:549–554
DeJongh J, Nordin-Andersson M, Ploeger BA, Forsby A (1999b) Estimation of systemic toxicity of acrylamide by integration of in vitro toxicity data with kinetic simulations. Toxicol Appl Pharmacol 158:261–268. https://doi.org/10.1006/taap.1999.8670
Dekant W (2009) The role of biotransformation and bioactivation in toxicity. In: Luch A (ed) Molecular, clinical and environmental toxicology vol 1. Birkhäuser Publishing, Basel, pp 57–86
Englund G, Lundquist P, Skogastierna C, Johansson J, Hoogstraate J, Afzelius L, Andersson TB, Projean D (2014) Cytochrome p450 inhibitory properties of common efflux transporter inhibitors. Drug Metab Dispos 42(3):441–447. https://doi.org/10.1124/dmd.113.054932
EPA (1998) Endocrine Disruptor Screening Program (EDSP) 1998 Federal Register Notices. https://www.epa.gov/endocrine-disruption/endocrine-disruptor-screening-program-edsp-1998-federal-register-notices. Accessed 20 Feb 2018
EPA (2009a) EPA guideline 890.1300. Endocrine Disruptor Screening Program Test Guidelines—OPPTS 890.1300: Estrogen Receptor Transcriptional Activation (Human Cell Line (HeLa-9903)) [EPA 740-C-09-006]. https://www.regulations.gov/document?D=EPA-HQ-OPPT-2009-0576-0006. Accessed 20 Feb 2018
EPA (2009b) EPA guideline 890.1450, endocrine disruptor screening program test guidelines—OPPTS 890.1450: pubertal development and thyroid function in intact juvenile/peripubertal female rats [EPA 740-C-09-009]. https://www.regulations.gov/document?D=EPA-HQ-OPPT-2009-0576-0009. Accessed 20 Feb 2018
EPA (2009c) EPA guideline 890.1500, endocrine disruptor screening program test guidelines—OPPTS 890.1500: pubertal development and thyroid function in intact juvenile/peripubertal male rats [EPA 740-C-09-012]. https://www.regulations.gov/document?D=EPA-HQ-OPPT-2009-0576-0010. Accessed 20 Feb 2018
Evans MV, Andersen ME (2000) Sensitivity analysis of a physiological model for 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD): assessing the impact of specific model parameters on sequestration in liver and fat in the rat. Toxicol Sci 54(1):71–80
Fabian E, Guth K, Zbranek R, van Ravenzwaay B, Landsiedel R (2015) Evaluation of physiologically based toxicokinetic (PTK) modelling for reverse dosimetry approaches. Toxicol Lett 238:168–169
FAO website (2018) FAO (Food and Agricultural Organization of the United Nations), fenarimol (191) identity ISO common name: fenarimol. http://www.fao.org/fileadmin/templates/agphome/documents/Pests_Pesticides/JMPR/Evaluation95/fenari.pdf. Accessed 20 Feb 2018
Fitch WL, Tran T, Young M, Liu L, Chen Y (2009) Revisiting the metabolism of ketoconazole using accurate mass. Drug Metab Lett 3(3):191–198
Forsby A, Blaauboer B (2007) Integration of in vitro neurotoxicity data with biokinetic modelling for the estimation of in vivo neurotoxicity. Hum Exp Toxicol 26:333–338
Ge S, Wei Y, Yin T, Xu B, Gao S, Hu M (2017) Transport-glucuronidation classification system and pbpk modeling: new approach to predict the impact of transporters on disposition of glucuronides. Mol Pharm 14(9):2884–2898. https://doi.org/10.1021/acs.molpharmaceut.6b00941
Gómez C, Pozo OJ, Marcos J, Segura J, Ventura R (2013) Alternative long-term markers for the detection of methyltestosterone misuse. Steroids 78(1):44–52. https://doi.org/10.1016/j.steroids.2012.10.008
Groothuis FA, Heringa MB, Nicol B, Hermens JL, Blaauboer BJ, Kramer NI (2013) Dose metric considerations in in vitro assays to improve quantitative in vitro-in vivo dose extrapolations. Toxicology 332:30–40. https://doi.org/10.1016/j.tox.2013.08.012
Gubbels-van Hal WM, Blaauboer BJ, Barentsen HM, Hoitink MA, Meerts IA, van der Hoeven JC (2005) An alternative approach for the safety evaluation of new and existing chemicals, an exercise in integrated testing. Regul Toxicol Pharmacol 42:284–295. https://doi.org/10.1016/j.yrtph.2005.05.002
Gundert-Remy U, Bernauer U, Blömeke B, Döring B, Fabian E, Goebel C, Petzinger E (2014) Extrahepatic metabolism at the body’s internal–external interfaces. Drug Metab Rev 46(3):291–324
Hamdy DA, Brocks DR (2008) A stereospecific high-performance liquid chromatographic assay for the determination of ketoconazole enantiomers in rat plasma. Biomed Chromatogr 22(5):542–547. https://doi.org/10.1002/bmc.967
Hayes KA, Brennan B, Chenery R, Houston JB (1995) In vivo disposition of caffeine predicted from hepatic microsomal and hepatocyte data. Drug Metab Dispos 23(3):349–353
Hayes AW, Thomas JA, Gardner DE, Eldridge JC, Stevens JT, Harvey PW, Everett DJ (2010) Endocrine toxicology. In: Eldridge JC, Stevens JT (eds) Organ toxicology series - endocrine toxicology, 3rd edn. Taylor & Francis Group, New York
Hintikka L, Kuuranne T, Aitio O, Thevis M, Schänzer W, Kostiainen R (2008) Enzyme-assisted synthesis and structure characterization of glucuronide conjugates of eleven anabolic steroid metabolites. Steroids 73(3):257–265. https://doi.org/10.1016/j.steroids.2007.10.008
Hirai S, Hussain A, Haddadin M, Smith RB (1981) First-pass metabolism of ethinyl estradiol in dogs and rats. J Pharm Sci 70:403–406
Hjelle JJ, Klaassen CD (1984) Glucuronidation and biliary excretion of acetaminophen in rats. J Pharmacol Exp Ther 228:407–413
Hou TJ, Zhang W, Xia K, Qiao XB, Xu XJ (2004) ADME evaluation in drug discovery. 5. Correlation of Caco-2 permeation with simple molecular properties. J Chem Inf Comput Sci 44(5):1585–1600
Houston B (1994) Utility of in vitro drug metabolism data in predicting in vivo metabolic clearance. Biochem Pharmacol 47:1469–1479. https://doi.org/10.1016/0006-2952(94)90520-7
Ito K, Houston JB (2004) Comparison of the use of liver models for predicting drug clearance using in vitro kinetic data from hepatic microsomes and isolated hepatocytes. Pharm Res 21(5):785–792. https://doi.org/10.1023/B:PHAM.0000026429.12114.7d
Jones H, Rowland-Yeo K (2013) Basic concepts in physiologically based pharmacokinetic modeling in drug discovery and development CPT. Pharmacomet Syst Pharmacol 14;2:e63. https://doi.org/10.1038/psp.2013.41
Kanno J, Onyon L, Haseman J, Fenner-Crisp P, Ashby J, Owens W, Organisation for Economic Co-operation and Development (2001) The OECD program to validate the rat Uterotrophic bioassay to screen compounds for in vivo estrogenic responses: phase 1. Environ Health Perspect 109(8):785–794. https://doi.org/10.2307/3454820
Kanno J, Onyon L, Peddada S, Ashby J, Jacob E, Owens W (2003) The OECD program to validate the rat Uterotrophic bioassay. Phase 2: dose–response studies. Environ Health Perspect 111(12):1530–1549. https://doi.org/10.1289/ehp.5780
Kilford PJ, Gertz M, Houston JB, Galetin A (2008) Hepatocellular binding of drugs: correction for unbound fraction in hepatocyte incubations using microsomal binding or drug lipophilicity data. Drug Metab Dispos 36(7):1194–1197. https://doi.org/10.1124/dmd.108.020834
Knaak JB, Sullivan LJ (1966) Metabolism of bisphenol A in the rat. Toxicol Appl Pharmacol 8(2):175–184
Kolle SN, Kamp HG, Huener HA, Knickel J, Verlohner A, Woitkowiak C, Landsiedel R, van Ravenzwaay B (2010) In house validation of recombinant yeast estrogen and androgen receptor agonist and antagonist screening assays. Toxicol In Vitro 24(7):2030–2040. https://doi.org/10.1016/j.tiv.2010.08.008
Kolle SN, Ramirez T, Kamp HG, Buesen R, Flick B, Strauss V, van Ravenzwaay B (2012) A testing strategy for the identification of mammalian, systemic endocrine disruptors with particular focus on steroids. Regul Toxicol Pharmacol 63(2):259–278
Kot M, Daniel WA (2008) Caffeine as a marker substrate for testing cytochrome P450 activity in human and rat. Pharmacol Rep 60(6):789–797
Landsiedel R, Fabian E, Tralau T, Luch A (2011) Chemical toxicity testing in vitro using cytochrome P450—expressing cell lines, such as human CYP1B1. Nat Protoc 6(5):677
Lelo A, Birkett DJ, Robson RA, Miners JO (1986) Comparative pharmacokinetics of caffeine and its primary demethylated metabolites paraxanthine, theobromine and theophylline in man. Br J Clin Pharmacol 22(2):177–182
Li H, Zhang M, Vervoort J, Rietjens IM, van Ravenzwaay B, Louisse J (2017) Use of physiologically based kinetic modeling-facilitated reverse dosimetry of in vitro toxicity data for prediction of in vivo developmental toxicity of tebuconazole in rats. Toxicol Lett 266:85–93. https://doi.org/10.1016/j.toxlet.2016.11.017
Louisse J, de Jong E, van de Sandt JJ, Blaauboer BJ, Woutersen RA, Piersma AH, Rietjens IM, Verwei M (2010) The use of in vitro toxicity data and physiologically based kinetic modeling to predict dose-response curves for in vivo developmental toxicity of glycol ethers in rat and man. Toxicol Sci 118:470–484. https://doi.org/10.1093/toxsci/kfq270
Louisse J, Bosgra S, Blaauboer BJ, Rietjens IM, Verwei M (2015) Prediction of in vivo developmental toxicity of all-trans-retinoic acid based on in vitro toxicity data and in silico physiologically based kinetic modeling. Arch Toxicol 89(7):1135–1148
Louisse J, Beekmann K, Rietjens IMCM (2017) Use of physiologically based kinetic modeling-based reverse dosimetry to predict in vivo toxicity from in vitro data. Chem Res Toxicol 30:114–125. https://doi.org/10.1021/acs.chemrestox.6b00302
Lu Y, Kim S, Park K (2011) In vitro–in vivo correlation: perspectives on model development. Int J Pharm 418(1):142–148. https://doi.org/10.1016/j.ijpharm.2011.01.010
Macey R, Oster G, Zahnley T (2009) Berkeley Madonna user’s guide. Berkeley Madonna Webpage, Berkeley
Manov I, Bashenko Y, Hirsh M, Iancu TC (2006) Involvement of the multidrug resistance P-glycoprotein in acetaminophen-induced toxicity in hepatoma-derived HepG2 and Hep3B cells. Basic Clin Pharmacol Toxicol 99(3):213–224. https://doi.org/10.1111/j.1742-7843.2006.pto_443.x
Matthew D, Brennan B, Zomorodi K, Houston JB (1993) Disposition of azole antifungal agents. I. Nonlinearities in ketoconazole clearance and binding in rat liver. Pharm Res 351(3):418–422
Mazur CS, Marchitti SA, Dimova M, Kenneke JF, Lumen A, Fisher J (2012) Human and rat ABC transporter efflux of bisphenol a and bisphenol a glucuronide: interspecies comparison and implications for pharmacokinetic assessment. Toxicol Sci 128(2):317–325. https://doi.org/10.1093/toxsci/kfs167
Naritomi Y (2003) Utility of hepatocytes in predicting drug metabolism: comparison of hepatic intrinsic clearance in rats and humans in vivo and in vitro. Drug Metab Dispos 31(5):580–588. https://doi.org/10.1124/dmd.31.5.580
OECD (1983) Test no. 415: one-generation reproduction toxicity study, OECD guidelines for the testing of chemicals, sect. 4. OECD Publishing, Paris. https://doi.org/10.1787/9789264070844-en
OECD (2001) Test no. 416: two-generation reproduction toxicity, OECD guidelines for the testing of chemicals, sect. 4. OECD Publishing, Paris. https://doi.org/10.1787/9789264070868-en
OECD (2007) Test no. 440: uterotrophic bioassay in rodents: a short-term screening test for oestrogenic properties, OECD guidelines for the testing of chemicals, sect, 4. OECD Publishing, Paris. https://doi.org/10.1787/9789264067417-en
OECD (2009a) Test no. 455: the stably transfected human estrogen receptor-alpha transcriptional activation assay for detection of estrogenic agonist-activity of chemicals. OECD Publishing, Paris. https://doi.org/10.1787/9789264076372-en
OECD (2009b) Test no. 441: hershberger bioassay in rats: a short-term screening assay for (Anti)androgenic properties, OECD guidelines for the testing of chemicals, sect, 4. OECD Publishing, Paris. https://doi.org/10.1787/9789264076334-en
OECD (2011) Test no. 456: H295R steroidogenesis assay, OECD guidelines for the testing of chemicals, Sect. 4. OECD Publishing, Paris. https://doi.org/10.1787/9789264122642-en
OECD (2012) Environment, Health and Safety Publications Series on Testing and Assessment No. 150: Guidance document on standardised test guidelines for evaluating chemicals for endocrine Disruption, ENV/JM/MONO(2012)22
OECD (2018a) Revised Guidance Document 150 on Standardised Test Guidelines for Evaluating Chemicals for Endocrine Disruption, OECD Series on Testing and Assessment, OECD Publishing, Paris. https://doi.org/10.1787/9789264304741-en
OECD (2018b) Test no. 443: extended one-generation reproductive toxicity study, OECD guidelines for the testing of chemicals, sect. 4. OECD Publishing, Paris. https://doi.org/10.1787/9789264185371-en
Oesch F, Fabian E, Landsiedel R (2018) Xenobiotica-metabolizing enzymes in the skin of rat, mouse, pig, guinea pig, man, and in human skin models. Arch Toxicol 92(8):2411–2456. https://doi.org/10.1007/s00204-018-2232-x
Owens W, Gray LE, Zeiger E, Walker M, Yamasaki K, Ashby J, Jacob E (2007) The OECD program to validate the rat Hershberger bioassay to screen compounds for in vivo androgen and antiandrogen responses: phase 2 dose–response studies. Environ Health Perspect 115(5):671–678. https://doi.org/10.1289/ehp.9666
Paini A, Punt A, Viton F, Scholz G, Delatour T, Marin-Kuan M, Schilter B, van Bladeren PJ, Rietjens IM (2010) A physiologically based biodynamic (PBBD) model for estragole DNA binding in rat liver based on in vitro kinetic data and estragole DNA adduct formation in primary hepatocytes. Toxicol Appl Pharmacol 245:57–66
Paini A, Sala Benito JV, Bessems J, Worth AP (2017) From in vitro to in vivo: integration of the virtual cell based assay with physiologically based kinetic modelling. Toxicol In Vitro 45(Pt 2):241–248. https://doi.org/10.1016/j.tiv.2017.06.015
Pottenger LH, Domoradzki JY, Markham DA, Hansen SC, Cagen SZ, Waechter JM Jr (2000) The relative bioavailability and metabolism of bisphenol A in rats is dependent upon the route of administration. Toxicol Sci 54(1):3–18
Pottier J, Cousty C, Heitzman RJ, Reynolds IP (1981) Differences in the biotransformation of a 17 beta-hydroxylated steroid, trenbolone acetate, in rat and cow. Xenobiotica 11(7):489–500
PubMed public database (2018) https://pubchem.ncbi.nlm.nih.gov. Accessed 20 Feb 2018
Punt A, Schiffelers MJ, Jean Horbach G, van de Sandt JJ, Groothuis GM, Rietjens IM, Blaauboer BJ (2011) Evaluation of research activities and research needs to increase the impact and applicability of alternative testing strategies in risk assessment practice. Regul Toxicol Pharmacol 61(1):105–114. https://doi.org/10.1016/j.yrtph.2011.06.007
Punt A, Brand W, Murk AJ, van Wezel AP, Schriks M, Heringa MB (2013) Effect of combining in vitro estrogenicity data with kinetic characteristics of estrogenic compounds on the in vivo predictive value. Toxicol In Vitro 27(1):44–51. https://doi.org/10.1016/j.tiv.2012.09.014
Remmel RP, Amoh K, Abdel-Monem MM (1987) The disposition and pharmacokinetics of ketoconazole in the rat. Drug Metab Dispos 15(6):735–739
Rietjens IM, Louisse J, Punt A (2011) Tutorial on physiologically based kinetic modeling in molecular nutrition and food research. Mol Nutr Food Res 55(6):941–956. https://doi.org/10.1002/mnfr.201000655
Routledge EJ, Sumpter JP (1996) Estrogenic activity of surfactants and some of their degradation products assessed using a recombinant yeast screen. Environ Toxicol Chem 15(3):241–248
Russell WMS, Burch RL, Hume CW (1959) The principles of humane experimental technique (vol 238); Chap. 5 replacement. Methuen, London
Saypol DC, Howards SS, Turner TT, Miller ED Jr (1981) Influence of surgically induced varicocele on testicular blood flow, temperature, and histology in adult rats and dogs. J Clin Investig 68(1):39–45
Schwenk M, Schiemenz C, Lopez del Pino V, Remmer H (1982) First pass biotransformation of ethinylestradiol in rat small intestine in situ. Naunyn Schmiedebergs Arch Pharmacol 321:223–225
Sfakianos J, Coward L, Kirk M, Barnes S (1997) Intestinal uptake and biliary excretion of the isoflavone genistein in rats. J Nutr 127:1260–1268. https://doi.org/10.1093/jn/127.7.1260
Shin JH, Moon HJ, Kang IH, Kim TS, Lee SJ, Ahn JY, Bae H, Jeung EB, Han SY (2007) OECD validation of the rodent Hershberger assay using three reference chemicals; 17alpha-methyltestosterone, procymidone, and p,p′-DDE. Arch Toxicol 81(5):309–318. https://doi.org/10.1007/s00204-006-0174-1
Smith DA, Di L, Kerns EH (2010) The effect of plasma protein binding on in vivo efficacy: misconceptions in drug discovery. Nat Rev Drug Discov 9(12):929–939. https://doi.org/10.1038/nrd3287
Snyder RW, Maness SC, Gaido KW, Welsch F, Sumner SC, Fennell TR (2000) Metabolism and disposition of bisphenol A in female rats. Toxicol Appl Pharmacol 1 168(3):225–234. https://doi.org/10.1006/taap.2000.9051
Soucy NV (2005) Kinetics of genistein and its conjugated metabolites in pregnant Sprague-Dawley rats following single and repeated genistein administration. Toxicol Sci 90(1):230–240. https://doi.org/10.1093/toxsci/kfj077
Strikwold M, Spenkelink B, Woutersen RA, Rietjens IM, Punt A (2013) Combining in vitro embryotoxicity data with physiologically based kinetic (PBK) modelling to define in vivo dose-response curves for developmental toxicity of phenol in rat and human. Arch Toxicol 87(9):1709–1723. https://doi.org/10.1007/s00204-013-1107-4
Strikwold M, Spenkelink B, de Haan LHJ, Woutersen RA5, Punt A, Rietjens IMCM (2017) Integrating in vitro data and physiologically based kinetic (PBK) modelling to assess the in vivo potential developmental toxicity of a series of phenols. Arch Toxicol 91(5):2119–2133. https://doi.org/10.1007/s00204-016-1881-x
Sun H, Xu XL, Qu JH, Hong X, Wang YB, Xu LC, Wang XR (2008) 4-Alkylphenols and related chemicals show similar effect on the function of human and rat estrogen receptor alpha in reporter gene assay. Chemosphere 71(3):582–588
Sun H, Chen W, Xu X, Ding Z, Chen X, Wang X (2014) Pyrethroid and their metabolite, 3-phenoxybenzoic acid showed similar (anti)estrogenic activity in human and rat estrogen receptor α-mediated reporter gene assays. Environ Toxicol Pharmacol 37(1):371–377
Taxvig C, Vinggaard AM, Hass U, Axelstad M, Metzdorff S, Nellemann C (2008) Endocrine-disrupting properties in vivo of widely used azole fungicides. Int J Androl 31(2):170–177. https://doi.org/10.1111/j.1365-2605.2007.00838.x
Thermo Scientific (2012) Manual: RED Device Inserts. Retrieved November 3, 2015, from http://www.thermoscientific.com/pierce
Tice RR, Austin CP, Kavlock RJ, Bucher JR (2013) Improving the human hazard characterization of chemicals: a Tox21 update. Environ Health Perspect 121(7):756
Twaddle NC, Churchwell MI, Newbold RR, Delclos KB, Doerge DR (2003) Determination using liquid-chromatography-electrospray tandem mass spectroscopy of ethinylestradiol serum pharmacokinetics in adult Sprague-Dawley rats. J Chromatogr B Analyt Technol Biomed Life Sci 793(2):309–315
US Environmental Protection Agency (2007) Integrated summary report for validation of a test method for assessment of pubertal development and thyroid function in juvenile male rats as a potential screen in the endocrine disruptor screening program tier-1 battery. http://citeseerx.ist.psu.edu/viewdoc/download;jsessionid=400908E7B5886C560C0964E1DC343786?doi=10.1.1.368.8060&rep=rep1%26type=pdf. Accessed 20 Feb 2018
van Liempd S, Morrison D, Sysmans L, Nelis P, Mortishire-Smith R (2011) Development and validation of a higher-throughput equilibrium dialysis assay for plasma protein binding. J Lab Autom 16(1):56–67. https://doi.org/10.1016/j.jala.2010.06.002
Verwej M, van Burgsteden JA, Krul CA, van de Sandt JJ, Freidig AP (2006) Prediction of in vivo embryotoxic effect levels with a combination of in vitro studies and PBPK modelling. Toxicol Lett 165:79–87
Vinken M (2013) The adverse outcome pathway concept: a pragmatic tool in toxicology. Toxicology 312:158–165. https://doi.org/10.1016/j.tox.2013.08.011
Waters NJ, Jones R, Williams G, Sohal B (2008) Validation of a rapid equilibrium dialysis approach for the measurement of plasma protein binding. J Pharm Sci 97(10):4586–4595. https://doi.org/10.1002/jps.21317
Wetmore BA, Wambaugh JF, Ferguson SS, Sochaski MA, Rotroff DM, Freeman K, Clewell HJ 3rd, Dix DJ, Andersen ME, Houck KA, Allen B, Judson RS, Singh R, Kavlock RJ, Richard AM, Thomas RS (2012) Integration of dosimetry, exposure, and high-throughput screening data in chemical toxicity assessment. Toxicol Sci 125(1):157–174. https://doi.org/10.1093/toxsci/kfr254
World Health Organization (2012) An assessment of the state of the science of endocrine disruptors prepared by a group of experts for the United Nations Environment Programme and World Health Organization, State of the science of endocrine disrupting chemicals summary for decision-makers. In: Bergman Åke, Heindel JJ, Jobling S, Kidd KA, Thomas Zoeller R (eds) http://apps.who.int/iris/bitstream/handle/10665/78102/WHO_HSE_PHE_IHE_2013.1_eng.pdf;jsessionid=846FD9E7C8F5F6477D6990D55584860E?sequence=1. Accessed 20 Feb 2018
Yu LX, Amidon GL (1999) A compartmental absorption and transit model for estimating oral drug absorption. Int J Pharm 186:119–125
Zamek-Gliszczynski MJ, Day JS, Hillgren KM, Phillips DL (2011) Efflux transport is an important determinant of ethinylestradiol glucuronide and ethinylestradiol sulfate pharmacokinetics. Drug Metab Dispos 39(10):1794–1800
Zhang F, Xue J, Shao J, Jia L (2012) Compilation of 222 drugs’ plasma protein binding data and guidance for study designs. Drug Discov Today 17(9–10):475–485. https://doi.org/10.1016/j.drudis.2011.12.018
Zhang M, van Ravenzwaay B, Fabian E, Rietjens IMCM, Louisse J (2018) Towards a generic physiologically based kinetic model to predict in vivo uterotrophic responses in rats by reverse dosimetry of in vitro estrogenicity data. Arch Toxicol 92(3):1075–1088. https://doi.org/10.1007/s00204-017-2140-5
Zuo Z, Tam YK, Diakur J, Wiebe LI (2002) Hydroxypropyl-beta-cyclodextrin-flutamide inclusion complex. II. Oral and intravenous pharmacokinetics of flutamide in the rat. J Pharm Pharm Sci 5(3):292–298
Acknowledgements
We would like to thank Dr. Nicola Hewitt for her contribution to the preparation of this manuscript. We would also like to thank Dr. Ursula Müller-Vieira and Dr. Peter Ehmer, Pharmacelsus GmbH, Saarbrücken, Germany for analytical support of determination of plasma protein binding by rapid equilibrium dialysis. Funding was provided by BASF (KTC Alternative Toxicological Methods).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Fabian, E., Gomes, C., Birk, B. et al. In vitro-to-in vivo extrapolation (IVIVE) by PBTK modeling for animal-free risk assessment approaches of potential endocrine-disrupting compounds. Arch Toxicol 93, 401–416 (2019). https://doi.org/10.1007/s00204-018-2372-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00204-018-2372-z