
Abstract
The outstanding clinical success of immune checkpoint blockade has revived the interest in underlying mechanisms of the immune system that are capable of eliminating tumors even in advanced stages. In this scenario, CD4 and CD8 T cell responses are part of the cancer immune cycle and both populations significantly influence the clinical outcome. In general, the immune system has evolved several mechanisms to protect the host against cancer. Each of them has to be undermined or evaded during cancer development to enable tumor outgrowth. In this review, we give an overview of T lymphocyte-driven control of tumor growth and discuss the involved tumor-suppressive mechanisms of the immune system, such as senescence surveillance, cancer immunosurveillance, and cancer immunoediting with respect to recent clinical developments of immunotherapies. The main focus is on the currently existing knowledge about the CD4 and CD8 T lymphocyte interplay that mediates the control of tumor growth.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
After a long and intense debate there is now clear evidence that the immune system mediates tumor-suppressive effects in both humans and animals [1]. Using inbred mice with specific disruption of genes affecting critical components of the immune system or by selective ablation of immune cells and cytokines it has been shown that macrophages, natural killer T (NKT) cells, αβ T cells, γδ T cells, and natural killer (NK) cells as well as deletion or neutralization of cytokines such as interferon gamma (IFN-γ) and interleukin (IL)-12 all contribute to increased susceptibility of the host to develop tumors [2,3,4,5,6]. There is also evidence for cancer immunosurveillance in humans. Immunosuppressed transplant patients and individuals with immunodeficiencies have a significantly increased incidence of tumor development [7,8,9]. Additionally, immune checkpoint blockade has also been confirmed to be able to stimulate the immune system thereby mediating remarkable tumor remissions in subgroups of patients [10, 11]. These therapeutic responses are highly heterogeneous when comparing different tumor entities. Checkpoint blockade with programmed death-1 (PD-1) or programmed death ligand-1 (PD-L1) blocking antibodies induce tumor remissions and durable responses even upon treatment discontinuation in patients with melanoma, mismatch repair-deficient cancers, lung cancers and a variety of other malignancies [12,13,14]. The most reliable marker today to predict antitumor responses is upregulated PD-L1 expression within the cancer tissue [15, 16]. A high mutational load appears to facilitate immune recognition that can be amplified by checkpoint blockade [17, 18]. Consequently, tumors that are associated with viral transformation such as Hodgkin lymphoma [19] or tumors that frequently exhibit insertion-and-deletion-derived neoantigens such as renal carcinoma [20] are among the tumor entities that respond well to checkpoint blockade. Unfortunately, basic causes predicting individual antitumor responses still remain unknown. However, it is generally accepted that tumor immunogenicity is a main driver for effective checkpoint blockade [21, 22]. Tumor immunogenicity is a general term that describes the ability of the immune system to discern healthy and malignant cells and to trigger innate as well as adaptive immune responses counteracting tumor growth in untreated hosts or upon immunotherapeutic interventions.
Among those immune cells that contribute to tumor suppression, αβ T cells have attracted the attention of tumor immunologists and clinical scientists. This cell type is the central component of the adaptive immune system and its presence within tumor tissue in human colon carcinomas is correlated with a significant survival benefit [23]. Furthermore, these cells drive potent tumor regression in late stages of cancer in mouse models [21, 24,25,26] and upon immunotherapeutic applications in patients [12, 27, 28].
Although CD4 and CD8 T cell responses to cancer have been extensively described, it is still poorly understood how regulatory mechanisms and crosstalk between T lymphocyte subsets influence tumor immunity. Understanding these molecular mechanisms of T cell regulation in depth will be vital for the rational design and improvement of immunotherapies for the treatment of cancer. In the following sections we summarize briefly the main fundamentals of T cell immunity against cancer and discuss their potential and limitations to control cancer outgrowth.
Establishment of the term ‘cancer immunosurveillance’
When Paul Ehrlich presented his studies on carcinomas in 1908, he was one of the first scientists to come up with the idea that the immune system is capable of suppressing carcinogenesis, thus lowering the frequency of developing tumors [29]. In the following decades neither his hypothesis about athreptic immunity nor his results on tumor vaccinations were thoroughly pursued, probably due to the still unevolved field of immunology. In 1957 the hypothesis about cancer immunosurveillance was revisited by several scientists. Sir Macfarlane Burnet proposed that cancers possess new antigenic properties that can provoke antitumor immunity [30]. Lewis Thomas speculated that in long-lived organisms allograft rejection is rather a side effect of the immune system, whereas its main function could be the protection of the host from altered cells and malignancy [31]. At the same time, Prehn and Main described immunity to methylcholanthrene-induced sarcomas and a study performed by Klein and colleagues demonstrated resistance to this kind of tumors in autochthonous hosts [32, 33]. In 1968, Ingegerd Hellström found evidence that cellular and humoral components of the immune system mediated antitumor effects in patients. Despite these observations, most patients suffered from tumor progression, a finding termed “Hellström paradox” [34]. It describes the dichotomy of tumor-directed immune responses in cancer patients with obvious tumor progress. In the following years, Burnet and Thomas both assumed that lymphocytes play a major role in cancer surveillance [35]. However, the details of antigen presentation and recognition by T lymphocytes remained undiscovered at that time and were outlined only some years later by Zinkernagel and Doherty in 1974, when they published their pioneering work on the function of the major histocompatibility complex (MHC) [36, 37]. This was an essential discovery to understand adaptive cellular immunity to pathogens and malignant cells. It took another 20 years until conclusive evidence from mouse models became available which demonstrated the existence of cancer immunosurveillance [3, 38]. Apart from IFN-γ, lymphocytes were clearly identified to prevent primary tumor development: in a key study by Shankaran et al., chemically-induced sarcomas derived from immunodeficient RAG2−/− (recombination activating gene 2−/−) mice were rejected in immunocompetent mice, while sarcomas obtained from wildtype mice successfully engrafted indicating reduced tumor cell immunogenicity when tumors were induced in immunocompetent mice [39]. This study also led to a refinement of the hypothesis of cancer surveillance to cancer immunoediting. The authors showed that lymphocytes not only suppress tumor growth but at the same time also shape the tumor towards low immunogenicity, due to the immunoselection that is applied by this effective extrinsic tumor suppressor system to a growing and often genetically instable tumor.
Since then, the overall interest in tumor immunology has reemerged and an increasing number of mouse models has been established to investigate tumor-suppressive mechanisms [40]. The existence of cancer immunoediting is now generally accepted and tumor immune escape is regarded as an emerging hallmark of cancer [41].
The role of lymphocytes in tumor-suppressive mechanisms and tumor outgrowth
Intrinsic tumor-suppressive mechanisms of healthy cells are active before the immune system becomes involved in tumor surveillance [42]. Aberrant activation of oncogenes in normal cells induces a p53-dependent state of stable cell-cycle arrest called cellular senescence. Senescent cells exhibit a secretory phenotype called SASP (senescence-associated secretory phenotype) [43] that is characterized by secretion of various cytokines mediating the attraction of immune cells. These immune cells ultimately lead to clearance of senescent cells in a process termed senescence surveillance [6]. In this study of Kang et al., antigen-specific CD4 T cells showing a Th1-phenotype were detected that were directed against oncogenic ras-expressing pre-malignant hepatocytes. Senescence surveillance is restricted to the pre-malignant state and requires macrophages and CD4 T cells. Interestingly, it has been shown that CD8 T cells are not required in this mechanism as long as the pre-malignant cell can be identified by SASP. If senescence is no longer maintained due to the accumulation of additional genetic alterations, such as loss of p53-function, the cell finally becomes malignant, loses its SASP and is no longer recognized by senescence surveillance. At this point, CD8 T lymphocytes start to play a crucial role in mediating tumor growth control [44]. This mechanism minimizes the risk of CD8 T cell-driven autoimmunity, since CD8 T cells are only induced in the last stage of tumor development. Additionally, the reactivation of cellular senescence in tumors by Th1-cytokines derived from CD4 lymphocytes has been identified recently as an additional mechanism for controlling tumor growth [45].
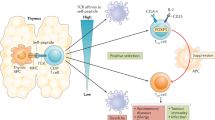
Tumor outgrowth is mainly controlled by CD4 and CD8 T cells [39]. During tumor development cancer immunoediting occurs with its three phases namely elimination, equilibrium and escape which have been described in great detail in other reviews and are therefore only briefly described here [1, 9, 40, 46]. After cellular transformation, nascent tumor lesions trigger an immune response that specifically eliminates these lesions, thus protecting the host from cancer; this refers to the elimination phase. When, however, the immune response is incapable of completely clearing the tumor cells during the equilibrium phase, but still prevents tumor outgrowth, the process of incomplete elimination promotes the generation of tumor cell variants with decreased immunogenicity. The existence of this phase and the ability of the immune system to maintain occult cancer in an equilibrium state have been demonstrated by Koebel et al. in mice [47]. Furthermore, issues with transplantation and neoantigen landscape studies indicate that this state also exists in humans [9, 48]. Following this selection process, sculpted tumor cells with low immunogenicity expand to a clinically manifest tumor. Yet, there is evidence that the immune system does not shape tumor immunogenicity towards complete tolerance. When tumor-infiltrating T cells were isolated from tumor tissue, expanded in vitro, and then retransferred in lymphopenic patients, this method showed impressive clinical effects [49]. The term “cancer immunosurveillance” is more applicable to early stages of host protection from neoplastic diseases. The concept of cancer immunoediting comprises not only events of early stages in tumor development, but also the consequences of this process on shaping the immunogenicity of a growing tumor. Figure 1 illustrates strategies of the host to facilitate tumor suppression throughout the whole course of tumor development. Furthermore, the graphic shows immune cells of vital importance for tumor-suppressive functions as described above in the pre-malignant and malignant state during tumor development.

Diverse strategies of the host have evolved to facilitate tumor suppression in different stages of malignancy. A dispensable healthy cell or a cell that acquired severe damage usually succumbs to apoptosis, a mechanism that is required to maintain cellular homeostasis. This can be regarded as an initial barrier of tumor development. Aberrant cell cycle activation leads to cellular senescence and initiates the extrinsic mechanism of senescence surveillance that limits cancer development. CD4 T cells and macrophages are the essential immune cells that mediate senescence surveillance of pre-malignant cells. When cells escape senescence surveillance and further proceed in their course of tumor development, they become malignant and are then subjected to cancer surveillance. In this phase, CD4 and CD8 T cell responses play a central role in mediating the elimination of malignant cells. T-cell-mediated cancer surveillance furthermore leads to cancer immunoediting that shapes tumors towards low immunogenicity
By the time a malignant disease becomes apparent, the tumor has escaped intrinsic growth control, senescence surveillance, and additionally, the tumor has reached the final state of cancer immunoediting, thus it has been sculptured towards low immunogenicity. The process of immunoediting has considerable impact on shaping the neoantigenic landscape of tumors for induction and expansion of T cell immune responses that determine accessibility to immunotherapies [17, 48, 50, 51].
Apart from T cell-induced selection of tumor cells towards those with reduced immunogenicity, another important mechanism affecting tumor-suppressive T cell responses is the induction of a dysfunctional state, anergy or even apoptosis of these cells, that can also have detrimental effects in immunotherapies [52]. Under certain conditions, suppression of cytotoxic T lymphocytes (CTLs) can be established upon direct cell-to-cell contact with their corresponding target cell. Tumors are capable of suppressing T cells once they migrate into tumor margin or infiltrate tumor tissue where they encounter the tumor microenvironment (TME). The TME consists of tumor cells, non-malignant stromal cells including the tumor vasculature, and stroma-infiltrating immune cells. Highly dynamic cell signaling networks, cross-talks, and adaptations within these components of the TME during tumor progression often confer resistance to tumor immunity and negatively influence therapeutic regimens by numerous mechanisms [53, 54]. A recent study by Katlinski and colleagues investigated the resistance of colorectal cancer (CRC) to CTL-mediated growth control [55]. They found that CRC cells downregulate the type I interferon receptor chain IFNAR1 in the stroma thus generating an immune-privileged niche. IFNAR1 regulates the viability of CTLs, and a low expression of this receptor confers TME stress on lymphocytes, leading to apoptosis. Other immunosuppressive mechanisms of the TME affect tumor-directed T cell responses and drive them to T cell exhaustion, a mechanism that has evolved avoiding immunopathology and is exploited by tumors to escape the immune attack. Exhausted T cells lose effector function and upregulate inhibitory receptors (reviewed in Refs. [56, 57]). This state is frequently associated with a loss of tumor growth control. Interestingly, exhaustion can be reversible. Using antagonistic antibodies targeting the exhaustion markers T cell immunoglobulin and mucin domain containing protein 3 (Tim-3) and PD-1, it has been shown that T cells can regain effector functions [58]. This has a particular clinical impact, because PD-1 blockade alone can lead to emergence of resistant tumor cell clones against immunotherapeutic treatment by loss-of-function mutations in genes encoding the interferon-receptor-associated Janus kinase 1, Janus kinase 2, or the gene encoding the antigen-presenting protein beta-2-microglobulin [59]. Some mutations also endow cancer cells with features that make them innately resistant against PD-1 blockade [60]. Development of PD-1 resistant tumors seems to involve other immune checkpoints such as Tim-3 that additionally regulate tumor directed T cell responses [61].
In models of chronic viral infections with antigen persistence much progress has been made understanding the regulation of exhausted CD8 T cells (Tex). In a recent study, Pauken et al. analyzed the cellular, transcriptional, and also epigenetic changes upon checkpoint blockade in a mouse model of chronic lymphocytic choriomeningitis virus (LCMV) infection [62]. Here, PD-L1 blockade led to a reinvigoration of Tex and induced a transcriptional re-engagement in the regulation of effector functions. However, this reinvigoration was lost again when antigen concentration remained high. Furthermore, these re-exhausted cells failed to generate memory T cells (Tmem) after antigen clearance. The results suggest that Tex is a distinct lineage of CD8 T cells. Most importantly, the authors indicate that epigenetic fate inflexibility may have limitations for immunotherapies, but moreover, treatment with IL-7 during the effector phase is able to prevent the development of Tex. Further investigations will reveal whether these findings will have impact on current immunotherapies of cancer. Sen et al. performed an in-depth analysis of the epigenetic landscape of T cell exhaustion [63]. To this end, the authors used chronic LCMV infection in mice to generate Tex and compared chromatin-accessible regions from Tex with functional Teff. This approach identified differential regulatory regions between the two groups that showed features of enhancing elements. Tex acquired a state-specific epigenetic landscape organized into functional modules of enhancers. In this study, this finding could also be validated in human antigen-specific CD8 T cells from subjects with chronic progressive HIV-1. Knowledge of functional enhancer maps may lead towards the development of regimens exploiting genome editing to alter gene expression in exhausted CD8 T cells. Thus, combining current checkpoint blockades with genome editing strategies to maintain T cell immunity to cancer cells would be an elegant approach to avoid acquired therapy resistance due to T cell exhaustion.
While T cell exhaustion for CD8 cells is quite well described, much less is known about exhaustion of CD4 T cells in cancer. Nevertheless, the phenotype of T cell exhaustion seems to occur in both T cell populations, as described in a rodent cancer model [64]. In another preclinical study by Goding et al., tumor-specific CD4 T cells were investigated and showed considerable upregulation of several co-inhibitory receptors (PD-1, TIM-3, lymphocyte-activation gene-3 (LAG-3), 2B4 and T cell immunoreceptor with Ig and ITIM domains (TIGIT)), accompanied by reduced effector cytokine expression [IFN-γ and tumor necrosis factor-α (TNF-α)] [65]. It should be noted that this dysfunction-associated phenotype occurred only in mice with relapsing melanoma and suggests that exhaustion of tumor-specific CD4 T cells contributed to recurrence of melanoma. In a clinically relevant approach, the researchers highlighted the necessity of combination therapy for the treatment of relapsing melanoma. The treatment of recurrent melanoma by combined checkpoint blockade against PD-L1 and LAG-3 turned out to be highly effective. This treatment led to a reversal of CD4 T cell exhaustion and restored the effector functions of tumor-specific CD4 T cells.
Cancer-induced T cell exhaustion consequently contributes to tumor progression by quenching remaining responses derived from residual tumor immunogenicity of immunoedited tumors. In addition, the aforementioned low immunogenicity of cancer cells is not always a result of immunoselection, but may also derive from a lack of suitable target structures for T cell recognition. As tumor-directed CD4 and CD8 T lymphocytes belong to the adaptive arm of the immune system they can only develop in dependence on their corresponding antigens. This makes tumor-specific antigens (TSAs) to be decisive molecular determinants for tumor growth control and the fundamental source of all T cell responses against cancer.
Immune targeting of cancer: tumor-specific antigens of CD4 and CD8 T cells
The major histocompatibility complex (MHC) is a highly variable region of the mammalian genome that encodes a large number of different loci comprising genes with many allelic variants [66]. The MHC class I and II genes of this polymorphic region encode the major proteins that are required for antigen presentation on the cellular surface. All MHC molecules share a common structure of a binding groove for peptides of different origin. Whereas all nucleated cells express MHC class I that bind peptides from degraded proteins of endogenous origin, MHC class II presents peptides of exogenous origin and is predominantly expressed by antigen-presenting cells, such as macrophages, B cells, dendritic cells, Langerhans, and Kupffer cells [67]. Some cells can show MHC class II expression under anomalous conditions, such as thyrocytes and intestinal epithelial cells in inflamed tissue [68, 69], as well as cancer cells [70, 71]. Furthermore, MHC class II expression can also vary depending on the location of the tumor cell: expression levels can be high in primary tumor lesions, but are often absent in metastases [72]. Abundant expression of MHC class II can also lead to potent tumor-specific CD4 T cell responses that dampen cytotoxic CD8 T cell responses in a TNF-related manner [73]. However, most tumor cells do not express MHC class II at all [74]. This has important implications for associated mechanisms of tumor-directed CD4 T cell responses that will be discussed in the next section. Preclinical studies investigating the role of CD4 T cell responses in MHC class II-positive tumors were performed in mouse models using the B16 melanoma cell line raising the question whether CD4 T cell responses are similarly regulated in other melanoma models and tumor types. Homet et al. addressed this issue by generating BRAFV600E-driven YUMM1.1 and YUMM2.1 cell lines derived from genetically engineered mice with melanocyte-specific mutant BRAF and PTEN knockout [75]. The authors comprehensively characterized the immunological properties of this model upon PD-1 blockade. Compared to the PD-1-responsive colon cancer control cell line MC38, both YUMM cell lines showed a considerably lower mutational load and distinct responsiveness to PD-1 immunotherapy. Antitumor activity was observed in YUMM2.1, but not YUMM1.1. In contrast to MC38, antibody-mediated depletion of CD8 cells only partially rescued therapeutic efficacy of PD-1 in YUMM2.1. Depleting YUMM2.1-bearing animals of CD4 cells completely abrogated the response to PD-1, suggesting a prominent role of CD4 T cells in this melanoma model. Although the MHC class II status of YUMM2.1 remains unknown, the results show differential immunological features to checkpoint blockade compared to B16 melanoma models. Also in other malignancies, apart from melanoma, MHC class II expression on tumor cells crucially affects tumor immunity. In a murine model of pancreatic ductal adenocarcinoma (PDA), two different cell lines were transduced with MHC class II transactivator (CIITA) and injected into syngeneic mice [76]. CIITA-positive PDA tumors were rejected, long-lasting memory responses were established, and recruitment of T cells to the tumor area was evident. In humans it has been shown that the magnitude of CD4 T cells in microsatellite-unstable (MSI-H) colon carcinomas is significantly higher in HLA class II-negative tumors harboring mutations in HLA class II-regulatory genes such as RFX5, CIITA, and RFXAP [77]. These data suggest that lacking HLA class II-expression on MSI-H colon carcinoma cells favored tumor progression in an environment of dense CD4 T cell infiltration. Taken together, these studies indicate that expression of MHC class II on tumor cells and corresponding CD4 T cell responses behave differently not only in a variety of tumor entities, but also in tumors of similar origin.
The frequency of tumor-specific MHC class II epitopes is usually much higher compared to MHC class I epitopes [78, 79]. This may be explained by the stringent sequence requirements and the defined length of peptides binding the MHC. MHC class II molecules have an open binding groove and peptides can protrude on both sides, making the length of binding peptides more flexible than MHC class I molecules, where the closed binding groove mainly binds peptides of 8–10 amino acids. Moreover, MHC class I is essential for CTL-mediated tumor elimination. It is therefore frequently downregulated in tumors, but a complete loss is a rare event, probably due to other counter regulations of the immune system [80] such as recognition of MHC I-depleted cells by NK cells.
Tumor-derived peptides that significantly shape tumor immunity can be divided into five classes of TSAs [81,82,83]: Overexpressed antigens (e.g. Human epidermal growth factor receptor 2 (HER-2/neu)); differentiation antigens (e.g. melanocyte differentiation antigens); mutational antigens, the vast majority of mutations are unique to each patient, hence the neoantigenome has a largely individual pattern; viral antigens [e.g. derived from Epstein–Barr virus (EBV)], and cancer-testis antigens [e.g. New York esophageal squamous cell carcinoma-1 (NY-ESO-1)]. Expression of cancer-testis antigens in adults is limited to germ cells in the testis. Tumor-associated re-expression of this class of antigens can be found in many tumor entities. Cancer-testis antigens are of particular interest for immunotherapy [84]. The classes of non-mutated antigens are associated with immune infiltrations and increased survival in patients with hepatocellular carcinoma [85, 86]. However, complete clinical remissions based on self-antigens have not been observed so far.
The TSAs that recently have come into focus are mutational antigens. Early studies using molecular methods already suggested that non-synonymous mutations are ideal candidates for the immune system to identify altered cells. Wolfel et al. identified a p16INK4a-insensitive cyclin-dependent kinase (CDK4)-R24C mutant in tumor cells of a melanoma patient. The epitope of this mutant form was targeted by CTLs and it was confirmed to occur in another cohort of melanoma patients. This mutation of CDK4 can generate a TSA and disrupt the cell-cycle regulation exerted by the tumor suppressor p16INK4a [87]. Later, it has been shown that the response of autologous T cells in melanoma is predominantly driven by mutated neoantigens [88]. Recognition of these neoantigens by the immune system is important for the therapeutic efficacy of checkpoint inhibitors [89]. Consistently, it has been shown that T cell responses raised by immune checkpoint blockade are mainly directed against mutant antigens [21, 27]. It has also been demonstrated that PD-1 is mainly upregulated on melanoma-specific T cell clones with high functional avidity [90]. In contrast, low avidity T cell clones had a substantially methylated PD-1 promotor region indicating epigenetic regulation of PD-1 expression and maintenance of peripheral blood cells in presence of PD-1 blocking antibodies led to selective expansion of high avidity T cell clones.
The effectiveness of immunotherapies against neoantigens has been convincingly demonstrated by induction of therapeutic CD4 and CD8 T cell responses in mouse models of melanoma, sarcoma, and colon cancer [21, 26, 91]. Therefore it is very likely that neoantigens will maintain their key role in future strategies of personalized immunotherapeutic applications.
In silico approaches of cancer neoantigen prediction have been used based on genomic sequencing data to generate putative peptide:MHC binders, such as SYFPEITHI [92] and NetMHC [93]. Using such prediction algorithms, identification of high affinity neoepitopes has been very successful in several studies [21, 25, 27, 50, 91]. It is known from reductionistic models using adoptive T cell therapies that binding affinities between antigenic peptides and MHC class I and also the binding affinity of the peptide:MHC complex to the corresponding T cell receptor (TCR) are critical determinants of antitumor T cell reactivity and the capability of T cell responses to reject a tumor [94]. However, there are several caveats to focus on single high affinity binding epitopes. First of all, neoepitope patterns are more or less unique among cancer patients with very little overlap. Immunotherapies such as DC-vaccinations targeting a specific neoepitope are therefore at best limited to a small number of patients. Even if more common neoepitopes bind to MHC class I with high affinity it is likely that such epitopes have undergone high selection pressure during immunoediting. Consistently, potent T cell responses against a particular epitope can also promote the occurrence of antigen-loss variants by epigenetic gene silencing of protein expression [95]. A study of Zhong et al. suggests that binding affinities above a certain threshold do not necessarily improve efficacy [96]. Additionally, neoepitope-directed CD8 T cell responses of lower affinity have also been shown to play a role in immunotherapeutic applications when regarding polyvalent responses [97, 98]. These responses would probably be best suited to prevent the generation of escape variants by the tumor. Future investigations will have to address the relevance of binding affinities in cancer neoepitope prediction in relevant models.
Taken together, occurrence of tumor-specific CD4 and CD8 T cells in tumor tissue is regarded as a good prognostic factor [23, 99], but tumor immunogenicity is not a general characteristic of tumor development [100]. Induction of tumor immunogenicity is a major aim in immune checkpoint blockade that is still achieved infrequently [15, 101].
T helper cell polarization, direct and indirect mechanisms of tumoricidal CD4 T cells, and regulatory T cells
So far we have discussed the frame conditions in which T cell responses against cancer cells arise. CD8 T cell responses are usually regarded as representing the immune cell type of paramount importance for control of tumor growth due to their potent cytotoxicity and the observation that tumors can prevent elimination by downregulation of MHC class I expression [102, 103]. In contrast, the role of CD4 T cell immunity to cancer is much less understood. It has now become increasingly clear that tumor-specific CD4 T cells display a complex biology and their roles are far beyond the mere task of providing helper signals to CD8 T cells [104]. Naïve CD4 T cells are able to differentiate into multiple effector subsets that can mediate various, even opposing functions. In one of the first studies showing that CD4 T cells have significant antitumor effects, Greenberg and colleagues performed an adoptive transfer of T cells obtained from syngeneic mice vaccinated with irradiated tumor cells that facilitated eradication of disseminated leukemia in recipients [105]. Within the last three decades several studies followed and further unraveled the mechanisms of CD4 T cells in tumor immunity.
The predominant helper cell subtypes are Th1 and Th2. Th1 commitment depends on local IL-12 secretion, whereas Th2 cells arise in dependence on IL-4 and in the absence of IL-12. Th2 cells have been reported to exert antitumor effects [106, 107]. Recently, it has been demonstrated that adoptively transferred Th2 cells were able to eradicate subcutaneous MHC class II-negative myeloma in mice [108]. The authors showed that persistence of Th2 cells in vivo correlates with long lasting immunity. The observed eradication of the tumor occurred in an antigen-specific manner and was independent of B cells, NK cells, CD8 cells, and IFN-γ. Transferred Th2 cells were able to induce a type II inflammation at the tumor site that interestingly involved M2 macrophages, which are usually regarded as a tumor-promoting macrophage subtype. Together these results highlight the potential of adoptive transfer of antigen-specific Th2 cells. Nevertheless, Th1 cells are regarded as the most important helper cell type for cancer immunity, being involved in the killing of tumor cells by secretion of cytokines that activate death receptors on the tumor cell surface and in the induction epitope spreading [109]. Th1 cells are also able to activate DC cytotoxic functions that eliminate tumor cells in an IFN-γ-dependent manner and provide a source of tumor-associated antigens derived from the killed tumor cells [110]. Furthermore, beside IFN-γ, cancer-specific CD4 T cells can also secrete IL-4 [106, 111,112,113], establish long-term memory immune responses to tumors [114, 115], and recruit eosinophils and macrophages [116, 117]. The study performed by Corthay and colleagues also comprehensively characterized the mechanism by which MHC class II-negative myeloma cells were identified and eliminated by CD4 T cells [117]. In this study, T cells from TCR-transgenic donors with a SCID-background were transferred to recipient mice and tumor challenge led to an activation of tumor-specific CD4 T cells in the draining lymph nodes. These T cells migrated to the tumor where they massively recruited and activated macrophages. CD4 T cell-depletion completely blocked macrophage activation. Moreover, MHC class II-blocking antibodies impaired the activation of tumor-specific CD4 T cells in the draining lymph nodes and inhibited migration to the tumor site and the activation of macrophages. In further experiments the authors injected TSA-positive myeloma cells in the right flank of mice and a TSA-negative control cell line in the left flank. Tumor-specific CD4 T cells became activated in the draining lymph nodes of myeloma tumors, but not in lymph nodes of the control tumors. This showed that the primary activation of tumor-specific CD4 T cells is locally restricted to the tumor site, rather than a systemic phenomenon. Lastly, the study again stressed the importance of IFN-γ in this mechanism of immunosurveillance that is released upon collaboration of tumor-specific CD4 T cells with macrophages. The indirect recognition mechanism of Th1-polarized CD4 T cells has been comprehensively investigated in a melanoma model as well [118]. Here, a direct killing was abolished by using an MHC class II-disparate model and clearance of tumor cells by CD4 T cells was critically reliant on IFN-γ. In general, these studies demonstrate that tumor-specific Th1-polarized T cells are capable of indirect tumor cell elimination by involving assistance of macrophages.
Another helper cell commitment distinct from Th1 and Th2 is the Th17 lineage, which is induced by TGF-β and IL-6 [119, 120]. T helper cell type 17 in mice and men have been associated with the production of IL-17 [121, 122] and expression of transcription factor retinoic acid-related orphan receptor gamma t (RORγt) [123, 124]. It has also been described that cyclophosphamide induces differentiation to Th17 cells in rodents and cancer patients [125]. This lineage has been associated with infectious diseases and development of experimental autoimmune encephalitis and collagen-induced arthritis in mouse models [126, 127]. Naturally occurring Th17 cells have also been identified in association with certain kinds of cancer, such as prostate cancer and cutaneous T cell lymphomas [128, 129]. A work conducted by Muranski et al. demonstrated eradication of an established melanoma by tumor-specific Th17 that was critically dependent on IFN-γ [71]. In this study Th0, Th1, and Th17 cells were generated in vitro and then adoptively transferred to tumor bearing mice. In contrast to Th17 cells, Th1-polarized cells were capable of secreting high levels of IFN-γ, but the investigators found that Th17 cells were superior in mediating destruction of advanced melanomas. It can only be speculated, why Th17 cells are highly dependent on IFN-γ. Indeed, Th17 cells secrete, apart from IL-17 and TNF-α, IFN-γ upon in vitro stimulation in this study and in models of autoimmunity [121], but the underlying mechanism of cytokine interplay for tumor rejection and autoimmunity is to be elucidated. Another important aspect of Th17-polarized T cells is their ability to mediate direct cytotoxicity on MHC class II-positive tumors [111, 112]. Adoptively transferred T cells from TCR-transgenic mice using Trp1-specific T cells in the context of MHC class II I-Ab have been used to demonstrate that these cells are able to eradicate B16 melanoma cells through a Granzyme-dependent mechanism [130]. As already mentioned, the recognition of non-secreted MHC class II antigens by CD4 T cells is mediated by macrophages. However, it has been shown that this is not sufficient to elicit direct cytotoxic effects of CD4 T cells [131].
In Table 1 we compare the key observations of animal experiments with the results of clinical studies investigating antitumor effects of Th2 and Th17 cells. While some of the key results are compatible with each other regardless of the host, there are also some other findings with remaining discrepancies. For example, while Th2 cells have been repeatedly shown to eradicate tumors in murine models [106,107,108], they have been observed to contribute to chronic inflammation in patients with metastases in clinical studies [132]. If Th2 responses are elicited in consequence of alternatively activated macrophages in the tumor tissue, it promotes the formation of an immunosuppressive microenvironment as shown in patients with esophageal cancer [133]. This was a central issue in a recent study by Ito et al., showing that blocking IL-4 changes the microenvironment of the tumor to the favor of Th1-polarization from Th2 in mice, suggesting a possible enhancement of human immunotherapy by IL-4 mAb treatment [134]. Pharmacologic interventions can also have a significant effect on Th17 responses in mouse models [135]. The use of a Th17 inhibitor SR1001 decreased the formation of micro-invasive prostate cancer in Pten-null mice. Furthermore, the compound also showed additional antitumor effects suggesting that Th17 responses can have a tumor supportive role. Also in humans there have been seemingly conflicting reports with regard to IL-17 expression by cells in tumor tissue and survival of patients [136, 137]. It is possible that the contribution by Th17 cells during the antitumor response and promotion of tumor growth is dependent on the cancer type or even the immune status of the host [138, 139]. In ovarian cancer [140], esophageal squamous cell carcinoma (ESCC) [141], head and neck squamous cell carcinoma (HNSCC) [137] the clinical data suggest Th17 cells to be involved in tumor suppression. In the latter study the data implicates Th17 cell migration towards the tumor site where these CD4 T cells inhibit the angiogenesis and suppress the proliferation of cancer cells. Additionally, a correlation between increased tumor-infiltration by IFN-γ producing CD4 and CD8 T cells, and the increased frequency of Th17 cells in the tumor tissue could be detected [140]. In contrast, higher numbers of IL-17 producing cells in breast cancer tissue are associated with shorter disease-free survival of patients [142]. Also the intratumor- and peritumor-enrichment of IL-17 producing cells is associated with shorter survival of patients with HCC [136].
In fact, the characterization of Th17 cells based on IL-17 expression alone can be delusive, since IL-17-expressing T cells in human blood and lymphatic tissue can also belong to immunosuppressive regulatory T cells (Tregs), a T cell lineage that will be introduced shortly [143]. Voo et al. could demonstrate that IL-17 expression was not limited to Th17 cells and suppressive Tregs expressed both lineage transcription factors RORγt (Th17) and forkhead box P3 (Foxp3). They further showed that the suppression of CD4 T cell proliferation could be in vitro induced by IL-17 positive Tregs and IL-17 negative Tregs, likewise. Furthermore, evidence suggests that a conversion of Th17 cells into immunosuppressive regulatory T cells is possible in the tumor microenvironment [144]. This finding may reflect the increased frequency of IL-17-producing cells in HCC, a cancer type where accumulation of Tregs is associated with cancer recurrence [145]. Overall, the plasticity of Th17 cells and seemingly contradictory findings in various cancers necessitate more detailed characterizations of this T helper cell type.
A CD4 T cell subtype that has been identified to play an important role in maintaining immunological tolerance to self- and non-self-antigens are regulatory T cells. Depletion of these cells causes several autoimmune disease-phenotypes in mice. Tregs are characterized by expression of the IL-2 receptor alpha chain (CD25) that was identified as a central marker by a seminal study of Sakaguchi et al. [146]. In addition, the forkhead transcription factor Foxp3 was later identified as an important regulatory transcription factor of Tregs [147, 148]. Upon characterization of these molecular markers, the link between Tregs contributing to tumor development and suppression of antitumor immunity was clearly established [149,150,151]. During cancer progression, an increase of Tregs was frequently observed independently of the thymus [152] in blood and tumor margins that suppress CD4 and CD8 T cell responses as well as dendritic cell function [153, 154]. Tregs located in the tumor microenvironment mediate immunosuppression by releasing cytokines such as TGF-β and IL-10 [155].
Several studies describe a conversion of conventional CD4 T cells to regulatory T cells in the tumor microenvironment. There is discordance whether thymus-derived Tregs mediate the conversion of conventional CD4 T cells to immunosuppressive Tregs by TGF-β and IL-10 [156, 157] or if peripherally-derived Tregs are accumulating in the tumor tissue [158, 159]. Regardless of their origin, thymus-derived Tregs and peripherally-derived Tregs independently contribute to tolerance in the tumor environment [160]. The infiltration of tumors by CD8 T cells is mostly considered as beneficial for the survival of the patient and the ratio of CD8 T cells/Tregs is regarded as a crucial prognostic factor for different types of cancer. A higher ratio of CD8 T cells/Tregs is usually associated with a favorable outcome as demonstrated in ovarian and liver cancer [145, 161]. A shift in the balance of tumor-infiltrating lymphocytes towards higher numbers of Tregs is associated with a poor prognosis in gastric and breast cancer [162, 163]. On the other hand, lymphomas and colorectal cancer account for malignancies in which occurrence of high numbers of Foxp3-positive Treg cells is associated with a good prognosis [164,165,166,167]. Unfortunately, the underlying mechanism of immune regulation of this opposing role for Tregs is not yet known.
A number of studies have already assessed suppressive mechanisms of Tregs. There is convincing evidence that the co-inhibitory receptor cytotoxic T lymphocyte antigen 4 (CTLA-4) is a potent mediator of Treg-induced immunosuppression [168]. In particular, during the interaction of Tregs and antigen-presenting cells, CTLA-4 is able to bind with high avidity to the CD80 and CD86 molecules that are present on the surface of antigen-presenting cells (APCs) [169]. This interaction leads to down-regulation and capture of CD80 and CD86 by trans-endocytosis from APCs, thereby depriving CD8 T cells of co-stimulatory signals that are required for sufficient activation of T cells [170, 171]. This process is mediated by CD80/CD86 ligation to the co-stimulatory CD28 receptor expressed on CD8 T cells. The impact of preventing interference by CTLA-4 has been demonstrated in melanoma patients where CTLA-4-blockade induced a broadening of tumor directed T cell responses [172]. Although the relevance of Tregs is mostly considered during initial T cell priming whereby Tregs inhibit T cell activation by CTLA-4 intervention, there is also evidence that Tregs apply other mechanisms to enforce peripheral tolerance in the tumor environment. In a model of acute myeloid leukemia (AML) only wild-type Tregs prevented tumor rejection, compared to Granzyme B knockout (Gzmb −/−) or Perforin1 knockout (Prf1 −/−) Tregs [173]. The regulatory T cells from the tumor microenvironment were also able to induce cell death of NK cells and CD8 T cells that required Granzyme B and perforin. In a different cancer model Bauer et al. highlighted that antigen recognition by Tregs in the tumor tissue is necessary to prevent tumor rejection [174]. These mechanisms of immunological tolerance induction reflect the role of Tregs in the tumor microenvironment by shifting the CD8 T cell/Treg ratio in favor of immunosuppression in various cancer types.
Furthermore, therapeutic agents have been described to influence the Treg-population: in cancer patients IL-2 increases the amount of Tregs [175, 176] and prostaglandin E2 can be used to induce Foxp3-expression and to increase suppressive activity of CD4 + CD25 + cells [177]. In contrast, low-dose cyclophosphamide eradicates Tregs and can thereby sensitize tumors to immunotherapy [178]. Hence, therapeutic interference with Treg-activity appears to be an important strategy to overcome immunosuppression of T cell responses in various immunotherapeutic applications.
Taken together, these studies show the complex regulation of immune responses by CD4 T cells that mediate antitumor immunity or promote tumor growth, depending on the context and the applied regimens. CD4 T cells are able to communicate with several types of immune cells and other cells of non-hematopoietic origin. Thus, they are a key component for modulation of tumor immunity. Finally, functional CD8 T cells are strictly dependent on CD4 T cell responses against cancer, a point that is often neglected in studies investigating cytotoxic CD8 T cell responses.
The interplay of CD4–CD8 T cells in controlling tumor growth
For cellular immunity the mutual relationship between CD4 and CD8 T cells for tumor suppression is a special feature of the immune system, since interactions between CD4 and CD8 T cells both derive from the adaptive arm of the immune system and require antigen experience for both T cell populations to trigger antitumor immunity. In a broader sense, the crosstalk of these T lymphocytes is part of the cancer immune cycle [179, 180]. This cycle describes the sequence of events that leads to the generation of tumor-directed T cell responses induced by dendritic cells that have captured TSAs, migrate to the lymph nodes and present their antigens to T cells. Tumor-specific T cells clonally expand and subsequently migrate through the lymphatic vessels and other tissues to the tumor, where malignant cells are successfully eliminated by these T lymphocytes. In the context of immunotherapies, all combined factors influencing the cycle of this complex process have also been described as the ‘cancer-immune set point’ [181]. Only a fully completed cycle enables the immune system to counteract tumor growth. To that effect, any interruption or defect within a single step of this cancer immune cycle can lead to failure of the whole process allowing the tumor to escape immune control. In this regard, the same rule is applicable to the CD4 and CD8 T cell interplay, as being part of the cancer immune cycle: any disruption of crucial cross-talk events results in a failure of tumor growth control. Incidents that can lead to a failure of the cancer immune cycle due to erroneous CD4 and CD8 T cell signaling are shown in Fig. 2.

Failure of tumor rejection is often due to a malfunction within any step of the cancer immune cycle. Central components of this cycle are CD4 and CD8 T cells that are involved in all steps of the cycle. Crucial steps of the cancer immune cycle are outlined by the arrows. Any disruption of mutual CD4/CD8 T cell interplay or other crucial steps of T-cell-signaling within this circle that have been demonstrated to abort the whole cancer immune cycle are noted within the graphic representation. The figure comprises studies that have been discussed in this review and therefore does not provide a complete overview. All descriptions include the reference to original studies
Several concepts and mechanisms for T cell crosstalk derive from mouse models using pathogens. Whereas rapid T cell contraction is generally observed after pathogen infections, frequently observed ongoing responses upon treatment discontinuation of immunotherapies in cancer patients challenge the view of the consistency in the regulation of pathogen- and cancer-derived T cell responses [11, 182]. Whether conclusions based on such pathogen models are generally applicable for cancer immunology is therefore an issue that needs further attention. The obvious differences in the requirements for the immune system to mediate pathogen clearance and to drive tumor remissions also suggest differences in immune regulations and T cell phenotypes. Accordingly, there is a general ambition to summarize the characteristics of cancer-specific T cells [183].
Also cells of non-hematopoietic origin appear to play a crucial role in cytokine-mediated tumor regression. Numerous studies emphasize the importance of IFN-γ in tumor suppression, a cytokine that is frequently released by activated CD4 and CD8 T cells. A recent study unraveled the effect of IFN-γ on the TME and identified cells of the tumor stroma that are involved in the rejection of large, established tumors [184]. Whereas T cells, other hematopoietic cells, or fibroblasts were not sufficient to induce IFN-γ-induced tumor regression, Kammertoens et al. showed that responsiveness to IFN-γ of endothelial cells was necessary and sufficient in this model. Further analysis revealed IFN-γ-induced regression of the tumor vasculature that was similar to non-hemorrhagic necrosis in ischemia. These results underline the complex regulation of the IFN-γ-pathway and suggest that T cells, as a source of IFN-γ, can also contribute to tumor remissions mediated by endothelial cells.
Until now only few studies investigated the contribution of both CD4 and CD8 T cell responses in tumor models with autologous immunity. Pioneering in vitro studies that addressed CD4/CD8 T cells interplay emphasized the importance of both populations for tumor immunity [185, 186]. The use of TCR transgenic mice with specificities for MHC class I and II as a source of naïve HA-specific T lymphocytes in a model of malignant mesothelioma showed a greatly enhanced T cell response and tumor rejection when CD4 and suboptimal numbers of CD8 lymphocytes were co-transferred, whereas adoptive transfer of CD8 T cells alone was not sufficient to induce tumor remissions [187]. Consistently, Church and colleagues demonstrated that tumor-specific CD4 T cells help to maintain functionality of tumor-directed CD8 T cells [188]. They observed that CD8 T cells expressed lower levels of PD-1 suggesting that the presence of CD4 T cells partially inhibits CD8 T cell exhaustion. In general, it has been shown that the presence of tumor-specific CD4 T cells enhanced recruitment, proliferation, and effector functions of CD8 T cells by IFN-γ-dependent production of chemokines and IL-2 [189]. Help provided by CD4 T cells was able to further improve the functionality of CD8 T cells with high affinity TCRs [190]. On the other hand, Wong et al. reported that CD4 T cells render the tumor microenvironment permissive for infiltration by low avidity CD8 T cells [191]. Hung et al. investigated the central role of CD4 T cells in antitumor immunity and revealed that these cells not only provide signals for CD8 T cells, but have a far broader role in orchestrating immune responses to the tumor [116].
These studies demonstrate that the use of adoptive transfer of T cells in mouse tumor models is a suitable method to shed light on mechanisms of T cell regulation and is one option for successful eradication of tumors. The majority of hitherto existing models, however, utilize transgenic T cells with high affinity model antigens that do not match with the nature of most human tumors. Furthermore, this method generally skips crucial steps of establishing T cell immunity in the cancer immune cycle. In humans, the individual mutanome give rise to neoantigens of various affinities and is itself subject to a high plasticity of de novo generation and elimination of neoantigens, thereby shaping the genomic landscape. This occurs due to the genetic instability of the tumor, especially in MSI-H tumors [192]. Theoretically, generation of high affinity T cell epitopes during advanced tumor stages by a subset of cells merely slows down tumor progression, since only a fraction of the whole tumor mass bearing the mutation would induce potent responses and be eliminated. Neoepitopes that arise early in tumor development should be present in most, if not all tumor cells. Regimens targeting these neoantigens are able to induce potent remissions, as observed in the clinic today. Comparing this to mouse models using adoptive transfer of monoclonal T cells, the latter approach appears feasible to study individual aspects of T cell immunology in models of solid tumors, but it apparently fails to describe the clinically relevant course of tumor onset. The cancer immune cycle that achieves a complete remission would require checkpoint blockade such as CTLA-4- or PD-1-blocking antibodies, as well as Treg manipulation. These interventions lower the threshold of self-tolerance and trigger immune responses that may be directed to neoepitopes derived from early stages of tumor transformation.
Orchestrating immune responses by CD4 and CD8 T cells was unraveled by a study carried out by the group of Hans Schreiber, interrogating the cooperation of CD4 and CD8 T cell responses in a model of bystander killing of cancer [193]. Here, potent model antigens were expressed by a MHC-disparate tumor and the antigens were found to be displayed on cross-presenting stromal cells of tumor tissues. The potent MHC class I and II antigens elicited autologous T cell responses that led to elimination of tumor-maintaining stromal cells and subsequently induced tumor remissions. They showed that co-expression of MHC class I and II antigens was required for bystander elimination of cancer cells. The bystander elimination of cancer cells turned out to be highly efficient, since even antigen-loss variants embedded in the TME were eliminated. The inoculation of a mixture of tumor cells with separated antigens for either MHC class I or II led, in sharp contrast to double MHC class I and II-positive cancer cells, to progressive growth of mosaic tumors. The required expression of MHC class I and class II antigens by the same cell clearly demonstrated a local cooperation of CD4 and CD8 T cells during the effector phase. Moreover, experiments using mice with double positive tumors on one flank and a mixed tumor with separated class I and II antigen expression on the contralateral flank led to outgrowth of the latter tumor. These observations again demonstrate that successful bystander killing of cancer cells requires local cooperation of CD4 and CD8 T cells not only during the effector phase but also during the induction phase of tumor rejection. These results also highlight that the orchestration of immune responses by CD4 T cells is not only limited to local tumors, but even suggest that CD4 T cells are able to differentiate between individual sub-clones within the tumor tissue that are class II TSA-positive or -negative.
One feature of cancer is a unique pattern of the mutational landscape. Unlike pathogens, malignant cells are not characterized by a conserved set of immunogenic antigens. In a recent study we took advantage of this feature to establish a rodent model with tailored tumor immunogenicity to further elucidate the interplay of autologous CD4 and CD8 T lymphocytes [194]. To this end, we took advantage of transposable elements expressing oncogenic Ras that we delivered by hydrodynamic tail vein-injection to induce liver cancer in mice. Oncogenic forms of K-, H-, and NRas rapidly induced CD8 T cell responses against co-delivered model antigens, whereas no CD8 T cell responses were detectable when expression of ras was absent or when wildtype- or dominant-negative forms of ras were used. This established a direct link between an oncogene and the induction of T cell responses. Tailored tumor immunogenicity was generated by concatenated short DNA fragments coding for pre-defined single epitopes. This allowed for dissecting MHC class I and II epitopes and also for comparison of epitopes derived from immunoedited tumors with potent rejection antigens. Tumor suppression only occurred when potent MHC class I and II epitopes were both expressed by the tumor cells. Expression of antigens restricted to MHC class I epitopes induced strong CD8 T cell responses that were not capable of eliminating tumor cells without tumor-specific CD4 T cells. When MHC class II epitopes were solely expressed by the tumor, no CD4 T cell responses were detected. The results of these experiments demonstrated the mutual dependence of CD4 and CD8 T cell responses, since these cells needed to act in concert to efficiently suppress tumor development and to allow for long term survival. The autologous tumor-specific T cells showed a Th1-polarization in this study with a low, but significant cytotoxicity. However, these CD4 T cells were not able to suppress outgrowth of liver tumors without significant amounts of tumor-specific CD8 T cells. Furthermore, this study also showed the limitations of mutated CD4 and CD8 neoepitopes for cancer surveillance: although these epitopes where high affinity binders to their MHC molecule and responded well upon application of immunotherapies in corresponding parental cell lines, the host’s immune system failed to mount an immune response able to inhibit cancer outgrowth.
A similar unresponsiveness has also been described for heavily glycosylated MUC1 and Her2/neu, which was not processed by DCs and remained long term in the early endosomes [195]. In contrast, a non-glycosylated form of MUC1 was efficiently processed by DCs. These results suggest that TSAs may often be inappropriate T cell targets for the immune system and require therapeutic interventions or modifications to overcome tolerance. The interplay of CD4 and CD8 T cell responses may also counteract tolerance induction by the tumor and lower the threshold of tumor immune recognition for CD8 T cell epitopes by induction of epitope spreading to antigens that do not trigger tumor remission without CD4 help. Surman et al. showed that adoptive transfer of Th1-polarized CD4 T cells induced tumor-specific CD8 T cell responses and tumor remission [196]. The authors suggested that lowering the threshold for immune recognition was achieved by CD4 T cells that enhanced the ability of APCs to trigger CTLs to a model antigen. Investigating the interactions between CD4 T cells with APCs revealed CD40-CD40L crosstalk to trigger effective CTLs in a CCL5-dependent manner [197]. In the same study, CCR5-positive DCs were attracted to the tumor site and were then licensed by CD4 T cells prior to the generation of CD8 T cell immunity. This CD4-mediated CD40-dependent licensing of DCs as a precondition for functional CTLs has also been shown in vitro for human cancers [198]. Another study demonstrating the importance of CD4 T cells in a murine model showed that CD8 T cell tolerance to the self-antigen MDM-2 could be overcome by adoptive transfer of TCR-modified CD4 T cells [199]. TCR-engineered CD8 T cells displayed an exhausted phenotype lacking cytotoxic function. Engineered CD4 T cells allowed for T cell help that facilitated a partial reversal of tolerance with the same MDM-2-specific TCR. Similar results of converting tolerized CD8 T cells were observed in a rodent TCR transgenic model with weakly immunogenic tumors [200]. Although transgenic CD4 and CD8 T cells were transferred and CD8 T cells were able to recognize their corresponding cross-presented antigen in draining lymph nodes, MHC class II antigen presentation on CD8-positive dendritic cells and subsequent priming of CD4 T cells was defective due to the influence of the TME. In accordance with studies mentioned above, in which CD8 T cell immunity was triggered or reactivated by CD4 lymphocytes, this work revealed that T cell immunity entirely failed to counteract tumor growth when the generation of CD4 T cell responses was interrupted.
On the one hand, these studies show the importance of T cell responses that may often dictate tumor regression or progress. On the other hand, CD4 and CD8 T cell responses frequently fail to maintain proper function during tumor remissions. Therapeutic intervention to sustain and promote T cell immune reactions directed to the tumor is therefore a major aim in clinical oncology. The rational design of such therapies will require a detailed understanding of CD4 and CD8 T cell regulation including their immune checkpoint signaling.
Interplay of CD4 and CD8 T cells in cancer immunotherapies
Cancer immunotherapies are designed to activate or reactivate a therapeutic antitumor activity in the immune system. There are different strategies to modulate the immune system and to induce or expand tumor-specific T cells: administration of therapeutic cancer vaccines against tumor-associated antigens [201,202,203]; application of cytokines, such as IL-2 [204]; cell based therapies including adoptive cell transfer of naturally occurring tumor-reactive lymphocytes [205] or genetically modified autologous T cells expressing chimeric antigen receptors (CARs) such as CD19 [206]; the use of oncolytic virotherapy to mediate tumor cell death and stimulate T cell-mediated tumor immunity [97, 207,208,209]; and immune checkpoint blockade targeting T cell regulatory pathways [210]. Albeit most pharmaceuticals are used off-the-shelf, the latter approach provides an attractive treatment option, resulting from the therapeutic effect on the immune system that stimulates T cell responses and subsequently target predominantly individual neoantigens of the tumor [211, 212].
In this regard, cancer mouse models in combination with next generation sequencing of murine cell lines were helpful to rapidly identify potent T cell epitopes that were of relevance for T-cell-mediated tumor clearance upon various immunotherapeutic regimens as described above. Valuable knowledge about molecular mechanisms of checkpoint blockade and regulation of the immune system arose from these models. However, with regard to checkpoint blockade, a critical differentiation between animal models and clinical studies in corresponding tumor entities appear reasonable. Whereas the neoantigenome of human cancers in individual patients are unique and within a certain range concerning accumulated number of mutations [213], mouse models are often based on well described cell lines with little variation and are, as such, biased. Also the method of tumor induction to generate cell lines is of vital importance here. The use of carcinogenic agents leads to abundant numbers of mutations that greatly exceed the mutation rates of comparable tumor types in humans. Compared to chemically-induced tumors, established tumor cell lines from genetically modified mouse models show a much lower mutation rate and may therefore be more suitable to investigate checkpoint blockade in rodents [75]. Additionally, there is a significant discrepancy between high affinity neoepitope occurrence in established mouse models and the frequency of high affinity antigens in the majority of patients. This is reflected by the overall response rate upon immune checkpoint blockade and the immune profiling data of van Rooij et al. that revealed a single ipilimumab-responsive MHC class I-restricted neoepitope candidate among 1075 nonsynonymous single nucleotide variants in a patient with melanoma [27]. Whether in vivo results from mouse models derived from a given cell line can be generalized to the corresponding tumor entity in humans remain, as mentioned above, questionable.
In 2011, the FDA approved ipilimumab, the first antibody targeting CTLA-4, for the treatment of metastatic melanoma [214]. Thus, clinical application required several years of research and development until the first evidence for antitumor immunity was established [215]. Since approval of ipilimumab, immune checkpoint therapies have established their position among the most effective cancer treatments available for patients today [216]. Unfortunately, despite of the high number of already approved drugs, immune-related adverse events are a significant problem. In the case of ipilimumab, a positive response is observable in one-fifth of the patients [217]. However, 10–35% of recipients also suffer from severe side effects [218,219,220]. In general, immune related adverse events of checkpoint inhibitors also depend on the target molecule. Compared to ipilimumab, the PD-1 checkpoint inhibitor pembrolizumab showed less high-grade toxicity in a large clinical study [221]. Until now, PD-1 checkpoint inhibitors such as nivolumab and pembrolizumab are regarded as the most effective immunotherapies with the best safety profile [222]. The majority of immune related adverse events are, if diagnosed timely, completely reversible upon proper management [223]. Compared to treatment of lung cancer with conventional regimens such as chemotherapy or radiation, immunotherapies generally are associated with less side effects and improve daily living [224]. Without much doubt similar results concerning this issue will be evident upon comparison of immunotherapy with conventional therapies in other tumor entities.
Indeed, melanoma patients with PD-L1 positive tumors that received a combination of nivolumab and ipilimumab showed the same progression-free survival as the patients in the nivolumab group [225]. In contrast, patients with PD-L1-negative tumors had longer progression-free survival in the combination therapy group, compared to nivolumab or ipilimumab alone. This shows the importance of the PD-L1 status of tumors as a biomarker for efficacy of immunotherapies, T cell responses and frequency of tumor remissions during checkpoint blockade. Although therapeutic interventions in cases of advanced melanoma and recurrent small-cell lung cancer (SCLC) using a combination of nivolumab and ipilimumab induces a considerably higher response rate compared to corresponding monotherapies, this combination also led to an increased higher number of treatment-related adverse events [182, 225, 226]. A phase 3 trial of patients with unresectable or metastatic melanoma who progressed after ipilimumab treatment demonstrated that some patients benefit from PD-1 checkpoint blockade using nivolumab [227]. Patients in this trial showed a better response rate after nivolumab treatment (31.7%), compared to patients treated with chemotherapy (10.6%). The reason why some patients responded to nivolumab again, while others were resistant to this immunotherapy remains obscure. However, these results demonstrate that acquired resistance to an immune checkpoint therapeutic is restricted to the target molecule and allows for effective continuation of immunotherapeutic treatment with other checkpoint targets. Thus, diverse immunological checkpoints enable continued adaptive immune response to cancer, which is mediated by cytotoxic T cells. Patients that received nivolumab had fewer treatment-related adverse events, compared to chemotherapy-treated patients. Weber et al. confirmed that patients with PD-L1-positive tumors also had a markedly better objective response (43.6%) compared to patients with PD-L1 negative tumors (20.3%).
There are a handful of studies that investigate T cell responses in recipients, providing valuable information on the human immune reactions. Such evidence suggests that CTLA-4 treatment induces a CD8 T cell response against new targets in melanoma patients, rather than boosting a pre-existing immune response [172]. The exact mechanism by which the T cells of patients during immune checkpoint blockade are triggered is yet to be elucidated. Initially, it was suggested that upon binding CTLA-4 the monoclonal antibody blocks its inhibitory effect on both effector and regulatory T cells, thereby increasing effector T cell activity sufficient for induction of tumor regression. Simpson et al. studied this mechanism and refined this view in a mouse model of cancer [228]. First, they showed that CD4 T cells are crucial for tumor rejection. Mice lacking CD4 T cells were unresponsive to CTLA-4 antibody treatment when challenged with a transplantable melanoma cell line. CTLA-4 treatment has been documented to enhance the ratio of intratumoral Teff and Tregs of the CD4 T cell compartment [112, 229, 230], an effect that was proposed to be important for the success of immunotherapy [231]. To elucidate the mechanisms responsible for this effect, the authors of this study injected Trp1-specific CD4 T cells into mice that were challenged with a melanoma cell line, and showed that CTLA-4 inhibition increased both Trp1-specific Teff and Treg cell numbers, while reducing intratumoral accumulation of Trp1-specific Tregs rapidly by depletion. After establishing that these results are neither observable in complement deficient nor in γ-chain knockout mice, they identified antibody-dependent cell-mediated cytotoxicity (ADCC) as the driving mechanism of tumor-residing Treg depletion, mediated by Fcγ receptors, mainly FcγRIV. They could also show that FcγRIV-positive macrophages facilitating Treg depletion were enriched in the microenvironment of tumors in mice responding to CTLA-4 treatment, underlining once again the importance of modifying the tumor microenvironment for a successful immune checkpoint blockade therapy.
The antitumor activity of CTLA-4 therapy is also strongly dependent on the isotype. Applications of identical CTLA-4 antibodies with diverse isotypes have shown profound differences in their efficacy and features to stimulate immunity throughout different mouse strains and MHC haplotypes [232]. In contrast to other isotypes, only IgG2a was highly effective in eradication of established tumors. It is important to note that antitumor activity of CTLA-4 IgG1 was comparable to the isotype control, stressing the importance of the isotype of monoclonal antibodies for therapeutic outcomes. Selby et al. also demonstrated the selective depletion of Tregs from the tumor site by CTLA-4-IgG2a and reported significant improvement of the Teff to Treg ratio. Interestingly, Tregs were expanded by all CTLA-4 isotypes at peripheral sites. However, constitutive expression of CTLA-4 was found only on tumor Tregs, providing a rationale for selective depletion of this cell type from tumors by ADCC or antibody-dependent cellular phagocytosis. It has to be noted that most human Fcγ receptors have the same CD and name as mouse Fcγ receptors. Their function in binding IgGs and their expression pattern is, however, quite different [233]. For instance human IgG1 binds to all activating Fcγ receptors, whereas mouse IgG1 binds only to activating FcγRIII. Hence, Fc receptor–immunoglobulin interactions and their subsequent effector signaling vary throughout different species and have to be considered accordingly when monoclonal antibodies for immunotherapeutic applications are developed and optimized. In clinical applications, examples for CTLA-4 antibodies are tremelimumab with an IgG2 isotype and ipilimumab with an IgG1 isotype. IgG2 in tremelimumab was chosen to minimize possible detrimental effects of cytotoxicity on activated T cells and cytokine release syndrome [234]. Moreover, tremelimumab did not affect Treg frequency. It instead increased the frequency of IL-2-secreting CD4 T cells and IFN-γ-secreting CD4 and CD8 T cells [235]. Hence, tremelimumab induces tumor remissions mainly by direct activation of Teff cells in responding patients. In case of ipilimumab the IgG1 isotype is capable of activating non-classical macrophages by binding to CD16 that leads to a selective depletion of Tregs in vitro [234]. Finally, these studies also demonstrate significant differences in monoclonal antibody-mediated effects determined by the isotype for cancer therapies in the human system. Whether patients can benefit from a combination of tumor evaluation and subsequent isotype selection of therapeutic antibodies remains to be determined.
The awareness that isotypes can have a crucial influence on therapeutic outcomes led to isotype engineering and optimization that has been performed on a CD25-depleting monoclonal antibody to increase responsiveness to PD-1 therapy by exploiting the mechanism of ADCC to deplete regulatory T cells [236]. Combining Fc-optimized anti-CD25 and PD-1 antibodies enhanced proliferation of effector T cells, CD4 and CD8 alike, and improved their IFN-γ production. Moreover, this markedly improved the CD8/Treg ratio in the tumor tissue in favor of tumor-infiltrating CD8 T cells. This combination led to efficacious tumor eradication of mouse cancer cells that were resistant against PD-1 checkpoint blockade alone. These findings clearly highlight the importance of immunotherapies that address several types of immune cells, thus preventing the emergence of cancer resistances and improving the responsiveness to treatment.
The PD-1-blocking antibody pembrolizumab has been shown to elicit an increased proliferation of both, Foxp3-negative and Foxp3-positive CD4 as well as CD8 T cells in stage IV melanoma patients, displaying the most significant effects in the PD-1 expressing cells of these populations [237]. Interestingly, while 74% of patients in this study displayed an increase in PD-1-positive CD8 T cell proliferation following PD-1 checkpoint blockade, a clinical response was apparent in only 38% of the patients. After immune profiling of peripheral blood samples, a population of circulating Tex cells could be identified as a major target of PD-1 blockade, showing a peak of increase in Tex cell proliferation or reinvigoration, generally about 3 weeks after starting an immunotherapeutic regimen. These cells contained T cell clones identical to tumor-infiltrating T cells. Antigen burden was estimated based on tumor lesion-size, and was found to correlate with Tex cell reinvigoration before and after PD-1 treatment. Consistent with the results of the aforementioned study, Kamphorst et al. have shown that following PD-1 targeted therapy in non-small cell lung cancer (NSCLC) patients, their CD4 and CD8 T cells also show an increase in Ki67 expression [238]. PD-1-positive CD8 T cells were again identified to be the most responsive immune cell subset, with a proliferative activation in 70% of the patients. Upon further analysis of these cells, they proved to have an effector-like phenotype and co-express CTLA-4 with PD-1 as well as the costimulatory molecules CD27, CD28, and inducible co-stimulator (ICOS). This immunological response translated into a clinical effect in 80% of the patients. The authors suggested that the Ki67-positive PD-1-positive CD8 T cells have mostly tumor-specific TCRs. Lack of knowledge about tumor-specific T cells made it difficult to prove this claim, but they showed that PD-1 targeted therapy does not change the proliferation rate of EBV-specific PD-1-positive CD8 T cells.
Nevertheless, both considering the immune regulatory factors that prevent the development of T cell responses and using appropriate markers that impact the efficacy of immunotherapy can directly translate to higher response rates of cancer patients, as demonstrated in a recent study of NSCLC [239].
To sum up, immunotherapies activate both CD4 and CD8 T cells, albeit to a different extent. Depending on their interplay and the combination of applied therapies, this effect can be sufficient to produce a positive clinical outcome, providing a valuable alternative to conventional cancer treatment options.
Conclusions
As we have seen from the literature, a malignant disease overcomes several tumor-suppressive mechanisms of the host by a plethora of adaptations. As such cancer is an ever evolving disease that also includes escape from counter-regulations of the immune system. In advanced stages of cancer, when the disease is usually detected, CD4 and CD8 T cell responses are often ineffective and at best only slow down tumor progression due to low tumor immunogenicity, influence of the TME, or an interruption of the cancer immune cycle. Consequently, the central aim in clinical oncology is to reactivate T cell responses or, in other words, to convert a ‘cold tumor’ into a ‘hot’ one. This colloquial term covers overall issues of checkpoint blockade for cancer therapy including unpredictability of treatment responses in individual patients and low response rates in general, developments of therapeutic resistance, and the need to establish additional supportive regimens to maintain or increase tumor immunogenicity during immunotherapy. Hence, development of novel approaches would not be based on monotherapies, but rather combine strategies that help to overcome any therapy-induced acquired resistances of the tumor in order to enable a continued therapeutic success.
Development of novel treatment approaches would include the use of checkpoint inhibitor combinations, or combinations with other available clinical drugs that demonstrate the best manageable safety profile and synergize with immunotherapy.
Establishing these kinds of approaches can only be successful upon a thorough understanding of molecular mechanisms of the immune system, in particular the detailed understanding of CD4 and CD8 T lymphocyte responses against tumors. Thus, characterization and investigation of the interplay of these responses should be performed in parallel with clinical trials for the rapid development of highly effective immunotherapies for the treatment of various types of cancers.
Abbreviations
- ADCC:
-
Antibody-dependent cell-mediated cytotoxicity
- AML:
-
Acute myeloid leukemia
- APC:
-
Antigen-presenting cell
- CAR:
-
Chimeric antigen receptor
- CD:
-
Cluster of differentiation
- CIITA:
-
MHC class II transactivator
- CDK4:
-
Cyclin-dependent kinase 4
- CRC:
-
Colorectal cancer
- CTL:
-
Cytotoxic T lymphocyte
- CTLA-4:
-
Cytotoxic T lymphocyte-associated protein 4
- DC:
-
Dendritic cell
- EBV:
-
Epstein–Barr virus
- ESCC:
-
Esophageal squamous cell carcinoma
- Fc:
-
Fragment, crystallizable
- FcγR:
-
Fc gamma receptor
- Foxp3:
-
Forkhead box P3
- Gzmb :
-
Granzyme B gene
- Her2/neu:
-
Human epidermal growth factor receptor 2
- HNSCC:
-
Head and neck squamous cell carcinoma
- HRas:
-
Harvey rat sarcoma
- ICOS:
-
Inducible costimulator
- IFNAR1:
-
Interferon-alpha/beta receptor alpha chain
- IFN-γ:
-
Interferon gamma
- IL:
-
Interleukin
- KRas:
-
Kirsten rat sarcoma
- LAG-3:
-
Lymphocyte-activation gene-3
- LCMV:
-
Lymphocytic choriomeningitis virus
- MHC:
-
Major histocompatibility complex
- NSCLC:
-
Non-small cell lung cancer
- NK cell:
-
Natural killer cell
- NKT cell:
-
Natural killer T cell
- NRas:
-
Neuroblastoma rat sarcoma
- NY-ESO-1:
-
New York esophageal squamous cell carcinoma-1
- p16INK4a :
-
Cyclin-dependent kinase inhibitor 2A
- PD-1:
-
Programmed death-1
- PDA:
-
Pancreatic ductal adenocarcinoma
- PD-L1:
-
Programmed death ligand-1
- Prf1 :
-
Perforin1 gene
- PTEN:
-
Phospatase and tensin homolog
- RAG2:
-
Recombination activating gene 2
- RORγt:
-
Retinoic acid-related orphan receptor gamma t
- SASP:
-
Senescence associated secretory phenotype
- SCLC:
-
Small-cell lung cancer
- TAA:
-
Tumor-associated antigen
- TCR:
-
T cell receptor
- Teff:
-
Effector T cell
- Tex:
-
Exhausted CD8 T cell
- TGF-β:
-
Transforming growth factor beta
- Th0/1/2/17:
-
T helper cell commitment 0/1/2/17
- TIGIT:
-
T cell immunoreceptor with Ig and ITIM domains
- TIM-3:
-
T cell immunoglobulin and mucin domain containing protein 3
- TME:
-
Tumor microenvironment
- Tmem:
-
Memory T cell
- TNF:
-
Tumor necrosis factor
- Treg:
-
Regulatory T cell
- TRP1:
-
Tyrosinase-related protein 1
- TSA:
-
Tumor-specific antigen
References
Dunn GP, Old LJ, Schreiber RD (2004) The three Es of cancer immunoediting. Annu Rev Immunol 22:329–360
Noguchi Y, Jungbluth A, Richards EC, Old LJ (1996) Effect of interleukin 12 on tumor induction by 3-methylcholanthrene. Proc Natl Acad Sci USA 93:11798–11801
Kaplan DH, Shankaran V, Dighe AS, Stockert E, Aguet M, Old LJ, Schreiber RD (1998) Demonstration of an interferon gamma-dependent tumor surveillance system in immunocompetent mice. Proc Natl Acad Sci USA 95:7556–7561
Girardi M, Oppenheim DE, Steele CR, Lewis JM, Glusac E, Filler R, Hobby P, Sutton B, Tigelaar RE, Hayday AC (2001) Regulation of cutaneous malignancy by gammadelta T cells. Science 294:605–609
Smyth MJ, Crowe NY, Godfrey DI (2001) NK cells and NKT cells collaborate in host protection from methylcholanthrene-induced fibrosarcoma. Int Immunol 13:459–463
Kang TW, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, Hohmeyer A, Gereke M, Rudalska R, Potapova A, Iken M, Vucur M, Weiss S, Heikenwalder M, Khan S, Gil J, Bruder D, Manns M, Schirmacher P, Tacke F, Ott M, Luedde T, Longerich T, Kubicka S, Zender L (2011) Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 479:547–551
Starzl TE, Penn I, Halgrimson CG (1970) Immunosuppression and malignant neoplasms. N Engl J Med 283:934
Gatti RA, Good RA (1971) Occurrence of malignancy in immunodeficiency diseases. A literature review. Cancer 28:89–98
Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD (2002) Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol 3:991–998
Maker AV, Phan GQ, Attia P, Yang JC, Sherry RM, Topalian SL, Kammula US, Royal RE, Haworth LR, Levy C, Kleiner D, Mavroukakis SA, Yellin M, Rosenberg SA (2005) Tumor regression and autoimmunity in patients treated with cytotoxic T lymphocyte-associated antigen 4 blockade and interleukin 2: a phase I/II study. Ann Surg Oncol 12:1005–1016
Gettinger SN, Horn L, Gandhi L, Spigel DR, Antonia SJ, Rizvi NA, Powderly JD, Heist RS, Carvajal RD, Jackman DM, Sequist LV, Smith DC, Leming P, Carbone DP, Pinder-Schenck MC, Topalian SL, Hodi FS, Sosman JA, Sznol M, McDermott DF, Pardoll DM, Sankar V, Ahlers CM, Salvati M, Wigginton JM, Hellmann MD, Kollia GD, Gupta AK, Brahmer JR (2015) Overall survival and long-term safety of nivolumab (anti-programmed death 1 antibody, BMS-936558, ONO-4538) in patients with previously treated advanced non-small-cell lung cancer. J Clin Oncol 33:2004–2012
Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, Horn L, Drake CG, Pardoll DM, Chen L, Sharfman WH, Anders RA, Taube JM, McMiller TL, Xu H, Korman AJ, Jure-Kunkel M, Agrawal S, McDonald D, Kollia GD, Gupta A, Wigginton JM, Sznol M (2012) Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 366:2443–2454
Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, Pitot HC, Hamid O, Bhatia S, Martins R, Eaton K, Chen S, Salay TM, Alaparthy S, Grosso JF, Korman AJ, Parker SM, Agrawal S, Goldberg SM, Pardoll DM, Gupta A, Wigginton JM (2012) Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 366:2455–2465
Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, Lu S, Kemberling H, Wilt C, Luber BS, Wong F, Azad NS, Rucki AA, Laheru D, Donehower R, Zaheer A, Fisher GA, Crocenzi TS, Lee JJ, Greten TF, Duffy AG, Ciombor KK, Eyring AD, Lam BH, Joe A, Kang SP, Holdhoff M, Danilova L, Cope L, Meyer C, Zhou S, Goldberg RM, Armstrong DK, Bever KM, Fader AN, Taube J, Housseau F, Spetzler D, Xiao N, Pardoll DM, Papadopoulos N, Kinzler KW, Eshleman JR, Vogelstein B, Anders RA, Diaz LA Jr (2017) Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 357:409–413
Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, Sosman JA, McDermott DF, Powderly JD, Gettinger SN, Kohrt HE, Horn L, Lawrence DP, Rost S, Leabman M, Xiao Y, Mokatrin A, Koeppen H, Hegde PS, Mellman I, Chen DS, Hodi FS (2014) Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 515:563–567
Taube JM, Klein A, Brahmer JR, Xu H, Pan X, Kim JH, Chen L, Pardoll DM, Topalian SL, Anders RA (2014) Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res 20:5064–5074
Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, Walsh LA, Postow MA, Wong P, Ho TS, Hollmann TJ, Bruggeman C, Kannan K, Li Y, Elipenahli C, Liu C, Harbison CT, Wang L, Ribas A, Wolchok JD, Chan TA (2014) Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med 371:2189–2199
Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, Schrock A, Campbell B, Shlien A, Chmielecki J, Huang F, He Y, Sun J, Tabori U, Kennedy M, Lieber DS, Roels S, White J, Otto GA, Ross JS, Garraway L, Miller VA, Stephens PJ, Frampton GM (2017) Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med 9:34
Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, Schuster SJ, Millenson MM, Cattry D, Freeman GJ, Rodig SJ, Chapuy B, Ligon AH, Zhu L, Grosso JF, Kim SY, Timmerman JM, Shipp MA, Armand P (2015) PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med 372:311–319
Turajlic S, Litchfield K, Xu H, Rosenthal R, McGranahan N, Reading JL, Wong YNS, Rowan A, Kanu N, Al BM, Chambers T, Salgado R, Savas P, Loi S, Birkbak NJ, Sansregret L, Gore M, Larkin J, Quezada SA, Swanton C (2017) Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: a pan-cancer analysis. Lancet Oncol 18:1009–1021
Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, Ivanova Y, Hundal J, Arthur CD, Krebber WJ, Mulder GE, Toebes M, Vesely MD, Lam SS, Korman AJ, Allison JP, Freeman GJ, Sharpe AH, Pearce EL, Schumacher TN, Aebersold R, Rammensee HG, Melief CJ, Mardis ER, Gillanders WE, Artyomov MN, Schreiber RD (2014) Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 515:577–581
Charoentong P, Finotello F, Angelova M, Mayer C, Efremova M, Rieder D, Hackl H, Trajanoski Z (2017) Pan-cancer immunogenomic analyses reveal genotype-immunophenotype relationships and predictors of response to checkpoint blockade. Cell Rep 18:248–262
Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, Tosolini M, Camus M, Berger A, Wind P, Zinzindohoue F, Bruneval P, Cugnenc PH, Trajanoski Z, Fridman WH, Pages F (2006) Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 313:1960–1964
Castle JC, Kreiter S, Diekmann J, Lower M, van de Roemer N, de Graaf J, Selmi A, Diken M, Boegel S, Paret C, Koslowski M, Kuhn AN, Britten CM, Huber C, Tureci O, Sahin U (2012) Exploiting the mutanome for tumor vaccination. Cancer Res 72:1081–1091
Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ, Arthur CD, White JM, Chen YS, Shea LK, Hundal J, Wendl MC, Demeter R, Wylie T, Allison JP, Smyth MJ, Old LJ, Mardis ER, Schreiber RD (2012) Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature 482:400–404
Kreiter S, Vormehr M, van de Roemer N, Diken M, Lower M, Diekmann J, Boegel S, Schrors B, Vascotto F, Castle JC, Tadmor AD, Schoenberger SP, Huber C, Tureci O, Sahin U (2015) Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature 520:692–696
van Rooij N, van Buuren MM, Philips D, Velds A, Toebes M, Heemskerk B, van Dijk LJ, Behjati S, Hilkmann H, El AD, Nieuwland M, Stratton MR, Kerkhoven RM, Kesmir C, Haanen JB, Kvistborg P, Schumacher TN (2013) Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J Clin Oncol 31:e439–e442
Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, Wunderlich JR, Somerville RP, Hogan K, Hinrichs CS, Parkhurst MR, Yang JC, Rosenberg SA (2014) Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 344:641–645
Ehrlich P (1909) About the current state of carcinoma research. Ned Tijdschr Geneeskd 5:273–290
Burnet M (1957) Cancer; a biological approach. I. The processes of control. Br Med J 1:779–786
Thomas L (1959) Cellular and humoral aspects of the hypersensitive states. Hoeber-Harper, New York, pp 529–532
Prehn RT, Main JM (1957) Immunity to methylcholanthrene-induced sarcomas. J Natl Cancer Inst 18:769–778
Klein G, Sjogren HO, Klein E, Hellstrom KE (1960) Demonstration of resistance against methylcholanthrene-induced sarcomas in the primary autochthonous host. Cancer Res 20:1561–1572
Hellstrom I, Hellstrom KE, Pierce GE, Yang JP (1968) Cellular and humoral immunity to different types of human neoplasms. Nature 220:1352–1354
Burnet FM (1970) The concept of immunological surveillance. Prog Exp Tumor Res 13:1–27
Zinkernagel RM, Doherty PC (1974) Restriction of in vitro T cell-mediated cytotoxicity in lymphocytic choriomeningitis within a syngeneic or semiallogeneic system. Nature 248:701–702
Zinkernagel RM (1974) Restriction by H-2 gene complex of transfer of cell-mediated immunity to Listeria monocytogenes. Nature 251:230–233
Dighe AS, Richards E, Old LJ, Schreiber RD (1994) Enhanced in vivo growth and resistance to rejection of tumor cells expressing dominant negative IFN gamma receptors. Immunity 1:447–456
Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, Schreiber RD (2001) IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 410:1107–1111
Mittal D, Gubin MM, Schreiber RD, Smyth MJ (2014) New insights into cancer immunoediting and its three component phases—elimination, equilibrium and escape. Curr Opin Immunol 27:16–25
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144:646–674
Lowe SW, Cepero E, Evan G (2004) Intrinsic tumour suppression. Nature 432:307–315
Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, Desmet CJ, Aarden LA, Mooi WJ, Peeper DS (2008) Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 133:1019–1031
Naito Y, Saito K, Shiiba K, Ohuchi A, Saigenji K, Nagura H, Ohtani H (1998) CD8+ T cells infiltrated within cancer cell nests as a prognostic factor in human colorectal cancer. Cancer Res 58:3491–3494
Braumuller H, Wieder T, Brenner E, Assmann S, Hahn M, Alkhaled M, Schilbach K, Essmann F, Kneilling M, Griessinger C, Ranta F, Ullrich S, Mocikat R, Braungart K, Mehra T, Fehrenbacher B, Berdel J, Niessner H, Meier F, van den Broek M, Haring HU, Handgretinger R, Quintanilla-Martinez L, Fend F, Pesic M, Bauer J, Zender L, Schaller M, Schulze-Osthoff K, Rocken M (2013) T-helper-1-cell cytokines drive cancer into senescence. Nature 494:361–365
Schreiber RD, Old LJ, Smyth MJ (2011) Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science 331:1565–1570
Koebel CM, Vermi W, Swann JB, Zerafa N, Rodig SJ, Old LJ, Smyth MJ, Schreiber RD (2007) Adaptive immunity maintains occult cancer in an equilibrium state. Nature 450:903–907
Verdegaal EM, de Miranda NF, Visser M, Harryvan T, van Buuren MM, Andersen RS, Hadrup SR, van der Minne CE, Schotte R, Spits H, Haanen JB, Kapiteijn EH, Schumacher TN, van der Burg SH (2016) Neoantigen landscape dynamics during human melanoma–T cell interactions. Nature 536:91–95
Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, Topalian SL, Sherry R, Restifo NP, Hubicki AM, Robinson MR, Raffeld M, Duray P, Seipp CA, Rogers-Freezer L, Morton KE, Mavroukakis SA, White DE, Rosenberg SA (2002) Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science 298:850–854
Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, Miller ML, Rekhtman N, Moreira AL, Ibrahim F, Bruggeman C, Gasmi B, Zappasodi R, Maeda Y, Sander C, Garon EB, Merghoub T, Wolchok JD, Schumacher TN, Chan TA (2015) Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348:124–128
Anagnostou V, Smith KN, Forde PM, Niknafs N, Bhattacharya R, White J, Zhang T, Adleff V, Phallen J, Wali N, Hruban C, Guthrie VB, Rodgers K, Naidoo J, Kang H, Sharfman WH, Georgiades C, Verde F, Illei P, Li QK, Gabrielson E, Brock MV, Zahnow CA, Baylin SB, Scharpf R, Brahmer JR, Karchin R, Pardoll DM, Velculescu VE (2017) Evolution of neoantigen landscape during immune checkpoint blockade in non-small cell lung cancer. Cancer Discov 7:264–276
Klemm F, Joyce JA (2015) Microenvironmental regulation of therapeutic response in cancer. Trends Cell Biol 25:198–213
Junttila MR, de Sauvage FJ (2013) Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 501:346–354
Banat GA, Tretyn A, Pullamsetti SS, Wilhelm J, Weigert A, Olesch C, Ebel K, Stiewe T, Grimminger F, Seeger W, Fink L, Savai R (2015) Immune and inflammatory cell composition of human lung cancer stroma. PLoS One 10:e0139073
Katlinski KV, Gui J, Katlinskaya YV, Ortiz A, Chakraborty R, Bhattacharya S, Carbone CJ, Beiting DP, Girondo MA, Peck AR, Pure E, Chatterji P, Rustgi AK, Diehl JA, Koumenis C, Rui H, Fuchs SY (2017) Inactivation of interferon receptor promotes the establishment of immune privileged tumor microenvironment. Cancer Cell 31:194–207
Wherry EJ, Kurachi M (2015) Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol 15:486–499
Catakovic K, Klieser E, Neureiter D, Geisberger R (2017) T cell exhaustion: from pathophysiological basics to tumor immunotherapy. Cell Commun Signal 15:1
Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC (2010) Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med 207:2187–2194
Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY, Abril-Rodriguez G, Sandoval S, Barthly L, Saco J, Homet MB, Mezzadra R, Chmielowski B, Ruchalski K, Shintaku IP, Sanchez PJ, Puig-Saus C, Cherry G, Seja E, Kong X, Pang J, Berent-Maoz B, Comin-Anduix B, Graeber TG, Tumeh PC, Schumacher TN, Lo RS, Ribas A (2016) Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med 375:819–829
Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, Berent-Maoz B, Pang J, Chmielowski B, Cherry G, Seja E, Lomeli S, Kong X, Kelley MC, Sosman JA, Johnson DB, Ribas A, Lo RS (2016) Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell 165:35–44
Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, Gandhi L, Redig AJ, Rodig SJ, Asahina H, Jones RE, Kulkarni MM, Kuraguchi M, Palakurthi S, Fecci PE, Johnson BE, Janne PA, Engelman JA, Gangadharan SP, Costa DB, Freeman GJ, Bueno R, Hodi FS, Dranoff G, Wong KK, Hammerman PS (2016) Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun 7:10501
Pauken KE, Sammons MA, Odorizzi PM, Manne S, Godec J, Khan O, Drake AM, Chen Z, Sen DR, Kurachi M, Barnitz RA, Bartman C, Bengsch B, Huang AC, Schenkel JM, Vahedi G, Haining WN, Berger SL, Wherry EJ (2016) Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 354:1160–1165
Sen DR, Kaminski J, Barnitz RA, Kurachi M, Gerdemann U, Yates KB, Tsao HW, Godec J, LaFleur MW, Brown FD, Tonnerre P, Chung RT, Tully DC, Allen TM, Frahm N, Lauer GM, Wherry EJ, Yosef N, Haining WN (2016) The epigenetic landscape of T cell exhaustion. Science 354:1165–1169
Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ, Bettini ML, Gravano DM, Vogel P, Liu CL, Tangsombatvisit S, Grosso JF, Netto G, Smeltzer MP, Chaux A, Utz PJ, Workman CJ, Pardoll DM, Korman AJ, Drake CG, Vignali DA (2012) Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res 72:917–927
Goding SR, Wilson KA, Xie Y, Harris KM, Baxi A, Akpinarli A, Fulton A, Tamada K, Strome SE, Antony PA (2013) Restoring immune function of tumor-specific CD4+ T cells during recurrence of melanoma. J Immunol 190:4899–4909
Bjorkman PJ, Saper MA, Samraoui B, Bennett WS, Strominger JL, Wiley DC (1987) Structure of the human class I histocompatibility antigen, HLA-A2. Nature 329:506–512
Neefjes J, Jongsma ML, Paul P, Bakke O (2011) Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat Rev Immunol 11:823–836
Hershberg RM, Cho DH, Youakim A, Bradley MB, Lee JS, Framson PE, Nepom GT (1998) Highly polarized HLA class II antigen processing and presentation by human intestinal epithelial cells. J Clin Investig 102:792–803
Wu Z, Biro PA, Mirakian R, Hammond L, Curcio F, Ambesi-Impiombato FS, Bottazzo GF (1999) HLA-DMB expression by thyrocytes: indication of the antigen-processing and possible presenting capability of thyroid cells. Clin Exp Immunol 116:62–69
Monach PA, Meredith SC, Siegel CT, Schreiber H (1995) A unique tumor antigen produced by a single amino acid substitution. Immunity 2:45–59
Muranski P, Boni A, Antony PA, Cassard L, Irvine KR, Kaiser A, Paulos CM, Palmer DC, Touloukian CE, Ptak K, Gattinoni L, Wrzesinski C, Hinrichs CS, Kerstann KW, Feigenbaum L, Chan CC, Restifo NP (2008) Tumor-specific Th17-polarized cells eradicate large established melanoma. Blood 112:362–373
Degenhardt Y, Huang J, Greshock J, Horiates G, Nathanson K, Yang X, Herlyn M, Weber B (2010) Distinct MHC gene expression patterns during progression of melanoma. Genes Chromosomes Cancer 49:144–154
Donia M, Andersen R, Kjeldsen JW, Fagone P, Munir S, Nicoletti F, Andersen MH, Thor SP, Svane IM (2015) Aberrant expression of MHC class II in melanoma attracts inflammatory tumor-specific CD4+ T-cells, which dampen CD8+ T-cell antitumor reactivity. Cancer Res 75:3747–3759
Cabrera T, Ruiz-Cabello F, Garrido F (1995) Biological implications of HLA-DR expression in tumours. Scand J Immunol 41:398–406
Homet MB, Zaretsky JM, Garcia-Diaz A, Tsoi J, Parisi G, Robert L, Meeth K, Ndoye A, Bosenberg M, Weeraratna AT, Graeber TG, Comin-Anduix B, Hu-Lieskovan S, Ribas A (2016) Response to programmed cell death-1 blockade in a murine melanoma syngeneic model requires costimulation, CD4, and CD8 T cells. Cancer Immunol Res 4:845–857
Ekkirala CR, Cappello P, Accolla RS, Giovarelli M, Romero I, Garrido C, Garcia-Lora AM, Novelli F (2014) Class II transactivator-induced MHC class II expression in pancreatic cancer cells leads to tumor rejection and a specific antitumor memory response. Pancreas 43:1066–1072
Surmann EM, Voigt AY, Michel S, Bauer K, Reuschenbach M, Ferrone S, von Knebel DM, Kloor M (2015) Association of high CD4-positive T cell infiltration with mutations in HLA class II-regulatory genes in microsatellite-unstable colorectal cancer. Cancer Immunol Immunother 64:357–366
Arnold PY, La Gruta NL, Miller T, Vignali KM, Adams PS, Woodland DL, Vignali DA (2002) The majority of immunogenic epitopes generate CD4+ T cells that are dependent on MHC class II-bound peptide-flanking residues. J Immunol 169:739–749
Linnemann C, van Buuren MM, Bies L, Verdegaal EM, Schotte R, Calis JJ, Behjati S, Velds A, Hilkmann H, Atmioui DE, Visser M, Stratton MR, Haanen JB, Spits H, van der Burg SH, Schumacher TN (2015) High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat Med 21:81–85
Giorda E, Sibilio L, Martayan A, Moretti S, Venturo I, Mottolese M, Ferrara GB, Cappellacci S, Eibenschutz L, Catricala C, Grammatico P, Giacomini P (2003) The antigen processing machinery of class I human leukocyte antigens: linked patterns of gene expression in neoplastic cells. Cancer Res 63:4119–4127
Boon T, van der Bruggen P (1996) Human tumor antigens recognized by T lymphocytes. J Exp Med 183:725–729
Old LJ, Chen YT (1998) New paths in human cancer serology. J Exp Med 187:1163–1167
Rosenberg SA (1999) A new era for cancer immunotherapy based on the genes that encode cancer antigens. Immunity 10:281–287
Scanlan MJ, Gure AO, Jungbluth AA, Old LJ, Chen YT (2002) Cancer/testis antigens: an expanding family of targets for cancer immunotherapy. Immunol Rev 188:22–32
Liang J, Ding T, Guo ZW, Yu XJ, Hu YZ, Zheng L, Xu J (2013) Expression pattern of tumour-associated antigens in hepatocellular carcinoma: association with immune infiltration and disease progression. Br J Cancer 109:1031–1039
Flecken T, Schmidt N, Hild S, Gostick E, Drognitz O, Zeiser R, Schemmer P, Bruns H, Eiermann T, Price DA, Blum HE, Neumann-Haefelin C, Thimme R (2014) Immunodominance and functional alterations of tumor-associated antigen-specific CD8+ T-cell responses in hepatocellular carcinoma. Hepatology 59:1415–1426
Wolfel T, Hauer M, Schneider J, Serrano M, Wolfel C, Klehmann-Hieb E, De PE, Hankeln T, Meyer zum Buschenfelde KH, Beach D (1995) A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science 269:1281–1284
Lennerz V, Fatho M, Gentilini C, Frye RA, Lifke A, Ferel D, Wolfel C, Huber C, Wolfel T (2005) The response of autologous T cells to a human melanoma is dominated by mutated neoantigens. Proc Natl Acad Sci USA 102:16013–16018
Postow MA, Callahan MK, Wolchok JD (2015) Immune checkpoint blockade in cancer therapy. J Clin Oncol 33:1974–1982
Simon S, Vignard V, Florenceau L, Dreno B, Khammari A, Lang F, Labarriere N (2016) PD-1 expression conditions T cell avidity within an antigen-specific repertoire. Oncoimmunology 5:e1104448
Yadav M, Jhunjhunwala S, Phung QT, Lupardus P, Tanguay J, Bumbaca S, Franci C, Cheung TK, Fritsche J, Weinschenk T, Modrusan Z, Mellman I, Lill JR, Delamarre L (2014) Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 515:572–576
Rammensee H, Bachmann J, Emmerich NP, Bachor OA, Stevanovic S (1999) SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics 50:213–219
Lundegaard C, Lamberth K, Harndahl M, Buus S, Lund O, Nielsen M (2008) NetMHC-3.0: accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8–11. Nucleic Acids Res 36:W509–W512
Engels B, Engelhard VH, Sidney J, Sette A, Binder DC, Liu RB, Kranz DM, Meredith SC, Rowley DA, Schreiber H (2013) Relapse or eradication of cancer is predicted by peptide-major histocompatibility complex affinity. Cancer Cell 23:516–526
Sanchez-Perez L, Kottke T, Diaz RM, Ahmed A, Thompson J, Chong H, Melcher A, Holmen S, Daniels G, Vile RG (2005) Potent selection of antigen loss variants of B16 melanoma following inflammatory killing of melanocytes in vivo. Cancer Res 65:2009–2017
Zhong S, Malecek K, Johnson LA, Yu Z, de Vega-Saenz ME, Darvishian F, McGary K, Huang K, Boyer J, Corse E, Shao Y, Rosenberg SA, Restifo NP, Osman I, Krogsgaard M (2013) T-cell receptor affinity and avidity defines antitumor response and autoimmunity in T-cell immunotherapy. Proc Natl Acad Sci USA 110:6973–6978
Woller N, Gurlevik E, Fleischmann-Mundt B, Schumacher A, Knocke S, Kloos AM, Saborowski M, Geffers R, Manns MP, Wirth TC, Kubicka S, Kuhnel F (2015) Viral infection of tumors overcomes resistance to PD-1-immunotherapy by broadening neoantigenome-directed T-cell responses. Mol Ther 23:1630–1640
Duan F, Duitama J, Al SS, Ayres CM, Corcelli SA, Pawashe AP, Blanchard T, McMahon D, Sidney J, Sette A, Baker BM, Mandoiu II, Srivastava PK (2014) Genomic and bioinformatic profiling of mutational neoepitopes reveals new rules to predict anticancer immunogenicity. J Exp Med 211:2231–2248
Schneider C, Teufel A, Yevsa T, Staib F, Hohmeyer A, Walenda G, Zimmermann HW, Vucur M, Huss S, Gassler N, Wasmuth HE, Lira SA, Zender L, Luedde T, Trautwein C, Tacke F (2012) Adaptive immunity suppresses formation and progression of diethylnitrosamine-induced liver cancer. Gut 61:1733–1743
DuPage M, Mazumdar C, Schmidt LM, Cheung AF, Jacks T (2012) Expression of tumour-specific antigens underlies cancer immunoediting. Nature 482:405–409
Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, West AN, Carmona M, Kivork C, Seja E, Cherry G, Gutierrez AJ, Grogan TR, Mateus C, Tomasic G, Glaspy JA, Emerson RO, Robins H, Pierce RH, Elashoff DA, Robert C, Ribas A (2014) PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515:568–571
Khong HT, Restifo NP (2002) Natural selection of tumor variants in the generation of “tumor escape” phenotypes. Nat Immunol 3:999–1005
Garrido F, Cabrera T, Aptsiauri N (2010) “Hard” and “soft” lesions underlying the HLA class I alterations in cancer cells: implications for immunotherapy. Int J Cancer 127:249–256
Hanson HL, Kang SS, Norian LA, Matsui K, O’Mara LA, Allen PM (2004) CD4-directed peptide vaccination augments an antitumor response, but efficacy is limited by the number of CD8+ T cell precursors. J Immunol 172:4215–4224
Greenberg PD, Cheever MA, Fefer A (1981) Eradication of disseminated murine leukemia by chemoimmunotherapy with cyclophosphamide and adoptively transferred immune syngeneic Lyt-1+2− lymphocytes. J Exp Med 154:952–963
Nishimura T, Iwakabe K, Sekimoto M, Ohmi Y, Yahata T, Nakui M, Sato T, Habu S, Tashiro H, Sato M, Ohta A (1999) Distinct role of antigen-specific T helper type 1 (Th1) and Th2 cells in tumor eradication in vivo. J Exp Med 190:617–627
Mattes J, Hulett M, Xie W, Hogan S, Rothenberg ME, Foster P, Parish C (2003) Immunotherapy of cytotoxic T cell-resistant tumors by T helper 2 cells: an eotaxin and STAT6-dependent process. J Exp Med 197:387–393
Lorvik KB, Hammarstrom C, Fauskanger M, Haabeth OA, Zangani M, Haraldsen G, Bogen B, Corthay A (2016) Adoptive transfer of tumor-specific Th2 cells eradicates tumors by triggering an in situ inflammatory immune response. Cancer Res 76:6864–6876
Knutson KL, Disis ML (2005) Tumor antigen-specific T helper cells in cancer immunity and immunotherapy. Cancer Immunol Immunother 54:721–728
LaCasse CJ, Janikashvili N, Larmonier CB, Alizadeh D, Hanke N, Kartchner J, Situ E, Centuori S, Har-Noy M, Bonnotte B, Katsanis E, Larmonier N (2011) Th-1 lymphocytes induce dendritic cell tumor killing activity by an IFN-gamma-dependent mechanism. J Immunol 187:6310–6317
Xie Y, Akpinarli A, Maris C, Hipkiss EL, Lane M, Kwon EK, Muranski P, Restifo NP, Antony PA (2010) Naive tumor-specific CD4(+) T cells differentiated in vivo eradicate established melanoma. J Exp Med 207:651–667
Quezada SA, Simpson TR, Peggs KS, Merghoub T, Vider J, Fan X, Blasberg R, Yagita H, Muranski P, Antony PA, Restifo NP, Allison JP (2010) Tumor-reactive CD4(+) T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J Exp Med 207:637–650
Tepper RI, Coffman RL, Leder P (1992) An eosinophil-dependent mechanism for the antitumor effect of interleukin-4. Science 257:548–551
Van WC, Urban JL, Rothstein JL, Ward PL, Schreiber H (1986) Highly malignant tumor variants retain tumor-specific antigens recognized by T helper cells. J Exp Med 164:1547–1565
Ossendorp F, Mengede E, Camps M, Filius R, Melief CJ (1998) Specific T helper cell requirement for optimal induction of cytotoxic T lymphocytes against major histocompatibility complex class II negative tumors. J Exp Med 187:693–702
Hung K, Hayashi R, Lafond-Walker A, Lowenstein C, Pardoll D, Levitsky H (1998) The central role of CD4(+) T cells in the antitumor immune response. J Exp Med 188:2357–2368
Corthay A, Skovseth DK, Lundin KU, Rosjo E, Omholt H, Hofgaard PO, Haraldsen G, Bogen B (2005) Primary antitumor immune response mediated by CD4+ T cells. Immunity 22:371–383
Shklovskaya E, Terry AM, Guy TV, Buckley A, Bolton HA, Zhu E, Holst J (2016) Fazekas dS: Tumour-specific CD4 T cells eradicate melanoma via indirect recognition of tumour-derived antigen. Immunol Cell Biol 94:593–603
Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver CT (2005) Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol 6:1123–1132
Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT (2006) Transforming growth factor-beta induces development of the T(H)17 lineage. Nature 441:231–234
Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q, Dong C (2005) A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol 6:1133–1141
Acosta-Rodriguez EV, Rivino L, Geginat J, Jarrossay D, Gattorno M, Lanzavecchia A, Sallusto F, Napolitani G (2007) Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat Immunol 8:639–646
Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR (2006) The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 126:1121–1133
Chen Z, Tato CM, Muul L, Laurence A, O’Shea JJ (2007) Distinct regulation of interleukin-17 in human T helper lymphocytes. Arthritis Rheum 56:2936–2946
Viaud S, Flament C, Zoubir M, Pautier P, LeCesne A, Ribrag V, Soria JC, Marty V, Vielh P, Robert C, Chaput N, Zitvogel L (2011) Cyclophosphamide induces differentiation of Th17 cells in cancer patients. Cancer Res 71:661–665
Weaver CT, Hatton RD, Mangan PR, Harrington LE (2007) IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu Rev Immunol 25:821–852
Bettelli E, Oukka M, Kuchroo VK (2007) T(H)-17 cells in the circle of immunity and autoimmunity. Nat Immunol 8:345–350
Steiner GE, Newman ME, Paikl D, Stix U, Memaran-Dagda N, Lee C, Marberger MJ (2003) Expression and function of pro-inflammatory interleukin IL-17 and IL-17 receptor in normal, benign hyperplastic, and malignant prostate. Prostate 56:171–182
Ciree A, Michel L, Camilleri-Broet S, Jean LF, Oster M, Flageul B, Senet P, Fossiez F, Fridman WH, Bachelez H, Tartour E (2004) Expression and activity of IL-17 in cutaneous T-cell lymphomas (mycosis fungoides and Sezary syndrome). Int J Cancer 112:113–120
Hirschhorn-Cymerman D, Budhu S, Kitano S, Liu C, Zhao F, Zhong H, Lesokhin AM, Avogadri-Connors F, Yuan J, Li Y, Houghton AN, Merghoub T, Wolchok JD (2012) Induction of tumoricidal function in CD4+ T cells is associated with concomitant memory and terminally differentiated phenotype. J Exp Med 209:2113–2126
Corthay A, Lundin KU, Lorvik KB, Hofgaard PO, Bogen B (2009) Secretion of tumor-specific antigen by myeloma cells is required for cancer immunosurveillance by CD4+ T cells. Cancer Res 69:5901–5907
Nevala WK, Vachon CM, Leontovich AA, Scott CG, Thompson MA, Markovic SN (2009) Evidence of systemic Th2-driven chronic inflammation in patients with metastatic melanoma. Clin Cancer Res 15:1931–1939
Gao J, Wu Y, Su Z, Amoah BP, Jiao Z, Bie Q, Lu L, Wang S, Xu H (2014) Infiltration of alternatively activated macrophages in cancer tissue is associated with MDSC and Th2 polarization in patients with esophageal cancer. PLoS One 9:e104453
Ito SE, Shirota H, Kasahara Y, Saijo K, Ishioka C (2017) IL-4 blockade alters the tumor microenvironment and augments the response to cancer immunotherapy in a mouse model. Cancer Immunol Immunother. doi:10.1007/s00262-017-2043-6
Zhang Q, Liu S, Ge D, Cunningham DM, Huang F, Ma L, Burris TP, You Z (2017) Targeting Th17-IL-17 pathway in prevention of micro-invasive prostate cancer in a mouse model. Prostate 77:888–899
Zhang JP, Yan J, Xu J, Pang XH, Chen MS, Li L, Wu C, Li SP, Zheng L (2009) Increased intratumoral IL-17-producing cells correlate with poor survival in hepatocellular carcinoma patients. J Hepatol 50:980–989
Kesselring R, Thiel A, Pries R, Trenkle T, Wollenberg B (2010) Human Th17 cells can be induced through head and neck cancer and have a functional impact on HNSCC development. Br J Cancer 103:1245–1254
Tartour E, Fossiez F, Joyeux I, Galinha A, Gey A, Claret E, Sastre-Garau X, Couturier J, Mosseri V, Vives V, Banchereau J, Fridman WH, Wijdenes J, Lebecque S, Sautes-Fridman C (1999) Interleukin 17, a T-cell-derived cytokine, promotes tumorigenicity of human cervical tumors in nude mice. Cancer Res 59:3698–3704
Benchetrit F, Ciree A, Vives V, Warnier G, Gey A, Sautes-Fridman C, Fossiez F, Haicheur N, Fridman WH, Tartour E (2002) Interleukin-17 inhibits tumor cell growth by means of a T-cell-dependent mechanism. Blood 99:2114–2121
Kryczek I, Banerjee M, Cheng P, Vatan L, Szeliga W, Wei S, Huang E, Finlayson E, Simeone D, Welling TH, Chang A, Coukos G, Liu R, Zou W (2009) Phenotype, distribution, generation, and functional and clinical relevance of Th17 cells in the human tumor environments. Blood 114:1141–1149
Lv L, Pan K, Li XD, She KL, Zhao JJ, Wang W, Chen JG, Chen YB, Yun JP, Xia JC (2011) The accumulation and prognosis value of tumor infiltrating IL-17 producing cells in esophageal squamous cell carcinoma. PLoS One 6:e18219
Chen WC, Lai YH, Chen HY, Guo HR, Su IJ, Chen HH (2013) Interleukin-17-producing cell infiltration in the breast cancer tumour microenvironment is a poor prognostic factor. Histopathology 63:225–233
Voo KS, Wang YH, Santori FR, Boggiano C, Wang YH, Arima K, Bover L, Hanabuchi S, Khalili J, Marinova E, Zheng B, Littman DR, Liu YJ (2009) Identification of IL-17-producing FOXP3+ regulatory T cells in humans. Proc Natl Acad Sci USA 106:4793–4798
Ye J, Su X, Hsueh EC, Zhang Y, Koenig JM, Hoft DF, Peng G (2011) Human tumor-infiltrating Th17 cells have the capacity to differentiate into IFN-gamma+ and FOXP3+ T cells with potent suppressive function. Eur J Immunol 41:936–951
Gao Q, Qiu SJ, Fan J, Zhou J, Wang XY, Xiao YS, Xu Y, Li YW, Tang ZY (2007) Intratumoral balance of regulatory and cytotoxic T cells is associated with prognosis of hepatocellular carcinoma after resection. J Clin Oncol 25:2586–2593
Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M (1995) Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol 155:1151–1164
Fontenot JD, Gavin MA, Rudensky AY (2003) Foxp3 programs the development and function of CD4+ CD25+ regulatory T cells. Nat Immunol 4:330–336
Sakaguchi S (2005) Naturally arising Foxp3-expressing CD25+ CD4+ regulatory T cells in immunological tolerance to self and non-self. Nat Immunol 6:345–352
Wei WZ, Morris GP, Kong YC (2004) Anti-tumor immunity and autoimmunity: a balancing act of regulatory T cells. Cancer Immunol Immunother 53:73–78
Viguier M, Lemaitre F, Verola O, Cho MS, Gorochov G, Dubertret L, Bachelez H, Kourilsky P, Ferradini L (2004) Foxp3 expressing CD4+ CD25(high) regulatory T cells are overrepresented in human metastatic melanoma lymph nodes and inhibit the function of infiltrating T cells. J Immunol 173:1444–1453
den Boer AT, van Mierlo GJ, Fransen MF, Melief CJ, Offringa R, Toes RE (2005) CD4+ T cells are able to promote tumor growth through inhibition of tumor-specific CD8+ T-cell responses in tumor-bearing hosts. Cancer Res 65:6984–6989
Valzasina B, Piconese S, Guiducci C, Colombo MP (2006) Tumor-induced expansion of regulatory T cells by conversion of CD4+ CD25− lymphocytes is thymus and proliferation independent. Cancer Res 66:4488–4495
Yang XH, Yamagiwa S, Ichida T, Matsuda Y, Sugahara S, Watanabe H, Sato Y, Abo T, Horwitz DA, Aoyagi Y (2006) Increase of CD4+ CD25+ regulatory T-cells in the liver of patients with hepatocellular carcinoma. J Hepatol 45:254–262
Larmonier N, Marron M, Zeng Y, Cantrell J, Romanoski A, Sepassi M, Thompson S, Chen X, Andreansky S, Katsanis E (2007) Tumor-derived CD4(+)CD25(+) regulatory T cell suppression of dendritic cell function involves TGF-beta and IL-10. Cancer Immunol Immunother 56:48–59
Jarnicki AG, Lysaght J, Todryk S, Mills KH (2006) Suppression of antitumor immunity by IL-10 and TGF-beta-producing T cells infiltrating the growing tumor: influence of tumor environment on the induction of CD4+ and CD8+ regulatory T cells. J Immunol 177:896–904
Zheng SG, Wang JH, Gray JD, Soucier H, Horwitz DA (2004) Natural and induced CD4+ CD25+ cells educate CD4+. J Immunol 172:5213–5221
Liu VC, Wong LY, Jang T, Shah AH, Park I, Yang X, Zhang Q, Lonning S, Teicher BA, Lee C (2007) Tumor evasion of the immune system by converting CD4+. J Immunol 178:2883–2892
Ghiringhelli F, Puig PE, Roux S, Parcellier A, Schmitt E, Solary E, Kroemer G, Martin F, Chauffert B, Zitvogel L (2005) Tumor cells convert immature myeloid dendritic cells into TGF-beta-secreting cells inducing CD4+ CD25+ regulatory T cell proliferation. J Exp Med 202:919–929
Hindley JP, Ferreira C, Jones E, Lauder SN, Ladell K, Wynn KK, Betts GJ, Singh Y, Price DA, Godkin AJ, Dyson J, Gallimore A (2011) Analysis of the T-cell receptor repertoires of tumor-infiltrating conventional and regulatory T cells reveals no evidence for conversion in carcinogen-induced tumors. Cancer Res 71:736–746
Zhou G, Levitsky HI (2007) Natural regulatory T cells and de novo-induced regulatory T cells contribute independently to tumor-specific tolerance. J Immunol 178:2155–2162
Sato E, Olson SH, Ahn J, Bundy B, Nishikawa H, Qian F, Jungbluth AA, Frosina D, Gnjatic S, Ambrosone C, Kepner J, Odunsi T, Ritter G, Lele S, Chen YT, Ohtani H, Old LJ, Odunsi K (2005) Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci USA 102:18538–18543
Bates GJ, Fox SB, Han C, Leek RD, Garcia JF, Harris AL, Banham AH (2006) Quantification of regulatory T cells enables the identification of high-risk breast cancer patients and those at risk of late relapse. J Clin Oncol 24:5373–5380
Shen Z, Zhou S, Wang Y, Li RL, Zhong C, Liang C, Sun Y (2010) Higher intratumoral infiltrated Foxp3+ Treg numbers and Foxp3+/CD8+ ratio are associated with adverse prognosis in resectable gastric cancer. J Cancer Res Clin Oncol 136:1585–1595
Carreras J, Lopez-Guillermo A, Fox BC, Colomo L, Martinez A, Roncador G, Montserrat E, Campo E, Banham AH (2006) High numbers of tumor-infiltrating FOXP3-positive regulatory T cells are associated with improved overall survival in follicular lymphoma. Blood 108:2957–2964
Garcia M, Bellosillo B, Sanchez-Gonzalez B, Garcia-Payarols F, Seoane A, Ferrer AM, Gimeno E, Barranco LE, Torner A, Sole F, Besses C, Serrano S, Salar A (2012) Study of regulatory T-cells in patients with gastric malt lymphoma: influence on treatment response and outcome. PLoS One 7:e51681
Salama P, Phillips M, Grieu F, Morris M, Zeps N, Joseph D, Platell C, Iacopetta B (2009) Tumor-infiltrating FOXP3+ T regulatory cells show strong prognostic significance in colorectal cancer. J Clin Oncol 27:186–192
Correale P, Rotundo MS, Del Vecchio MT, Remondo C, Migali C, Ginanneschi C, Tsang KY, Licchetta A, Mannucci S, Loiacono L, Tassone P, Francini G, Tagliaferri P (2010) Regulatory (FoxP3+) T-cell tumor infiltration is a favorable prognostic factor in advanced colon cancer patients undergoing chemo or chemoimmunotherapy. J Immunother 33:435–441
Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, Nomura T, Sakaguchi S (2008) CTLA-4 control over Foxp3+ regulatory T cell function. Science 322:271–275
Linsley PS, Brady W, Urnes M, Grosmaire LS, Damle NK, Ledbetter JA (1991) CTLA-4 is a second receptor for the B cell activation antigen B7. J Exp Med 174:561–569
Oderup C, Cederbom L, Makowska A, Cilio CM, Ivars F (2006) Cytotoxic T lymphocyte antigen-4-dependent down-modulation of costimulatory molecules on dendritic cells in CD4+ CD25+ regulatory T-cell-mediated suppression. Immunology 118:240–249
Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, Baker J, Jeffery LE, Kaur S, Briggs Z, Hou TZ, Futter CE, Anderson G, Walker LS, Sansom DM (2011) Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science 332:600–603
Kvistborg P, Philips D, Kelderman S, Hageman L, Ottensmeier C, Joseph-Pietras D, Welters MJ, van der Burg S, Kapiteijn E, Michielin O, Romano E, Linnemann C, Speiser D, Blank C, Haanen JB, Schumacher TN (2014) Anti-CTLA-4 therapy broadens the melanoma-reactive CD8+ T cell response. Sci Transl Med 6:254ra128
Cao X, Cai SF, Fehniger TA, Song J, Collins LI, Piwnica-Worms DR, Ley TJ (2007) Granzyme B and perforin are important for regulatory T cell-mediated suppression of tumor clearance. Immunity 27:635–646
Bauer CA, Kim EY, Marangoni F, Carrizosa E, Claudio NM, Mempel TR (2014) Dynamic Treg interactions with intratumoral APCs promote local CTL dysfunction. J Clin Investig 124:2425–2440
Ahmadzadeh M, Rosenberg SA (2006) IL-2 administration increases CD4 + CD25(hi) Foxp3+ regulatory T cells in cancer patients. Blood 107:2409–2414
Zorn E, Nelson EA, Mohseni M, Porcheray F, Kim H, Litsa D, Bellucci R, Raderschall E, Canning C, Soiffer RJ, Frank DA, Ritz J (2006) IL-2 regulates FOXP3 expression in human CD4+ CD25+ regulatory T cells through a STAT-dependent mechanism and induces the expansion of these cells in vivo. Blood 108:1571–1579
Baratelli F, Lin Y, Zhu L, Yang SC, Heuze-Vourc’h N, Zeng G, Reckamp K, Dohadwala M, Sharma S, Dubinett SM (2005) Prostaglandin E2 induces FOXP3 gene expression and T regulatory cell function in human CD4+ T cells. J Immunol 175:1483–1490
Ghiringhelli F, Larmonier N, Schmitt E, Parcellier A, Cathelin D, Garrido C, Chauffert B, Solary E, Bonnotte B, Martin F (2004) CD4+ CD25+ regulatory T cells suppress tumor immunity but are sensitive to cyclophosphamide which allows immunotherapy of established tumors to be curative. Eur J Immunol 34:336–344
Chen DS, Mellman I (2013) Oncology meets immunology: the cancer-immunity cycle. Immunity 39:1–10
Preusser M, Berghoff AS, Thallinger C, Zielinski CC (2016) Cancer immune cycle: a video introduction to the interaction between cancer and the immune system. ESMO Open 1:e000056
Chen DS, Mellman I (2017) Elements of cancer immunity and the cancer-immune set point. Nature 541:321–330
Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, Linette GP, Meyer N, Giguere JK, Agarwala SS, Shaheen M, Ernstoff MS, Minor D, Salama AK, Taylor M, Ott PA, Rollin LM, Horak C, Gagnier P, Wolchok JD, Hodi FS (2015) Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med 372:2006–2017
Apetoh L, Smyth MJ, Drake CG, Abastado JP, Apte RN, Ayyoub M, Blay JY, Bonneville M, Butterfield LH, Caignard A, Castelli C, Cavallo F, Celis E, Chen L, Colombo MP, Comin-Anduix B, Coukos G, Dhodapkar MV, Dranoff G, Frazer IH, Fridman WH, Gabrilovich DI, Gilboa E, Gnjatic S, Jager D, Kalinski P, Kaufman HL, Kiessling R, Kirkwood J, Knuth A, Liblau R, Lotze MT, Lugli E, Marincola F, Melero I, Melief CJ, Mempel TR, Mittendorf EA, Odun K, Overwijk WW, Palucka AK, Parmiani G, Ribas A, Romero P, Schreiber RD, Schuler G, Srivastava PK, Tartour E, Valmori D, van der Burg SH, van der Bruggen P, van den Eynde BJ, Wang E, Zou W, Whiteside TL, Speiser DE, Pardoll DM, Restifo NP, Anderson AC (2015) Consensus nomenclature for CD8 T cell phenotypes in cancer. Oncoimmunology 4:e998538
Kammertoens T, Friese C, Arina A, Idel C, Briesemeister D, Rothe M, Ivanov A, Szymborska A, Patone G, Kunz S, Sommermeyer D, Engels B, Leisegang M, Textor A, Joerg FH, Fruttiger M, Lohoff M, Herrmann A, Yu H, Weichselbaum R, Uckert W, Hubner N, Gerhardt H, Beule D, Schreiber H, Blankenstein T (2017) Tumour ischaemia by interferon-gamma resembles physiological blood vessel regression. Nature 545:98–102
Schirrmacher V, Schild HJ, Guckel B (1993) von HP: Tumour-specific CTL response requiring interactions of four different cell types and recognition of MHC class I and class II restricted tumour antigens. Immunol Cell Biol 71(Pt 4):311–326
Okada K, Yasumura S, Muller-Fleckenstein I, Fleckenstein B, Talib S, Koldovsky U, Whiteside TL (1997) Interactions between autologous CD4+ and CD8+ T lymphocytes and human squamous cell carcinoma of the head and neck. Cell Immunol 177:35–48
Marzo AL, Lake RA, Robinson BW, Scott B (1999) T-cell receptor transgenic analysis of tumor-specific CD8 and CD4 responses in the eradication of solid tumors. Cancer Res 59:1071–1079
Church SE, Jensen SM, Antony PA, Restifo NP, Fox BA (2014) Tumor-specific CD4+ T cells maintain effector and memory tumor-specific CD8+ T cells. Eur J Immunol 44:69–79
Bos R, Sherman LA (2010) CD4+ T-cell help in the tumor milieu is required for recruitment and cytolytic function of CD8+ T lymphocytes. Cancer Res 70:8368–8377
Bos R, Marquardt KL, Cheung J, Sherman LA (2012) Functional differences between low- and high-affinity CD8(+) T cells in the tumor environment. Oncoimmunology 1:1239–1247
Wong SB, Bos R, Sherman LA (2008) Tumor-specific CD4+ T cells render the tumor environment permissive for infiltration by low-avidity CD8+ T cells. J Immunol 180:3122–3131
Kwong LN, Zou L, Chagani S, Pedamallu CS, Liu M, Jiang S, Protopopov A, Zhang J, Getz G, Chin L (2017) Modeling genomic instability and selection pressure in a mouse model of melanoma. Cell Rep 19:1304–1312
Schietinger A, Philip M, Liu RB, Schreiber K, Schreiber H (2010) Bystander killing of cancer requires the cooperation of CD4(+) and CD8(+) T cells during the effector phase. J Exp Med 207:2469–2477
Knocke S, Fleischmann-Mundt B, Saborowski M, Manns MP, Kuhnel F, Wirth TC, Woller N (2016) Tailored tumor immunogenicity reveals regulation of CD4 and CD8 T cell responses against cancer. Cell Rep 17:2234–2246
Hiltbold EM, Vlad AM, Ciborowski P, Watkins SC, Finn OJ (2000) The mechanism of unresponsiveness to circulating tumor antigen MUC1 is a block in intracellular sorting and processing by dendritic cells. J Immunol 165:3730–3741
Surman DR, Dudley ME, Overwijk WW, Restifo NP (2000) Cutting edge: CD4+ T cell control of CD8+ T cell reactivity to a model tumor antigen. J Immunol 164:562–565
Nesbeth YC, Martinez DG, Toraya S, Scarlett UK, Cubillos-Ruiz JR, Rutkowski MR, Conejo-Garcia JR (2010) CD4+ T cells elicit host immune responses to MHC class II-negative ovarian cancer through CCL5 secretion and CD40-mediated licensing of dendritic cells. J Immunol 184:5654–5662
Baxevanis CN, Voutsas IF, Tsitsilonis OE, Gritzapis AD, Sotiriadou R, Papamichail M (2000) Tumor-specific CD4+ T lymphocytes from cancer patients are required for optimal induction of cytotoxic T cells against the autologous tumor. J Immunol 164:3902–3912
Ghorashian S, Velica P, Chua I, McNicol AM, Carpenter B, Holler A, Nicholson E, Ahmadi M, Zech M, Xue SA, Uckert W, Morris E, Chakraverty R, Stauss HJ (2015) CD8 T cell tolerance to a tumor-associated self-antigen is reversed by CD4 T cells engineered to express the same T cell receptor. J Immunol 194:1080–1089
Gerner MY, Casey KA, Mescher MF (2008) Defective MHC class II presentation by dendritic cells limits CD4 T cell help for antitumor CD8 T cell responses. J Immunol 181:155–164
Nimanong S, Ostroumov D, Wingerath J, Knocke S, Woller N, Gurlevik E, Falk CS, Manns MP, Kuhnel F, Wirth TC (2017) CD40 signaling drives potent cellular immune responses in heterologous cancer vaccinations. Cancer Res 77:1918–1926
Sayem MA, Tomita Y, Yuno A, Hirayama M, Irie A, Tsukamoto H, Senju S, Yuba E, Yoshikawa T, Kono K, Nakatsura T, Nishimura Y (2016) Identification of glypican-3-derived long peptides activating both CD8+ and CD4+ T cells; prolonged overall survival in cancer patients with Th cell response. Oncoimmunology 5:e1062209
Melero I, Gaudernack G, Gerritsen W, Huber C, Parmiani G, Scholl S, Thatcher N, Wagstaff J, Zielinski C, Faulkner I, Mellstedt H (2014) Therapeutic vaccines for cancer: an overview of clinical trials. Nat Rev Clin Oncol 11:509–524
Sim GC, Radvanyi L (2014) The IL-2 cytokine family in cancer immunotherapy. Cytokine Growth Factor Rev 25:377–390
Rosenberg SA, Restifo NP (2015) Adoptive cell transfer as personalized immunotherapy for human cancer. Science 348:62–68
Brentjens RJ, Riviere I, Park JH, Davila ML, Wang X, Stefanski J, Taylor C, Yeh R, Bartido S, Borquez-Ojeda O, Olszewska M, Bernal Y, Pegram H, Przybylowski M, Hollyman D, Usachenko Y, Pirraglia D, Hosey J, Santos E, Halton E, Maslak P, Scheinberg D, Jurcic J, Heaney M, Heller G, Frattini M, Sadelain M (2011) Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood 118:4817–4828
Zamarin D, Holmgaard RB, Subudhi SK, Park JS, Mansour M, Palese P, Merghoub T, Wolchok JD, Allison JP (2014) Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci Transl Med 6:226ra32
Babiker HM, Riaz IB, Husnain M, Borad MJ (2017) Oncolytic virotherapy including Rigvir and standard therapies in malignant melanoma. Oncolytic Virother 6:11–18
Bartee MY, Dunlap KM, Bartee E (2017) Tumor-localized secretion of soluble PD1 enhances oncolytic virotherapy. Cancer Res 77:2952–2963
Sharma P, Allison JP (2015) The future of immune checkpoint therapy. Science 348:56–61
Bobisse S, Foukas PG, Coukos G, Harari A (2016) Neoantigen-based cancer immunotherapy. Ann Transl Med 4:262
Schumacher TN, Schreiber RD (2015) Neoantigens in cancer immunotherapy. Science 348:69–74
Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW (2013) Cancer genome landscapes. Science 339:1546–1558
Wolchok JD, Hodi FS, Weber JS, Allison JP, Urba WJ, Robert C, O’Day SJ, Hoos A, Humphrey R, Berman DM, Lonberg N, Korman AJ (2013) Development of ipilimumab: a novel immunotherapeutic approach for the treatment of advanced melanoma. Ann N Y Acad Sci 1291:1–13
Leach DR, Krummel MF, Allison JP (1996) Enhancement of antitumor immunity by CTLA-4 blockade. Science 271:1734–1736
Farkona S, Diamandis EP, Blasutig IM (2016) Cancer immunotherapy: the beginning of the end of cancer? BMC Med 14:73
Schadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O, Patt D, Chen TT, Berman DM, Wolchok JD (2015) Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol 33:1889–1894
Abdel-Wahab N, Shah M, Suarez-Almazor ME (2016) Adverse events associated with immune checkpoint blockade in patients with cancer: a systematic review of case reports. PLoS One 11:e0160221
Ascierto PA, Del VM, Robert C, Mackiewicz A, Chiarion-Sileni V, Arance A, Lebbe C, Bastholt L, Hamid O, Rutkowski P, McNeil C, Garbe C, Loquai C, Dreno B, Thomas L, Grob JJ, Liszkay G, Nyakas M, Gutzmer R, Pikiel J, Grange F, Hoeller C, Ferraresi V, Smylie M, Schadendorf D, Mortier L, Svane IM, Hennicken D, Qureshi A, Maio M (2017) Ipilimumab 10 mg/kg versus ipilimumab 3 mg/kg in patients with unresectable or metastatic melanoma: a randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol 18:611–622
Eggermont AM, Chiarion-Sileni V, Grob JJ, Dummer R, Wolchok JD, Schmidt H, Hamid O, Robert C, Ascierto PA, Richards JM, Lebbe C, Ferraresi V, Smylie M, Weber JS, Maio M, Konto C, Hoos A, de Pril V, Gurunath RK, Suciu S, Testori A (2015) Adjuvant ipilimumab versus placebo after complete resection of high-risk stage III melanoma (EORTC 18071): a randomised, double-blind, phase 3 trial. Lancet Oncol 16:522–530
Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, Daud A, Carlino MS, McNeil C, Lotem M, Larkin J, Lorigan P, Neyns B, Blank CU, Hamid O, Mateus C, Shapira-Frommer R, Kosh M, Zhou H, Ibrahim N, Ebbinghaus S, Ribas A (2015) Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med 372:2521–2532
O’Reilly A, Larkin J (2017) The safety of nivolumab for the treatment of metastatic melanoma. Expert Opin Drug Saf 16:955–961
Kumar V, Chaudhary N, Garg M, Floudas CS, Soni P, Chandra AB (2017) Current diagnosis and management of immune related adverse events (irAEs) induced by immune checkpoint inhibitor therapy. Front Pharmacol 8:49
Yu DP, Cheng X, Liu ZD, Xu SF (2017) Comparative beneficiary effects of immunotherapy against chemotherapy in patients with advanced NSCLC: meta-analysis and systematic review. Oncol Lett 14:1568–1580
Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, Ferrucci PF, Hill A, Wagstaff J, Carlino MS, Haanen JB, Maio M, Marquez-Rodas I, McArthur GA, Ascierto PA, Long GV, Callahan MK, Postow MA, Grossmann K, Sznol M, Dreno B, Bastholt L, Yang A, Rollin LM, Horak C, Hodi FS, Wolchok JD (2015) Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med 373:23–34
Antonia SJ, Lopez-Martin JA, Bendell J, Ott PA, Taylor M, Eder JP, Jager D, Pietanza MC, Le DT, Morse MA, Ascierto PA, Horn L, Amin A, Pillai RN, Evans J, Chau I, Bono P, Atmaca A, Sharma P, Harbison CT, Lin CS, Christensen O, Calvo E (2016) Nivolumab alone and nivolumab plus ipilimumab in recurrent small-cell lung cancer (CheckMate 032): a multicentre, open-label, phase 1/2 trial. Lancet Oncol 17:883–895
Weber JS, D’Angelo SP, Minor D, Hodi FS, Gutzmer R, Neyns B, Hoeller C, Khushalani NI, Miller WH Jr, Lao CD, Linette GP, Thomas L, Lorigan P, Grossmann KF, Hassel JC, Maio M, Sznol M, Ascierto PA, Mohr P, Chmielowski B, Bryce A, Svane IM, Grob JJ, Krackhardt AM, Horak C, Lambert A, Yang AS, Larkin J (2015) Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol 16:375–384
Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, Roddie C, Henry JY, Yagita H, Wolchok JD, Peggs KS, Ravetch JV, Allison JP, Quezada SA (2013) Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med 210:1695–1710
Quezada SA, Peggs KS, Curran MA, Allison JP (2006) CTLA4 blockade and GM-CSF combination immunotherapy alters the intratumor balance of effector and regulatory T cells. J Clin Investig 116:1935–1945
Liakou CI, Kamat A, Tang DN, Chen H, Sun J, Troncoso P, Logothetis C, Sharma P (2008) CTLA-4 blockade increases IFNgamma-producing CD4+ ICOShi cells to shift the ratio of effector to regulatory T cells in cancer patients. Proc Natl Acad Sci USA 105:14987–14992
Quezada SA, Peggs KS, Simpson TR, Allison JP (2011) Shifting the equilibrium in cancer immunoediting: from tumor tolerance to eradication. Immunol Rev 241:104–118
Selby MJ, Engelhardt JJ, Quigley M, Henning KA, Chen T, Srinivasan M, Korman AJ (2013) Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol Res 1:32–42
Bruhns P (2012) Properties of mouse and human IgG receptors and their contribution to disease models. Blood 119:5640–5649
Romano E, Kusio-Kobialka M, Foukas PG, Baumgaertner P, Meyer C, Ballabeni P, Michielin O, Weide B, Romero P, Speiser DE (2015) Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proc Natl Acad Sci USA 112:6140–6145
Khan S, Burt DJ, Ralph C, Thistlethwaite FC, Hawkins RE, Elkord E (2011) Tremelimumab (anti-CTLA4) mediates immune responses mainly by direct activation of T effector cells rather than by affecting T regulatory cells. Clin Immunol 138:85–96
Arce VF, Furness AJS, Solomon I, Joshi K, Mekkaoui L, Lesko MH, Miranda RE, Dahan R, Georgiou A, Sledzinska A, Ben AA, Franz D, Werner SM, Wong YNS, Henry JY, O’Brien T, Nicol D, Challacombe B, Beers SA, Turajlic S, Gore M, Larkin J, Swanton C, Chester KA, Pule M, Ravetch JV, Marafioti T, Peggs KS, Quezada SA (2017) Fc-optimized anti-CD25 depletes tumor-infiltrating regulatory T cells and synergizes with PD-1 blockade to eradicate established tumors. Immunity 46:577–586
Huang AC, Postow MA, Orlowski RJ, Mick R, Bengsch B, Manne S, Xu W, Harmon S, Giles JR, Wenz B, Adamow M, Kuk D, Panageas KS, Carrera C, Wong P, Quagliarello F, Wubbenhorst B, D’Andrea K, Pauken KE, Herati RS, Staupe RP, Schenkel JM, McGettigan S, Kothari S, George SM, Vonderheide RH, Amaravadi RK, Karakousis GC, Schuchter LM, Xu X, Nathanson KL, Wolchok JD, Gangadhar TC, Wherry EJ (2017) T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 545:60–65
Kamphorst AO, Pillai RN, Yang S, Nasti TH, Akondy RS, Wieland A, Sica GL, Yu K, Koenig L, Patel NT, Behera M, Wu H, McCausland M, Chen Z, Zhang C, Khuri FR, Owonikoko TK, Ahmed R, Ramalingam SS (2017) Proliferation of PD-1+ CD8 T cells in peripheral blood after PD-1-targeted therapy in lung cancer patients. Proc Natl Acad Sci USA 114:4993–4998
Reck M, Rodriguez-Abreu D, Robinson AG, Hui R, Csoszi T, Fulop A, Gottfried M, Peled N, Tafreshi A, Cuffe S, O’Brien M, Rao S, Hotta K, Leiby MA, Lubiniecki GM, Shentu Y, Rangwala R, Brahmer JR (2016) Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med 375:1823–1833
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Ostroumov, D., Fekete-Drimusz, N., Saborowski, M. et al. CD4 and CD8 T lymphocyte interplay in controlling tumor growth. Cell. Mol. Life Sci. 75, 689–713 (2018). https://doi.org/10.1007/s00018-017-2686-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-017-2686-7