
Abstract
Objective and design
The aim was to investigate the signaling mechanisms and regulation of bradykinin (BK)-induced inflammation in rat knee joint.
Materials and methods
Knee joints of anesthetized rats were perfused with BK (0.1–1.0 μM), and synovial plasma extravasation (PE) was evaluated by spectrophotometrical measurement of Evans Blue leakage. To examine the signaling pathway, B1 antagonist [des-Arg10]-HOE140 (0.1–1.0 μM) and B2 antagonist HOE140 (0.05–1.0 μM), calcitonin gene-related peptide (CGRP) antagonist CGRP8-37 (0.5–1.0 μM), prostaglandin E2 antagonist AH-6809 (0.1–1.0 μM), and histamine H1 antagonist mepyramine (0.1–1.0 μM) were used. Nociceptin (0.0001–1.0 μM) and antagonist J-113397 were tested for modulation of BK-induced PE. The analyses were compared side-by-side with 5-hydroxytryptamine-induced PE.
Results
BK perfusion dose-dependently induced PE, which was blocked by HOE140, CGRP8-37, AH-6809, and mepyramine. It was also inhibited by nociceptin, which could be reversed by antagonist J-113397. In contrast, 5-hydroxytryptamine-induced PE was biphasically regulated by nociceptin and was not antagonized by CGRP8-37.
Conclusions
BK-induced PE is mediated by B2 receptors and may involve CGRP, prostaglandin, and histamine pathways. BK-induced PE is inhibited by nociceptin through the activation of ORL1 receptors. There are differences between BK- and 5-hydroxytryptamine-induced inflammation in signaling and modulation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Inflammation as well as its associated pain and plasma extravasation (PE, the cause of swelling) in the joint—characteristics of arthritis—can be produced and mediated by multiple neurobiochemical molecules, such as bradykinin (BK), 5-hydroxytryptamine (5-HT), histamine, ATP, prostaglandins (PGs), and cytokines [1–3]. BK and 5-HT, the two naturally occurring neurotransmitters, are among the strongest inflammatory mediators released by peripheral nervous terminals, platelets, and mast cells upon noxious stimulations and synovial tissue injury, causing PE and pain in the joints [4–6]. Studies suggest that the mechanisms underlying the BK- and 5-HT-induced PE have significant similarities. Both BK and 5-HT stimulate PE by activating sympathetic efferent terminals in the synovia. Selective lesion of the lumbar sympathetic ganglia, but not the lesion of the C-fiber afferents, substantially decreases BK- and 5-HT-induced PE [4–6]. Other mechanisms by which BK and 5-HT could act on primary sensory neurons or other cells to produce PE may also come into play however. There have been studies to address the receptor mediation and the involvement of other mediators in 5-HT-induced inflammation in the knee joints [6–8]. Some studies have suggested that BK is able to stimulate the release of the neurotransmitters calcitonin gene-related peptide (CGRP) and PGs [9, 10]. However, more thorough investigations are required to address the receptor-signaling mechanisms of BK in inducing inflammation in the joints.
The process of inflammation is tightly regulated by several neurotransmitters. There has been a significant amount of literature reporting the peripheral regulatory effect of opioids in the knee joints both in animal models and in human clinical studies [11, 12]. The majority of these studies have mainly focused on three classical types (μ, δ, and κ) of opioids and receptors. Recent studies have revealed that nociceptin (also named orphanin FQ) and its receptor ORL1 (opioid-receptor-like receptor 1, also named NOR), considered as the fourth and atypical member of the endogenous opioid family [13, 14], play a significant role in the modulation of inflammation and nociception [15, 16]. Nociceptin and the ORL1 receptors are widely distributed throughout the central and peripheral nervous systems, including sensory and sympathetic ganglia [17–19]. Studies have suggested that nociceptin and ORL1 could mediate opioid-like and anti-opioid effects [20]. Some studies have shown that nociceptin induces nociception [13], while others have demonstrated an inhibitory modulation of pain and inflammation [21, 22]. A study by Grond et al. [23] demonstrated that nociceptin bi-directionally modulates 5-HT-induced PE in the rat knee joint, with lower doses of nociceptin potentiating and higher doses inhibiting the effect of 5-HT. However, the regulation of BK-induced PE in the knee joint by nociceptin has not been studied.
In the present study, we employed the rat knee-joint perfusion model, a well-established experimental animal model for inflammation [4–6, 24], to examine the signaling pathways of BK-induced inflammation and its modulation by nociceptin. We applied selective antagonists to analyze the receptor pathways and other mediators involved. We compared the analyses with those of 5-HT-induced PE under the same experimental conditions.
Materials and methods
Animals
All experiments that employed animals were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of California, San Francisco. Guidelines and policies set by the National Institutes of Health and the Public Health Service on the care and use of laboratory animals were followed at all times. Every effort was made to minimize animal suffering.
Male Sprague-Dawley rats (Bantin and Kingman, Fremont, CA) weighing 320–350 g were used in the knee-joint perfusion experiments. Rats were housed individually in cages at room temperature (approximately 25°C) under controlled lighting conditions (lights on from 6 a.m. to 6 p.m.) with free access to food and water.
Chemicals and reagents
BK, BK receptor B1-selective antagonist [des-Arg10]-HOE 140, B2-selective antagonist HOE 140, and 5-HT (Sigma-RBI, St. Louis, MO); 5-HT1A/1B receptor selective antagonist (-)pindolol, 5-HT2A receptor selective antagonist ketanserin, CGRP receptor antagonist CGRP(8-37), and histamine H1 receptor selective antagonist mepyramine (Tocris Bioscience, Ellisville, MO); prostaglandin E2 (and D2) receptor antagonist AH-6809 (Cayman, Ann Arbor, MI); and nociceptin (Bachem, Torrance, CA), were used in rat knee-joint perfusion. The ORL1 receptor antagonist J-113397 was generously provided by Banyu Pharmaceutical (Tsukuba, Ibaraki, Japan). These chemicals were dissolved in normal saline and were freshly prepared prior to performing perfusion experiments. Evans Blue dye (Sigma-RBI) was also prepared in saline at a concentration of 20 mg/ml. All working solutions were used within 12 h after preparation and would not be stored for re-use.
Intra-articular perfusion and measurement of PE in the rat knee joint
The rat knee-joint perfusion and PE measurement were performed as previously described [6]. Briefly, rats were anesthetized with intraperitoneal injection of sodium pentobarbital (65 mg/kg body weight) (Abbott, Chicago, IL), and then Evans Blue dye (50 mg 2.5 ml−1 kg−1 body weight) was injected into the tail vein as an indicator for PE. The skin overlying the knee was excised to expose the joint capsule, and a 30-gauge, 1.25-cm-long needle was inserted into the knee joint for infusion of solution. After infusion of about 100 μl of saline, a 25-gauge needle was placed into the joint space for outflow of solution, with the tips of the two needles separated approximately 3 mm apart. The rates of both infusion and outflow were controlled at 200 μl/min by a push–pull syringe pump (SP120p, WPI, Sarasota, FL). The perfusion outflow was continuously collected in microcentrifuge tubes at 1 ml/tube every 5 min for a total of 14 tubes (70 min). The samples were immediately centrifuged to remove any red blood cells if present; only blood-free samples were used to measure the optical density (OD, proportional to the concentration of Evans Blue dye) at 620 nm on a spectrometer Spectronic 21D (Spectronic Instruments, Rochester, NY) [25]. Rat knees that had evidence of physical damage, swelling, or improperly aligned inflow and outflow needles were excluded from the study. The number of rat knees that were successfully perfused in each group was ≥6 or as indicated in the figures. The baseline value of PE was determined by averaging OD620 measurements of three continuous perfusion samples with 0.9% saline in the first 15 min. Another 10-min period was given for the application of antagonists or modulators prior to the perfusion of the primary PE inducer (BK or 5-HT). Perfusion of saline for the entire 70-min period was performed as a control.
To assess the dose–response of BK-induced PE, five concentrations of BK (0.1–1.0 μM) were perfused for 45 min after the 25-min saline infusion [3]. For 5-HT-induced PE, a concentration of 1 μM of 5-HT was chosen based on our own previous studies [6, 23]. To examine the modulatory effect of nociceptin on BK- or 5-HT-induced PE, various concentrations of nociceptin (0.0001–1.0 μM) were perfused 10 min before the perfusion of BK or 5-HT.
To evaluate whether the effects of BK, 5-HT, and nociceptin observed in this study were specifically receptor-mediated, and whether other mediators were involved, selective antagonists for the respective receptors and receptor subtypes were used 10 min before the application of BK, 5-HT, or nociceptin. Dosages of the antagonists—[des-Arg10]-HOE 140 (0.5–1.0 μM), HOE 140 (0.05–1.0 μM), (−)pindolol (0.5–1.0 μM), ketanserin (0.1–1.0 μM), CGRP8-37 (0.5–1.0 μM), AH-6809 (0.1–1.0 μM), mepyramine (0.1–1.0 μM), and J-113397 (0.005, 1.0 μM)—were chosen based on their binding affinities for respective receptors and in vivo doses reported in the literature [7, 23, 26–32], and with additional consideration of possible degradation during perfusion.
Statistical analysis
Data are presented as mean ± standard error of the mean (SEM) for the complete curves of dose–response and time–course measurements, as well as for measurements of the area under the curve (AUC) from 30 to 70 min. Statistical differences between two groups were analyzed by a Student’s t-test, and those between different groups and doses were analyzed by ANOVA with Fisher’s post-hoc test.
Results
Dose response and time course of BK-induced PE
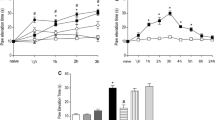
Intra-articular perfusion of BK produced significant PE in the rat knee joints in a dose-dependent manner, with 0.1 μM BK inducing small but significant PE as compared with saline (P < 0.05) and 0.25 μM BK doubling it (P < 0.01). At a concentration of 0.5 μM, BK induced a PE that was threefold that of 0.1 μM BK (P < 0.001) (Fig. 1a). As a negative control, perfusion with saline alone did not produce PE in the knee joints. At a concentration of 1 μM, BK induced the maximum PE. The ED50 for BK in stimulating PE was estimated to be 0.32 μM (Fig. 1b). The significant increase in PE occurred after 10 min of BK perfusion (35 min in the time scale in Fig. 1a), reached a peak level after 20 min of BK perfusion, and declined slightly over time. The effect of 0.5 μM of BK was comparable to that of 1 μM of 5-HT. The concentrations of 0.5 μM of BK and 1 μM of 5-HT were chosen in the subsequent experiments for analyzing their signaling and modulation mechanisms in inducing PE in the rat knee joints.

BK-induced PE in the rat knee joint. a The dose response and time course of BK-induced PE. The starting points of perfusion of BK or saline are indicated. b The dose-dependent effect and the estimation of ED50 of BK in producing PE in the rat knee joint, calculated as the area under the curve (AUC, OD 620 nm) between the time points of 30 and 70 min. The number of rat knee joints perfused in each group was n ≥ 6. Data are presented as the mean ± SEM
Specific receptor mediation of BK- and 5-HT-induced PE
To examine which subtypes of receptors mediate BK- and 5-HT-induced PE, receptor subtype-selective antagonists were perfused 10 min before the perfusion of BK and 5-HT, respectively. The BK receptor B1-selective antagonist [des-Arg10]-HOE 140 (0.5–1 μM) did not show any blocking effect on BK-induced PE. B2-selective antagonist HOE 140, at concentrations of 0.05–0.5 μM, displayed a dose-dependent blockade effect (Fig. 2a); response of 1.0 μM of HOE140 was equal to that of 0.5 μM (Fig. 3a).

Effects of receptor subtype-selective antagonists on BK- and 5-HT-induced PE. PE is calculated as the area under the curve (OD 620 nm) between the time points 30 and 70 min. Different concentrations of B1 antagonist [des-Arg10]-HOE140 and B2 antagonist HOE140 were applied 10 min before the perfusion of BK (a), and 5-HT1A/1B antagonist (−)pindolol and 5-HT2A antagonist ketanserin were applied 10 min before the perfusion of 5-HT (b). Only the effect of the highest concentration (1 μM) of [des-Arg10]-HOE140 and (−)pindolol is shown (column 2 in a and b). Data are presented as the mean ± SEM (n ≥ 6). *P < 0.05, **P < 0.01, as compared with the control group (BK or 5-HT alone)

The involvement of CGRP, PGE2, and histamine in BK and 5-HT-induced PE. The individual or the combination of the antagonists was applied 10 min prior to the perfusion of BK (a) or 5-HT (b). *P < 0.05, **P < 0.01, and ***P < 0.001, as compared with the saline control. Number of knee joints n ≥ 6 in each group
There are seven types of 5-HT receptors, some of them even have subtypes. As a more complete investigation was conducted in our previous study using the same experimental model [6], the present study only used two antagonists to demonstrate the receptor mediation. In 5-HT-induced PE, the 5-HT1A/1B antagonist (−)pindolol at 0.5 and 1 μM concentrations showed very little antagonizing effect; however, the 5-HT2A antagonist ketanserin significantly blocked PE in a dose-dependent manner (Fig. 2b). These results indicate that the observed PE produced by either BK or 5-HT is mediated by specific subtype(s) of receptors.
Involvement of CGRP, prostaglandins, and histamine in BK- and 5-HT-induced PE
To investigate whether other known inflammation mediators and neurotransmitters are involved in the signaling pathways of BK- or 5-HT-induced PE, we applied specific antagonists of the CGRP, PGE2, and histamine H1 receptors. These receptors have been shown to be involved in inflammation [7, 33, 34]. CGRP receptor antagonist CGRP8-37, PGE2 receptor antagonist AH-6809, and histamine H1 receptor antagonist mepyramine each partially inhibited BK-induced PE in a dose-dependent manner; when applied together, they were able to reduce BK-induced PE to the extent near (but not ≥) that of BK B2-selective antagonist HOE140 alone. When all four antagonists (i.e., CGRP8-37, AH-6809, mepyramine, and HOE140) were co-perfused, they markedly inhibited the BK-induced PE, which exceeded the blocking effect of HOE140 alone (Fig. 3a). In contrast, in 5-HT-induced PE, only AH-6809 and mepyramine showed a partial inhibitory effect, whereas CGRP8-37 had no effect (Fig. 3b). The results suggest that CGRP, PGs, and histamine are all involved in the signaling of BK-induced PE; and that PGs and histamine, but not CGRP, are involved in 5-HT-induced PE.
Effect of nociceptin on BK- and 5-HT-induced PE
To examine the effect of nociceptin on BK- and 5-HT-induced PE, we perfused nociceptin without or with the ORL1 receptor antagonist J-113397 prior to the application of the primary PE inducer BK or 5-HT. At subnanomolar concentrations (<10−3 μM), nociceptin had very little effect on BK-induced PE. With an increase in concentration (≥10−3 μM), nociceptin inhibited BK-induced PE dose-dependently, with 0.01 μM of nociceptin showing an inhibition by approximately 32% (AUC, 0.82 ± 0.2, P > 0.05), 0.1 μM by 44% (AUC, 0.67 ± 0.18, P < 0.05), and 1 μM by 51% (AUC, 0.58 ± 0.07, P < 0.01). Nociceptin did not facilitate BK-produced PE at any concentration. The inhibitory effect of nociceptin on BK-produced PE was effectively blocked by 1 μM of antagonist J-113397, indicating that it was specifically mediated by ORL1 receptors (Fig. 4a, b). Nociceptin or J-113397 alone did not produce PE in the rat knee joints (data not shown).

The effect of nociceptin on BK- and 5-HT-induced PE. a The dose-dependent effect of nociceptin on BK-induced PE and its reversal by ORL1-antagonist J-113397, as presented by time course curves. The starting time points of perfusion of different agents are indicated. b and c The comparison of dose-response of nociceptin and its antagonist J-113397 on BK- and 5-HT-induced PE, presented as AUC (OD 620 nm) between time points of 30 and 70 min. Two concentrations (0.005 and 1 μM) of J-113397 were used to block the effect of low and high concentrations of nociceptin, respectively. Data are presented as the mean ± SEM (n ≥ 6). *P < 0.05, **P < 0.01, as compared with the control group (BK or 5-HT alone)
The effect of nociceptin on 5-HT-induced PE was dose-related and bi-directional. Lower concentrations of nociceptin (10−4 and 10−3 μM) potentiated 5-HT-induced PE by 45 and 50% (AUC, 1.50 ± 0.17 and 1.55 ± 0.12, P < 0.05), respectively. Higher concentrations of nociceptin (0.1 and 1 μM) inhibited 5-HT-induced PE. At a concentration of 1 μM, nociceptin inhibited 5-HT-induced PE by 45% (AUC, 0.57 ± 0.14, P < 0.05). At a concentration of 0.01 μM, nociceptin showed neither potentiation nor inhibition. Antagonist J-113397 blocked both potentiation and inhibition effects produced by low and high concentrations of nociceptin on 5-HT-produced PE, respectively, indicating that both effects were specifically mediated by ORL1 receptors (Fig. 4b).
Discussion
BK and 5-HT are two of the most potent stimulants and mediators of inflammation in the joints. Therefore, it is important to understand the processes and pathways in BK- and 5-HT-induced inflammation. Previous studies showed that both BK and 5-HT produce PE through an activation of sympathetic nerve terminals [4–6].
The present study has investigated the signaling mechanism of BK-induced PE and its modulation using the rat knee-joint perfusion model, and compared them with those of 5-HT-induced PE. Our experiments clearly demonstrate that BK-induced PE in the rat knee is dose-dependent, and is specifically mediated by B2 subtype receptors, but not by B1, as indicated by antagonist blockade results. These results are in agreement with previous reports using similar animal models [3, 35, 36]. Early studies showed that B2 receptors are expressed in human synovial tissues [37] and that BK can be released from peripheral nerve terminals, as well as from peripheral tissues and cells including endothelial cells and neutrophils, upon nociceptive stimulation [37–40]. These findings indicate an important role of BK and B2 receptors in knee-joint inflammation. However, our study could not rule out the contribution of the B1 subtype receptors. One reason is the conditions and timing of the B1 receptor expression. While B2 receptors are normally expressed in many tissues, the B1 receptors have little or very low expression under normal conditions. The expression of B1 could, however, be upregulated over a period of hours following a chronic exposure to noxious stimuli or inflammation factors [41]. Our experimental knee perfusion model was designed to analyze acute inflammation events within 1 h. Previous studies have reported that B1 receptors can be induced and activated in chronic inflammation [36]. Another possible reason is that our present study only used BK in its full-length peptide form, which is selective for the B2 receptors and much less selective for the B1 [41]. Without using a B1-selective agonist to induce PE, any assessment of the contribution of B1 receptors would be inconclusive.
Our study also confirms that 5-HT induces PE via specific 5-HT2A subtype of serotonin receptor, which is consistent with previous studies [6].
Using specific receptor antagonists, the study demonstrates that other inflammation mediators and neurotransmitters are involved in the processes of BK- and 5-HT-induced PE. Results show that CGRP, PGs, and histamine are involved in the signaling pathway of BK-induced PE. As the blocking effects of each or the combination of the three antagonists (CGRP8-37, AH-6809, and mepyramine) are smaller than the effects of the B2 antagonist HOE 140, CGRP, PG, and histamine are likely to be activated downstream of the activation of B2 receptors. Our results also suggest that these mediators can in turn augment the BK effect, as the three antagonists and B2 antagonist together dramatically blocked BK-induced PE, an effect significantly greater than that of B2 antagonist alone. There is evidence that BK is able to induce the release of CGRP, PGs, and histamine in a variety of tissues and cells [9, 10, 42], and these secondary mediators in turn can cause or enhance inflammation [7, 43–45]. In comparison, our results show that in 5-HT-stimulated PE, PGs and histamine also play important roles, which is in agreement with our previous studies [7, 46], but CGRP is apparently not involved.
A previous study by Cambridge and Brain investigated the signaling mechanisms of BK-induced PE in a similar knee-joint perfusion model [35]. There are agreements and differences between their findings and ours. One important difference is whether secondary mediators, such as neuropeptides and histamine, are involved in BK-induced PE. Their results suggested no secondary release of inflammation mediators is involved. One possible explanation is an obvious difference in the perfusion scheme in the two studies. The study by Cambridge and Brain perfused testing agents for 5 min and analyzed a single sample of 1 ml collected 10 min after, whereas our study carried out continuous perfusion for 45 min and collected samples every 5 min for the whole length of time. Therefore, our present study might be able to obtain results different from those of the early study.
The present study demonstrated that BK- and 5-HT-induced PE are differentially regulated by nociceptin. Our data show that the modulation by nociceptin of 5-HT-stimulated PE is bi-phased: lower concentrations of nociceptin potentiate and higher concentrations inhibit the PE. In contrast, the modulation by nociceptin of BK-stimulated PE is mono-phased: only a dose-dependent inhibition exists. One possible explanation is that there might be a significant difference in nociceptin regulation of the BK and 5-HT signaling systems, including the regulation of presynaptic and post-synaptic events. Nociceptin may differentially regulate the release of 5-HT and BK. It has been reported that nociceptin bi-directionally regulates the release of 5-HT in neurons [47]. Secondly, nociceptin may differentially regulate the second mediators downstream of BK or 5-HT receptor activation. For example, nociceptin can inhibit the release of CGRP [15, 48]; the latter is an important mediator in BK-induced PE, but not in 5-HT-induced PE, as demonstrated by this study. In addition, it is known that while BK is degraded by peptidases after action, the effect of 5-HT can be reduced and terminated by pre-synaptic serotonin transporters, which re-uptake and remove 5-HT from the synaptic clefts. A previous study reported that nociceptin at micromolar concentrations inhibits dopamine and GABA transporters and has little effect on serotonin transporters [49]. The effect of lower concentrations (nanomolar and subnanomolar) of nociceptin was not examined however. There is a possibility that nociceptin may have a dose-dependent effect on serotonin transporters and thus differentially regulates the quantity of 5-HT available for activating post-synaptic serotonin receptors in the knee joints.
In summary, BK- and 5-HT-induced PE in the rat knee joints are mediated by their specific receptors and downstream inflammation mediators. There are fine differences between BK- and 5-HT-induced PE in their signaling pathways and their regulation by nociceptin. The understanding of these mechanisms could help to develop new therapies for acute and chronic inflammation in the joints.
References
Scott DT, Lam FY, Ferrell WR. Acute joint inflammation—mechanisms and mediators. Gen Pharmacol. 1994;25:1285–96.
Vane J, Botting R. Inflammation and the mechanism of action of anti-inflammatory drugs. FASEB J. 1987;1:89–96.
Green PG, Luo J, Heller PH, Levine JD. Neurogenic and non-neurogenic mechanisms of plasma extravasation in the rat. Neuroscience. 1993;52:735–43.
Green PG, Luo J, Heller P, Levine JD. Modulation of bradykinin-induced plasma extravasation in the rat knee joint by sympathetic co-transmitters. Neuroscience. 1993;52:451–8.
Miao FJ, Jänig W, Levine J. Role of sympathetic postganglionic neurons in synovial plasma extravasation induced by bradykinin. J Neurophysiol. 1996;75:715–24.
Pierce PA, Xie GX, Peroutka SJ, Green PG, Levine JD. 5-Hydroxytryptamine-induced synovial plasma extravasation is mediated via 5-hydroxytryptamine2A receptors on sympathetic efferent terminals. J Pharmacol Exp Ther. 1995;275:502–8.
Xie G, Wang Y, Sharma M, Gabriel A, Mitchell J, Xing Y, et al. 5-Hydroxytryptamine-induced plasma extravasation in the rat knee joint is mediated by multiple prostaglandins. Inflamm Res. 2003;52:32–8.
Wang Y, Mitchell J, Sharma M, Gabriel A, Moriyama K, Palmer PP. Leukotrienes mediate 5-hydroxytryptamine-induced plasma extravasation in the rat knee joint via CysLT-type receptors. Inflamm Res. 2004;53:66–71.
Averbeck B, Reeh PW. Interactions of inflammatory mediators stimulating release of calcitonin gene-related peptide, substance P and prostaglandin E(2) from isolated rat skin. Neuropharmacology. 2001;40:416–23.
Hua XY, Jinno S, Back SM, Tam EK, Yaksh TL. Multiple mechanisms for the effects of capsaicin, bradykinin and nicotine on CGRP release from tracheal afferent nerves: role of prostaglandins, sympathetic nerves and mast cells. Neuropharmacology. 1994;33:1147–54.
Green PG, Levine JD. Delta- and kappa-opioid agonists inhibit plasma extravasation induced by bradykinin in the knee joint of the rat. Neuroscience. 1992;49:129–33.
Stein C, Comisel K, Haimerl E, Yassouridis A, Lehrberger K, Herz A, et al. Analgesic effect of intraarticular morphine after arthroscopic knee surgery. N Engl J Med. 1991;325:1123–6.
Meunier JC, Mollereau C, Toll L, Suaudeau C, Moisand C, Alvinerie P, et al. Isolation and structure of the endogenous agonist of opioid receptor-like ORL1 receptor. Nature. 1995;377:532–5.
Reinscheid RK, Nothacker HP, Bourson A, Ardati A, Henningsen RA, Bunzow JR, et al. Orphanin FQ: a neuropeptide that activates an opioidlike G protein-coupled receptor. Science. 1995;270:792–4.
Helyes Z, Németh J, Pintér E, Szolcsányi J. Inhibition by nociceptin of neurogenic inflammation and the release of SP and CGRP from sensory nerve terminals. Br J Pharmacol. 1997;121:613–5.
Inoue M, Kawashima T, Takeshima H, Calo G, Inoue A, Nakata Y, et al. In vivo pain-inhibitory role of nociceptin/orphanin FQ in spinal cord. J Pharmacol Exp Ther. 2003;305:495–501.
Neal CR Jr, Mansour A, Reinscheid R, Nothacker HP, Civelli O, Watson SJ Jr. Localization of orphanin FQ (nociceptin) peptide and messenger RNA in the central nervous system of the rat. J Comp Neurol. 1999;406:503–47.
Pettersson LM, Sundler F, Danielsen N. Expression of orphanin FQ/nociceptin and its receptor in rat peripheral ganglia and spinal cord. Brain Res. 2002;945:266–75.
Xie GX, Meuser T, Pietruck C, Sharma M, Palmer PP. Presence of opioid receptor like (ORL1) receptor mRNA splice variants in peripheral sensory and sympathetic neuronal ganglia. Life Sci. 1999;64:2029–37.
Tian JH, Xu W, Fang Y, Mogil JS, Grisel JE, Grandy DK, et al. Bidirectional modulatory effect of orphanin FQ on morphine-induced analgesia: antagonism in brain and potentiation in spinal cord of the rat. Br J Pharmacol. 1997;120:676–80.
Li N, Wei SY, Yu LC, Moriyama K, Mitchell J, Palmer PP. Role of nociceptin in the modulation of nociception in the arcuate nucleus of rats. Brain Res. 2004;1025:67–74.
Carpenter KJ, Vithlani M, Dickenson AH. Unaltered peripheral excitatory actions of nociceptin contrast with enhanced spinal inhibitory effects after carrageenan inflammation: an electrophysiological study in the rat. Pain. 2000;85:433–41.
Grond S, Gabriel A, Pietruck C, Yu LC, Xie GX, Pierce Palmer P. Bi-directional modulation of 5-hydroxytryptamine-induced plasma extravasation in the rat knee joint by nociceptin. Neuroscience. 2001;103:1085–92.
Barton NJ, Stevens DA, Hughes JP, Rossi AG, Chessell IP, Reeve AJ, et al. Demonstration of a novel technique to quantitatively assess inflammatory mediators and cells in rat knee joints. J Inflamm (Lond). 2007;4:13–20.
Carr J, Wilhelm DL. The evaluation of increased vascular permeability in the skin of guinea-pigs. Aust J Exp Biol Med Sci. 1964;42:511–22.
Regoli D, Nsa Allogho S, Rizzi A, Gobeil FJ. Bradykinin receptors and their antagonists. Eur J Pharmacol. 1998;348:1–10.
Rhaleb NE, Rouissi N, Jukic D, Regoli D, Henke S, Breipohl G, et al. Pharmacological characterization of a new highly potent B2 receptor antagonist (HOE 140: D-Arg-[Hyp3,Thi5,D-Tic7,Qic8]bradykinin). Eur J Pharmacol. 1992;210:115–20.
Corradetti R, Laaris N, Hanoun N, Laporte AM, Le Poul E, Hamon M, et al. Antagonist properties of (-)-pindolol and WAY 100635 at somatodendritic and postsynaptic 5-HT1A receptors in the rat brain. Br J Pharmacol. 1998;123:449–62.
de Clerck F, David JL, Janssen PA. Inhibition of 5-hydroxytryptamine-induced and -amplified human platelet aggregation by ketanserin (R 41 468), a selective 5-HT2-receptor antagonist. Agents Actions. 1982;12:388–97.
Dennis T, Fournier A, Cadieux A, Pomerleau F, Jolicoeur FB, St Pierre S, et al. hCGRP8-37, a calcitonin gene-related peptide antagonist revealing calcitonin gene-related peptide receptor heterogeneity in brain and periphery. J Pharmacol Exp Ther. 1990;254:123–8.
Van Rossum D, Menard DP, Fournier A, St-Pierre S, Quirion R. Binding profile of a selective calcitonin gene-related peptide (CGRP) receptor antagonist ligand, [125I-Tyr]hCGRP8-37, in rat brain and peripheral tissues. J Pharmacol Exp Ther. 1994;269:846–53.
Hill SJ, Daum P, Young JM. Affinities of histamine H1-antagonists in guinea pig brain: similarity of values determined from [3H]mepyramine binding and from inhibition of a functional response. J Neurochem. 1981;37:1357–60.
Cruwys SC, Kidd BL, Mapp PI, Walsh DA, Blake DR. The effects of calcitonin gene-related peptide on formation of intra-articular oedema by inflammatory mediators. Br J Pharmacol. 1992;107:116–9.
Taylor DJ, Woolley DE. Histamine H1 receptors on adherent rheumatoid synovial cells in culture: demonstration by radioligand binding and inhibition of histamine-stimulated prostaglandin E production by histamine H1 antagonists. Ann Rheum Dis. 1987;46:425–30.
Cambridge H, Brain SD. Mechanism of bradykinin-induced plasma extravasation in the rat knee joint. Br J Pharmacol. 1995;115:641–7.
Cao T, Brain SD, Khodr B, Khalil Z. B1 and B2 antagonists and bradykinin-induced blood flow in rat skin inflammation. Inflamm Res. 2002;51:295–9.
Cassim B, Naidoo S, Ramsaroop R, Bhoola KD. Immunolocalization of bradykinin receptors on human synovial tissue. Immunopharmacology. 1997;36:121–5.
Raidoo DM, Bhoola KD. Pathophysiology of the kallikrein-kinin system in mammalian nervous tissue. Pharmacol Ther. 1998;79:105–27.
Linardi A, Costa SK, da Silva GR, Antunes E. Involvement of kinins, mast cells and sensory neurons in the plasma exudation and paw oedema induced by staphylococcal enterotoxin B in the mouse. Eur J Pharmacol. 2000;399:235–42.
Hargreaves KM, Costello A. Glucocorticoids suppress levels of immunoreactive bradykinin in inflamed tissue as evaluated by microdialysis probes. Clin Pharmacol Ther. 1990;48:168–78.
Couture R, Harrisson M, Vianna RM, Cloutier F. Kinin receptors in pain and inflammation. Eur J Pharmacol. 2001;429:161–76.
Vietinghoff G, Paegelow I, Reissmann S. Induction of histamine release from rat mast cells by bradykinin analogues. Peptides. 1996;17:1467–70.
Cambridge H, Brain SD. Calcitonin gene-related peptide increases blood flow and potentiates plasma protein extravasation in the rat knee joint. Br J Pharmacol. 1992;106:746–50.
Holden NS, Gong W, King EM, Kaur M, Giembycz MA, Newton R. Potentiation of NF-kappaB-dependent transcription and inflammatory mediator release by histamine in human airway epithelial cells. Br J Pharmacol. 2007;152:891–902.
Bileviciute I, Theodorsson E, Lundeberg T. Effects of histamine on neuropeptide release into the knee joint perfusate and cerebrospinal fluid in rats. Neurosci Lett. 1997;226:9–12.
Grond S, Demopulos G, Herz J, Pierce Palmer P. Inhibition of synovial plasma extravasation by preemptive administration of an antiinflammatory irrigation solution in the rat knee. Anesth Analg. 2001;92:1301–6.
Mela F, Marti M, Ulazzi L, Vaccari E, Zucchini S, Trapella C, et al. Pharmacological profile of nociceptin/orphanin FQ receptors regulating 5-hydroxytryptamine release in the mouse neocortex. Eur J NeuroSci. 2004;19:1317–24.
Németh J, Helyes Z, Oroszi G, Thán M, Pintér E, Szolcsányi J. Inhibition of nociceptin on sensory neuropeptide release and mast cell-mediated plasma extravasation in rats. Eur J Pharmacol. 1998;347:101–4.
Liu Z, Wang Y, Zhang J, Ding J, Guo L, Cui D, et al. Orphanin FQ: an endogenous antagonist of rat brain dopamine transporter. NeuroReport. 2001;12:699–702.
Acknowledgments
This study was supported by grants NS56256, AR45570, and NS042593 from the National Institutes of Health (to Pamela Pierce Palmer).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible Editor: J. Di Battista.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Moriyama, K., Liu, J., Jang, Y. et al. Receptor mediation and nociceptin inhibition of bradykinin-induced plasma extravasation in the knee joint of the rat. Inflamm. Res. 58, 873–880 (2009). https://doi.org/10.1007/s00011-009-0058-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00011-009-0058-y