
Abstract
The numbers of insulin-secreting pancreatic beta cells are reduced in people with type 1 and type 2 diabetes. Driving beta cell regeneration in the pancreases of people with diabetes would be an attractive approach to reversing diabetes. While adult human beta cells have long been believed to be terminally differentiated and, therefore, irreversibly quiescent, it has become clear over recent years that this is not true. More specifically, both candidate and unbiased high-throughput screen approaches have revealed several classes of molecules that are clearly able to induce human beta cell proliferation. Here, we review recent approaches and accomplishments in human beta cell regenerative drug discovery. We also list the challenges that this rapidly moving field must confront to translate beta cell regenerative therapy from the laboratory to the clinic.
Similar content being viewed by others



Introduction
Type 1 and type 2 diabetes result entirely or in part from inadequate numbers of normally functioning pancreatic beta cells [1,2,3,4]. It therefore follows that restoration of normal beta cell mass and function should reverse diabetes, a concept that underlies islet transplantation [5] and whole pancreas transplantation [6] and stem cell-based strategies for transplantable beta cells [7,8,9] in type 1 and 2 diabetes, as well as efforts to induce alpha cells to transdifferentiate into beta cells [10], and re-differentiation of de-differentiated beta cells [11,12,13]. Our group, however, focuses on an alternative approach, developing methods for inducing proliferation of residual human beta cells [3, 14], which is the theme of the current review.
Beta cell replication
Since some beta cells remain in most people with either type 1 or type 2 diabetes [1,2,3,4], it is reasonable to assume that if one were able to develop drugs able to induce beta cell replication, beta cell mass could be restored. This raises the question as to if and when beta cells replicate under normal conditions. The answer is that human beta cells are able to replicate, but only at low rates, in the range of 2%/day, and only in the first few years of life [3, 15,16,17,18,19]. Historically, pharmacological attempts to induce adult human beta cells to replicate have been unsuccessful. This changed in 2015 with the discovery of the harmine-dual-specificity tyrosine phosphorylation-regulated kinase (DYRK)1A inhibitor class of drugs [14, 20,21,22,23], discussed in detail below. However, even with use of this new class of drugs, the rates of beta cell replication observed remain low, in the range of 1–3%/day [14, 20,21,22,23].
What is the optimal rate of therapeutic human beta cell replication? The answer to this question is unknown, of course, since we have no way at present to reliably assess beta cell mass in humans, and since no human trials have been performed in relation to this. Moreover, current methods used to assess human beta cell proliferation employ markers of S- and/or G2M-phase entry (bromo-deoxyuridine [BrdU], 5-ethynyl-2′-deoxyuridine [EdU], Ki67, phospho-histone-H3), but do not unequivocally confirm completion of the cell cycle and the birth of two new daughter cells. Thus, the actual rate of new beta cell accrual in short-term human beta cell cultures is difficult to assess. With these constraints, a person with type 1 diabetes and few remaining beta cells would require long-term treatment with a drug that increases beta cell mass by 1–2%/day in [2, 4], whereas beta cell mass may actually normalise with this rate of proliferation in a person with type 2 diabetes and 50% of their beta cells remaining [1]. Thus, to clarify the quantitative requirements for therapeutic human beta cell replication, long-term studies of transplanted human islets in immunodeficient mice and, ultimately, human clinical trials are needed.
To make matters more complex, there is also the issue of beta cell heterogeneity [23,24,25,26,27,28]. It has long been known [24, 25], and is becoming even clearer recently from conventional flow cytometry [26], CyTOF (a modification of conventional flow cytometry that employs time-of-flight [TOF] mass spectroscopy using heavy metal-tagged antibodies to assess multiple internal and surface antigens on single cells) [23], advanced imaging [28] and single cell RNA sequencing [25, 28] studies, that all beta cells are not the same. In particular, it remains unknown if all beta cells are capable of proliferation, or if only a subset has mitogenic potential. It is also unknown whether and for how long human beta cells that have divided once, remain refractory to undergoing a second, third, fourth round of replication. As a corollary, if some beta cells have innate proliferative capacities, while others are terminally quiescent, it would be optimal to target drugs to those with replicative potential.
Finally, there is the teleological question as to why adult beta cells have evolved to become so remarkably resistant to replication, as compared, for example, with lymphocytes, intestinal enterocytes, keratinocytes and hepatocytes. One might suppose that an evolutionary advantage accrues in mammals by avoiding the hyperinsulinaemia and consequent hypoglycaemia that might accompany inappropriately expanded beta cell mass. Human insulinomas (see below) and congenital hyperinsulinism–hypoglycaemia syndromes provide clear examples of the notion that having too many inappropriately active beta cells may adversely affect species propagation.
Obstacles and impediments to human beta cell proliferation
What cellular mechanisms render adult beta cells so resistant to proliferation? This topic has been reviewed recently [3, 29, 30] and will not be discussed in detail, except to say that multiple mechanisms likely apply. One apparent mechanism relates to cell cycle molecule trafficking within beta cells. For example, key cell cycle regulatory molecules, such as the cyclins and cyclin-dependent kinases (CDKs), are constrained to the cytoplasm in beta cells for reasons that remain unknown [31, 32]. In contrast, overexpressing cyclins and CDKs causes them to shuttle into the nucleus and activate cell cycle entry [31, 32]. Why and how cell cycle molecules are constrained to the cytoplasm in beta cells remains unknown.
It is also possible that adult beta cells lack, or have functional barriers in, key mitogenic pathway molecules, such as growth factors, cell surface receptors, signalling and scaffold molecules, and activated or de-repressed promoters of the cell cycle or related genes required for proliferation [3, 33,34,35]. It is also possible that cytokines, growth inhibitors, such as TGF-β superfamily ligands, and/or cytokine-mediated glucotoxic, lipotoxic, replicative and/or endoplasmic reticulum stress may block cell cycle entry [2, 33,34,35].
Increasingly, however, epigenetic factors, including DNA methylation and chromatin-modifying histone modifications, have been shown to underlie the enforced quiescence in adult human beta cells. For example, Avrahami et al have shown that, in juvenile beta cells, the CDKN2A gene, encoding the cell cycle inhibitor p16INK4A, is repressed by methylation, but is later de-methylated, and therefore de-repressed, in adult beta cells [36]. Further, Kaestner’s group [37] and Arda et al [38] defined alpha and beta cell-specific open chromatin and gene expression signatures using a combination of assay for transposase-accessible chromatin sequencing (ATAC-seq), histone methylation marks and RNA sequencing (RNA-seq). Our own studies on the genomics and transcriptomics of human insulinoma indicate that, essentially, all human insulinomas display mutation, copy number loss, amplification and/or misexpression of key chromatin-modifying enzymes, with resultant misexpression of cell cycle activators and inhibitors, permitting proliferation [39]. Collectively, these growth factor, cytokine, receptor, signalling pathway and epigenetic scenarios described above make the point that there are likely multiple pharmacological opportunities for therapeutic human beta cell replication.
Drug discovery strategies
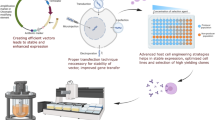
A number of strategies have been used in attempts to identify drugs or growth factors able to induce human beta cell replication (Fig. 1). This area has been reviewed recently by Wagner’s group [29] and Shirakawa and Kulkarni [30]. In broad terms, strategies include both candidate approaches and unbiased screens. To give a few examples, candidate approaches to beta cell regenerative drug discovery have suggested that gamma aminobutyric acid (GABA) [40], glucagon-like peptide-1 (GLP-1) [41] osteoprotegerin/denosumab, inhibitors of the receptor activator of nuclear kappa-B ligand (RANKL) [42], the TGF-β superfamily [43,44,45,46], serpin B1 [47] and a V-growth factor (VGF)-derived peptide called TLQP-21 [48] may have mitogenic effects on beta cells.

Strategies for beta cell regenerative drug discovery. Drug discovery may be candidate-based, or may involve unbiased high- or medium-throughput screens. Candidates may be derived from the literature, from large datasets or from data mines. Examples of human beta cell mitogenic compounds discovered through these processes are shown in yellow boxes. These include DYRK1A, harmine analogues, gamma aminobutyric acid (GABA), glucagon-like peptide-1 (GLP-1), prolactin and placental lactogens (PRL/PL), osteoprotegerin, and its mimetic, the monoclonal antibody denosumab (OPG), TLQP-21 (a small fragment of a parent peptide called V-growth factor [VGF]), TGF-β, p18 and p21 (cell cycle inhibitors encoded by CDKN2C and CDKN1A, respectively), lysine demethylase 6A (KDM6A) and CDKN1C (encoding p57Kip2). The goal of human beta cell regenerative studies, beta cell proliferation, is illustrated by showing a small, beta cell-deficient islet becoming a larger islet with a higher number of cells. The different coloured cells represent some of the different endocrine cell types within the human islet: beta cells are shown in blue; glucagon-producing alpha cells are shown in red; and somatostatin-producing delta cells are shown in green. This figure is available as part of a downloadable slideset
A broad range of unbiased high- or low-throughput screens have also been developed by many investigators, employing proteomics [47, 48], small interfering RNA (siRNA) libraries [49] on zebrafish [50], mouse and rat islets [20, 21, 51, 52], human islets [14, 21, 53] and surrogate cell lines [14, 20] (Fig. 1). For example, we used a luciferase reporter-based high-throughput screen in human HepG2 cells to identify the harmine family (DYRK1A inhibitors) as a potential target for beta cell replication [14]. In line with this, Laffitte’s group used a rat insulinoma cell line [20] and Annes et al used primary rat cells [51] to support the role of harmine and related drugs in beta cell proliferation.
Finally, since the highest rates of human beta cell proliferation in vivo are observed in juvenile beta cells and human insulinomas, we and others have used these cell types as data mines for human beta cell replication [39, 54].
Collectively, these approaches have yielded many putative pathways that may act as drug targets, and classes of drugs (Fig. 1). Among these, the DYRK1A inhibitor findings have been most widely replicated [14, 20,21,22,23] and these inhibitors provide the highest proliferation rates in human beta cells.
The DYRK1A inhibitor family
The DYRK1A inhibitor family includes a variety of chemical entities, including harmine (a beta-carboline) [14], INDY (derived from ‘INhibitor of DYrk1a’; a benzothiazole) [14, 55], 5-iodo-tubericidin (5-IT; an adenosine analogue) [21] and GNF4877 (an aminopyrazine) [20]. All were reported in 2015, or shortly thereafter, to drive human beta cell replication by serving as reversible inhibitors of DYRK1A [13, 17,18,19,20]. In general, the ‘rate’ of human beta cell proliferation with use of these inhibitors has been reported in the 1–3% range [14, 22, 23], but higher labelling indices of approximately 5% have been observed with multi-day BrdU or EdU labelling protocols [20, 21].
That DYRK1A inhibition is responsible for cell proliferation has been documented by the observation that each of the small molecule DYRK1A inhibitors described above bind with great specificity to the DYRK1A protein in kinome screens [21, 55, 56], and genetic silencing of DYRK1A in human islets induces beta cell proliferation [14, 21]. Conversely, overexpressing DYRK1A abolishes the proliferative effects of harmine in human beta cells [14]. Of course, kinome screens reveal other lower affinity targets of harmine, INDY, GNF4877 and/or 5-IT, notably other DYRKs (DYRK1B, DYRK2, DYRK3 and DYRK4), glycogen synthase kinase (GSK)3α and GSKβ, and cell-division cycle (CDC)-like kinases (CLKs), CLK1, CLK2 and CLK4 [20, 21, 55, 56]. Among these candidates, only silencing of DYRK1A, but not the other kinases, induces proliferation, making it clear that DYRK1A is the principal target relating to beta cell proliferation. It is also possible, however, that other so-far unknown but important non-kinase targets exist and, therefore, would not be detected in kinome screens. Finally, it is important to note that the increases in beta cell proliferation with DYRK1A inhibitors have been observed, not only in culture settings in vitro, but also in human islets transplanted into immunodeficient mouse models in vivo [14, 21].
In mechanistic terms, the nuclear factor activated in T cells (NFaT) family of transcription factors has been shown to bind to, and transactivate, cell cycle activating genes, such as CCNE, CCNA (encoding cyclins E and A) and CDK1, and to repress cell cycle inhibitor genes, such as CDKN1C, CDKN2A and CDKN2B, thereby activating cell cycle progression [14]. In order to gain nuclear entry, NFaTs must be dephosphorylated by calcineurin. DYRK1A serves as a nuclear kinase that re-phosphorylates nuclear NFaTs, terminating their mitogenic signal [14]. Thus, DYRK1A inhibitors appear to act by preventing NFaT re-phosphorylation, allowing continued stimulation of cell cycle activation (Fig. 2).

The calcium-calmodulin-calcineurn-NFaT-DYRK1A pathway to human beta cell proliferation. Increases in intracellular calcium (Ca++), induced, for example, by glucose, sulfonylureas and the glucagon-like peptide-1 (GLP-1) family of drugs, activate calmodulin (CAM), which in turn activates the calcineurins (CnA and CnB). CnA and CnB form a phosphatase complex, which de-phosphorylates a number of substrates, including the NFaT family of transcription factors that, in their phosphorylated state, are tethered to 14-3-3 scaffold proteins in the cytoplasm. De-phosphorylation by calcineurin allows these transcription factors to translocate into the nuclear compartment, where they bind to and activate promoters of cyclins E and A, and cyclin-dependent kinase 1 (CDK1), and repress promoters of the cell cycle inhibitor genes, CDKN2A, CDKN2B and CDKN1C, encoding p16INK4, p15INK4 and p57Kip2, respectively. Collectively these events result in cell cycle entry. The kinase DYRK1A serves as the normal termination mechanism in this process by re-phosphorylating NFaT members. This results in their return to the cytoplasm, and return to quiescence. Thus, DYRK1A is the ‘brake’ on cell cycle entry or proliferation. Harmine, INDY, 5-iodo-tubericidin (5-IT) and GNF4877 are all inhibitors of DYRK1A and, in essence, function by disabling the DYRK1A ‘brake’, permitting continued proliferation [14, 20, 21]. VDCC, voltage-dependent calcium channel. Figure adapted from [14]. This figure is available as part of a downloadable slideset
The pros and cons of DYRK1A inhibitor use for beta cell proliferation
One might worry that driving beta cells to replicate might lead them to de-differentiate [57]. Remarkably, however, it appears that the reverse may be true when using harmine to promote beta cell replication; harmine-induced proliferation also leads to increases in markers of beta cell differentiation, such as the transcription factors pancreas-duodenum homeobox protein (PDX1), NK6 homeobox 1 (NKX6.1) and MafA [14]. Moreover, harmine treatment improves glucose tolerance in immunodeficient mice transplanted with human islets [14]. For type 1 and type 2 diabetes, in which beta cells are reported to de-differentiate [11,12,13], this may prove particularly important. Of course, these studies have been performed over days to a week and, so, longer term studies are required to determine whether this enhanced differentiation persists over time.
While the discovery of the role of harmine in beta cell proliferation represents an important advance in this field, the harmine class is not perfect for four principal reasons. First, the DYRK1A enzyme is ubiquitous, predicting adverse effects of harmine in many organs. For example, harmine is a well-known hallucinogen [58] and activates proliferation not only in beta cells, but also in alpha cells and ductal cells in the islet [14, 21, 23] and likely others outside the islet. Thus, there is an urgent need to specifically target harmine analogues to the beta cell, to avoid off-target adverse effects. Perhaps this can be obviated by the use of harmine-related compounds ex vivo, to expand cadaveric or induced pluripotent stem cell (iPS)-derived beta cells destined for transplant, an area that has not been explored. Second, rates of beta cell proliferation higher than 1–2%/day would likely be preferable. This is particularly true for type 1 diabetes, in which beta cell mass is depleted to a greater degree than in type 2 diabetes [2, 4], in which lower rates of proliferation might be acceptable. Thus, there is a need for more potent beta cell mitogenic compounds. Third, the current generation of DYRK1A inhibitors are not entirely DYRK1A-specific or ‘clean’. Further medicinal chemistry optimisation is therefore needed. Finally, beta cell regenerative strategies are doomed to failure in type 1 diabetes unless they are accompanied by measures to block or reverse beta cell autoimmunity.
Lessons from human insulinomas
Perhaps the most important lesson from the harmine–DYRK1A story is that it is finally possible to identify small molecule drugs that are able to coerce the previously refractory human beta cell into cell cycle activation. As one corollary, it also suggests that there may be additional, complimentary, alternate, equally or more effective pathways that may be exploited to drive human beta cells to proliferate.
As alluded to above, insulinomas are rare tumours of the beta cell of the pancreas that come to medical attention because of the hyperinsulinaemia and hypoglycaemia they cause, which may lead to seizures, unconsciousness and death. Although a small percentage (2–4%) may be malignant, the large majority are benign and cause morbidity and mortality solely through hypoglycaemia. Once discovered, they are readily removed by laparoscopic surgery and the patient cured. Since they are not a common oncological public health problem, insulinomas have not been studied in detail with the next-generation sequencing tools now widely applied to common cancers. However, we viewed insulinomas through a different lens, seeing them as an ideal ‘data mine’ that might reveal novel pathways that may act as drug targets for human beta cell proliferation [39].
With this goal in mind, we have built an extensive human insulinoma biorepository, containing almost 100 samples, and have recently reported the status of the first 38 of these insulinomas based on paired whole-exome DNA sequencing, RNA-seq, DNA methylation status and comparisons of insulinoma gene expression with that of normal FACS-sorted human beta cells [39]. Space does not permit a detailed description of the findings, but they can be succinctly summarised as follows: (1) there was an enormous number of mutations: 278 mutations in 38 insulinomas, giving an average of approximately ten mutations per insulinoma; (2) although few of these mutations occurred in more than one insulinoma, almost every insulinoma had multiple mutations in three or four families of genes that control the three-dimensional structure of DNA and determine whether specific chromosomal regions are ‘open’ and accessible to transcription factors, or are ‘closed’ and, therefore, silenced. Examples of these mutated gene families include the trithorax members (MEN1, KDM6A), the polycomb member EZH2, the polycomb target H3F3A and the general mitogenic and chromatin remodelling factor MYC, along with many others; (3) we found thousands of genes that were differentially expressed between normal beta cells and insulinomas, and high on the list was DYRK1A; (4) bioinformatic analysis of the aggregate data suggested that, at the cellular level, the CDKN1C gene, which encodes the cell cycle inhibitor p57Kip2, is a key downstream target of chromatin-modifying genes and their associated pathways; (5) frequent regional deletions and methylation abnormalities were discovered in or near the canonical imprinted region of chromosome 11, which contains not only CDKN1C, but also MEN1 (encoding menin), ABCC8 (encoding the sulfonylurea receptor type 1 [SUR1]) and KCNJ11 (encoding the potassium channel, Kir6.2), all of which are implicated in human beta cell proliferation; (6) we showed that genetically or pharmacologically manipulating chromatin-modifying genes and their downstream pathways, exemplified by CDKN1C, EZH2, DYRK1A and KDM6A, can lead to human beta cell proliferation; a finding that validates the utility of the insulinoma data mine as a drug discovery resource [39]. We are currently in the process of diving deeper into this insulinoma data mine.
Challenges ahead
Although progress in therapeutic human beta cell replication has been remarkable, additional difficult challenges remain, as summarised in the text box. As noted above, generating higher rates of proliferation, developing tools to allow beta cell-specific drug targeting and imaging in humans, and overcoming or preventing continuing autoimmunity are all hurdles that will need to be surmounted. Further, one must also worry that, if regenerative pathways are driven too aggressively, there may be oncogenic consequences. Of course, the pathways being driven (for example, reducing cell cycle inhibitors and increasing cell cycle activators) are shared in normal developmental physiology and oncogenesis. We would argue that whereas oncogenic mutations are permanent and irreversible, developmental and drug-driven cell cycle induction are temporally restricted. These concepts predict that, following drug removal, beta cells will return to baseline quiescence. It is also important to highlight that, while harmine family members have been shown to increase proliferation in vitro and in vivo in transplanted human islets, it has yet to be demonstrated that this translates into greater numbers of human beta cells. Demonstrating this will likely require long-term human islet transplant studies with higher rates of proliferation. Similarly, while data available suggest that harmine family members may enhance beta cell differentiation, longer term in vivo studies using human islets transplanted into animal models of type 1 and type 2 diabetes are required to determine whether this effect occurs in intact animals and persists over time.
Overall, while daunting challenges remain, what has transpired in the field of human beta cell regeneration over the past few years is outstanding. Together, these findings provide optimism that the next group of challenges can be overcome, and that preclinical and clinical trials are on the horizon.
Abbreviations
- BrdU:
-
Bromo-deoxyuridine
- CDK:
-
Cyclin-dependent kinase
- CLK:
-
Cell-division cycle-like kinases
- DYRK:
-
Dual-specificity tyrosine phosphorylation-regulated kinase
- EdU:
-
5-ethynyl-2′-deoxyuridine
- GSK:
-
Glycogen synthase kinase
- NFaT:
-
Nuclear factor activated in T cells
- RNA-seq:
-
RNA sequencing
References
Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC (2003) Beta cell deficit and increased beta cell apoptosis in humans with diabetes. Diabetes 52:102–110
Meier JJ, Bhushan A, Butler AE, Rizza RA, Butler PC (2005) Sustained beta cell apoptosis in longstanding type 1 diabetes: indirect evidence for islet regeneration? Diabetologia 48:2221–2228
Wang P, Fiaschi-Taesch NM, Vasavada RC, Scott DK, Garcia-Ocaña A, Stewart AF (2015) Advances and challenges in human beta cell proliferation for diabetes. Nat Rev Endocrinol 11:201–212
Campbell-Thompson M, Fu A, Kaddis JS et al (2016) Insulitis and beta cell mass in the natural history of type 1 diabetes. Diabetes 65:719–731
Hering BJ, Clarke WR, Bridges ND et al (2016) Phase 3 trial or transplantation of human islets in type 1 diabetes complicated by severe hypoglycemia. Diabetes Care 39:1230–1240
Van Dellen D, Worthington J, Mitu-Pretorian OM et al (2013) Mortality in diabetes: pancreas transplantation is associated with significant survival benefit. Nephrol Diab Transplant 28:1315–1322
Millman JR, Xie C, van Dervort A, Gurtler M, Pagliucia FW, Melton DA (2016) Generation of stem cell-derived β-cells from patients with type 1 diabetes. Nat Commun 7:11463
Zhu S, Russ HA, Wang X et al (2016) Human pancreatic beta-like cells converted form fibroblasts. Nat Commun 7:10080
Rezania A, Bruin JE, Arora P et al (2014) Reversal of diabetes with insulin-producing cells derived from human pluripotent stem cells. Nat Biotechnol 32:1121–1133
Thorel F, Nepote V, Avril I et al (2010) Conversion of adult alpha cells to beta cells after extreme beta cell loss. Nature 464:1149–1154
Talchai C, Xuan S, Lin HV, Sussel L, AcciIi D (2012) Pancreatic beta cell dedifferentiation as a mechanism of diabetic beta cell failure. Cell 150:1223–1234
Cinti F, Bouchi R, Kim-Muller JY et al (2016) Evidence of beta cell dedifferentiation in human type 2 diabetes. J Clin Endocrinol Metab 101:1044–1054
Rui J, Deng S, Arzai A, Perdigoto AL, Liu Z, Herold KC (2017) Beta cell that resist immunological attack develop during progression of autoimmune diabetes in NOD mice. Cell Metab 25:727–738
Wang P, Felsenfeld DP, Liu H et al (2015) A high-throughput chemical screen reveals that harmine-mediated inhibition of DYRK1A increases human pancreatic beta cell replication. Nat Med 21:383–388
Meier JJ, Butler AE, Saisho Y et al (2008) Beta cell replication is the primary mechanism subserving the postnatal expansion of beta cell mass in humans. Diabetes 57:1584–1594
Kassem SA, Ariel I, Thornton PS, Scheimberg I, Glaser B (2000) Beta-cell proliferation and apoptosis in the developing normal human pancreas and in hyperinsulinism of infancy. Diabetes 49:1325–1333
Gregg BE, Moore PC, Demozay D et al (2012) Formation of a human beta cell population within pancreatic islets is set early in life. J Clin Endocrinol Metab 97:3197–3206
Perl S, Kushner JA, Buchholtz BA et al (2010) Significant human ß-cell turnover is limited to the first three decades of life as determined by in vivo thymidine analog incorporation and radiocarbon dating. J Clin Endocrinol Metab 95:E234–E239
Cnop M, Hughes SJ, Igoillo-Esteve M et al (2010) The long lifespan and low turnover of human islet beta cells estimated by mathematical modeling of lipofuscin accumulation. Diabetologia 53:321–330
Shen W, Taylor B, Jin Q et al (2015) Inhibition of DYRK1A and GSK3B induces human beta cell proliferation. Nat Commun 6:8372
Dirice E, Walpita D, Vetere A et al (2016) Inhibition of DYRK1A stimulates human beta cell proliferation. Diabetes 65:1660–1671
Aamodt KI, Aramandia R, Brown J et al (2016) Development of a reliable automated screening system to identify small molecules and biologics that promote human beta cell regeneration. AJP Endorinol Metab 311:E859–E868
Wang YJ, Golson ML, Schug J et al (2016) Singe cell mass cytometry analysis of human endocrine pancreas. Cell Metab 24:616–626
Salomon D, Maeda P (1986) Heterogeneity and contact-dependent regulation of hormone secretion by individual beta cells. Exp Cell Res 162:507–520
Avrhami D, Klochendler A, Dor Y, Glaser B (2017) Beta cell heterogeneity: an evolving concept. Diabetologia 60:1363–1369
Dorrell C, Schug J, Canaday PS et al (2016) Human islets contain four distinct subtypes of beta cells. Nat Commun 7:11756
Johnston NR, Mitchell RK, Haythorne E et al (2016) Beta cell hubs dictate pancreatic islet responses to glucose. Cell Metab 24:389–401
Enge M, Arda HE, Mignardi M et al (2017) Single cell analysis of human pancreas reveals transcriptional signatures of aging and somatic mutation patterns. Cell 171:321–330
Vetere A, Choudhary A, Burns SM, Wagner BK (2014) Targeting the pancreatic beta cell to treat diabetes. Nat Rev Drug Discov 13:278–289
Shirakawa J, Kulkarni RN (2016) Novel factors modulating human beta cell proliferation. Diabetes Obes Metab 18(Suppl1):71–77
Fiaschi-Taesch NM, Kleinberger JW, Salim F et al (2013) Developing a human pancreatic beta cell G1/S molecule atlas. Diabetes 62:2450–2459
Fiaschi-Taesch NM, Kleinberger JW, Salim F et al (2013) Cytoplasmic-nuclear trafficking of G1/S cell cycle molecules and adult human beta cell replication: a revised model of human beta cell G1/S control. Diabetes 62:2460–2470
Kulkarni RN, Bernal-Mizrachi E, Garcia-Ocaña A, Stewart AF (2012) Human ß-cell proliferation and intracellular signaling: driving in the dark without a roadmap. Diabetes 61:2205–2213
Bernal-Mizrachi E, Kulkarni RN, Stewart AF, Garcia-Ocaña A (2014) Human β-cell proliferation and intracellular signaling part 2: still driving in the dark without a roadmap. Diabetes 63:819–831
Stewart AF, Hussain MA, Garcia-Ocana A et al (2015) Human beta cell proliferation and intracellular signaling: part 3. Diabetes 54:1872–1885
Avrahami D, Li C, Zhang J et al (2015) Aging-dependent demethylation of regulatory elements correlates with chromatin state and improved beta cell function. Cell Metab 22:619–632
Ackermann AM, Wang Z, Schug J, Naji A, Kaestner KH (2016) Integration of ATAC-seq and RNA-seq identifies human alpha and beta cell signature genes. Mol Metab 3:233–244
Arda HE, Li L, Tsai J et al (2016) Age-dependent pancreatic gene regulation reveals mechanisms governing human beta cell function. Cell Metab 23:909–920
Wang H, Bender A, Wang P et al (2017) Insights into beta cell regeneration for diabetes via integration of molecular landscapes in human insulinomas. Nat Commun 8:767
Purwana I, Zheng J, Li X et al (2014) GABA promotes human beta cell proliferation and modulates glucose homeostasis. Diabetes 63:4197–4205
Drucker DJ (2018) Mechanisms of action and therapeutic application of glucagon-like peptide-1. Cell Metab 27:740–756
Kondegowda NG, Fenutria R, Pollack IR et al (2015) Osteoprotegerin and denosumab stimulate human beta cell proliferation through inhibition of the receptor activator of NFkB ligand pathway. Cell Metab 22:77–85
Dhawan S, Dirice E, Kulkarni RN, Bhushan A (2016) Inhibition of TGF-beta signaling promotes human pancreatic beta cell replication. Diabetes 65:1208–1218
Brown ML, Schneyer AL (2010) Emerging roles for the TGFb superfamily in pancreatic beta cell homeostasis. Trends Endocrinol Metab 21:441–448
Smart NG, Apelqvist AA, Gu X et al (2006) Conditional expression of Smad7 in pancreatic beta cells disrupts TGF-beta signaling and induces reversible diabetes mellitus. PLoS Biol 4:e39
El-Gohary Y, Tulachan S, Wiersch J et al (2014) A smad signaling network regulates islet proliferation. Diabetes 63:224–236
El Ouaamari A, Dirice E, Gedeon N et al (2016) Serpin B1 promotes pancreatic beta cell proliferation. Cell Metab 23:1–12
Stephens SB, Schisler JC, Hohmeier H et al (2012) A VGF-derived peptide attenuates development of type 2 diabetes via enhancement of islet beta cell survival and function. Cell Metab 16:33–43
Robitaille K, Rourke JL, McBane JE et al (2016) High-throughput functional genomics identifies regulators of primary human beta cell proliferation. J Biol Chem 291:4614–4625
Andersson O, Adams BA, Yoo D et al (2012) Adenosine signaling promotes regeneration of pancreatic beta cells in vivo. Cell Metab 15:885–894
Annes JP, Ryu JH, Lam K et al (2012) Adenosine kinase inhibition selectively promotes rodent and porcine islet beta cell proliferation. Proc Natl Acad Sci U S A 109:3915–3920
Wang W, Walker JR, Wang X et al (2009) Identification of small molecule inducers of pancreatic beta cell expansion. Proc Natl Acad Sci U S A 106:1427–1432
Walpita D, Hasaka T, Spoonamore J et al (2012) A human islet cell culture system for high-throughput screening. J Biomol Screen 17:509–518
Dai C, Hang Y, Shostak A et al (2017) Age-dependent human beta cell proliferation induced by glucagon-like peptide-1 and calcineurin signaling. J Clin Invest 127:3835–3844
Ogawa Y, Nonaka Y, Goto T et al (2010) Development of a novel selective inhibitor of the Down syndrome-related kinase Dyrk1A. Nat Commun 1:86
Tahtouh T, Elkins JM, Filippakopoulos P et al (2012) Selectivity, co-crystal structures and neuroprotective properties of leucettines, a family of protein kinase inhibitors derived from the marine sponge alkaloid leucettamine B. J Med Chem 55:9312–9330
Leibowitz G, Beattie GM, Kafri T et al (1999) Gene transfer to human pancreatic endocrine cells using viral vectors. Diabetes 48:1013–1019
Brierley DI, Davidson C (2012) Developments in harmine pharmacology – implications for ayahuasca use and drug-dependence treatment. Prog Neuropsychopharmacol Biol Psychiatry 39:263–272
Acknowledgements
We thank A. Garcia-Ocaña, D. Scott, M. Donovan, R. DeVita, R. Sanchez, C. Argmann and E. Schadt, all at the Icahn School of Medicine at Mount Sinai (New York, NY, USA), for invaluable discussions while the work described herein unfolded. We also apologise in advance to authors whose work we were unable to cite or discuss because of space limitations.
Funding
This work was supported by the Foundation for Diabetes Research (FDR), the Human Islet and Adenoviral Core (HIAC) of the Einstein-Sinai Diabetes Research Centre (E-S DRC), the Human Islet Research Network (HIRN) and by NIH/NIDDK grants (P-30 020541, UC-4 104211, R-01 105015, R-01 55023) and a JDRF grant (2-SRA-2015-62).
Author information
Authors and Affiliations
Contributions
All authors were responsible for drafting the article and revising it critically for important intellectual content. All authors approved the version to be published.
Corresponding author
Ethics declarations
The authors declare that there is no duality of interest associated with this manuscript.
Electronic supplementary material
ESM Downloadable slideset
(PPTX 405 kb)
Rights and permissions
About this article

Cite this article
Karakose, E., Ackeifi, C., Wang, P. et al. Advances in drug discovery for human beta cell regeneration. Diabetologia 61, 1693–1699 (2018). https://doi.org/10.1007/s00125-018-4639-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-018-4639-6