
Abstract
Background
Amyloid beta (Aβ) oligomers play a critical role in the pathogenesis of Alzheimer’s disease (AD) and represent a promising target for drug development. Tramiprosate is a small-molecule Aβ anti-aggregation agent that was evaluated in phase III clinical trials for AD but did not meet the primary efficacy endpoints; however, a pre-specified subgroup analysis revealed robust, sustained, and clinically meaningful cognitive and functional effects in patients with AD homozygous for the ε4 allele of apolipoprotein E4 (APOE4/4 homozygotes), who carry an increased risk for the disease. Therefore, to build on this important efficacy attribute and to further improve its pharmaceutical properties, we have developed a prodrug of tramiprosate ALZ-801 that is in advanced stages of clinical development. To elucidate how tramiprosate works, we investigated its molecular mechanism of action (MOA) and the translation to observed clinical outcomes.
Objective
The two main objectives of this research were to (1) elucidate and characterize the MOA of tramiprosate via an integrated application of three independent molecular methodologies and (2) present an integrated translational analysis that links the MOA, conformation of the target, stoichiometry, and pharmacokinetic dose exposure to the observed clinical outcome in APOE4/4 homozygote subjects.
Method
We used three molecular analytical methods—ion mobility spectrometry–mass spectrometry (IMS–MS), nuclear magnetic resonance (NMR), and molecular dynamics—to characterize the concentration-related interactions of tramiprosate versus Aβ42 monomers and the resultant conformational alterations affecting aggregation into oligomers. The molecular stoichiometry of the tramiprosate versus Aβ42 interaction was further analyzed in the context of clinical pharmacokinetic dose exposure and central nervous system Aβ42 levels (i.e., pharmacokinetic–pharmacodynamic translation in humans).
Results
We observed a multi-ligand interaction of tramiprosate with monomeric Aβ42, which differs from the traditional 1:1 binding. This resulted in the stabilization of Aβ42 monomers and inhibition of oligomer formation and elongation, as demonstrated by IMS–MS and molecular dynamics. Using NMR spectroscopy and molecular dynamics, we also showed that tramiprosate bound to Lys16, Lys28, and Asp23, the key amino acid side chains of Aβ42 that are responsible for both conformational seed formation and neuronal toxicity. The projected molar excess of tramiprosate versus Aβ42 in humans using the dose effective in patients with AD aligned with the molecular stoichiometry of the interaction, providing a clear clinical translation of the MOA. A consistent alignment of these preclinical-to-clinical elements describes a unique example of translational medicine and supports the efficacy seen in symptomatic patients with AD. This unique “enveloping mechanism” of tramiprosate also provides a potential basis for tramiprosate dose selection for patients with homozygous AD at earlier stages of disease.
Conclusion
We have identified the molecular mechanism that may account for the observed clinical efficacy of tramiprosate in patients with APOE4/4 homozygous AD. In addition, the integrated application of the molecular methodologies (i.e., IMS-MS, NMR, and thermodynamics analysis) indicates that it is feasible to modulate and control the Aβ42 conformational dynamics landscape by a small molecule, resulting in a favorable Aβ42 conformational change that leads to a clinically relevant amyloid anti-aggregation effect and inhibition of oligomer formation. This novel enveloping MOA of tramiprosate has potential utility in the development of disease-modifying therapies for AD and other neurodegenerative diseases caused by misfolded proteins.
Similar content being viewed by others


We have elucidated and characterized the molecular mechanism of action of tramiprosate. |
Tramiprosate modulates conformational flexibility of amyloid beta Aβ42, leading to the prevention of oligomer seed formation and thus aggregation. |
Translational analysis shows an alignment of the three described molecular effects of Aβ42 with pharmacokinetic and published clinical data. |
1 Introduction
Alzheimer’s disease (AD) is the most prevalent neurodegenerative disorder, affecting a large number of elderly people worldwide. It is widely accepted that amyloid beta (Aβ) is one of the key pathogenic causes for AD [1, 2]. The level of soluble, non-fibrillar Aβ oligomers in the brain correlates strongly with the severity of the disease [1, 3, 4], suggesting that soluble oligomeric species of Aβ, rather than the fibrillary form within amyloid plaques, likely play a pivotal role in AD pathophysiology.
Aβ peptides, particularly Aβ42, have a strong intrinsic tendency to self-assemble and form aggregates that constitute neurotoxic oligomeric species [5]. Monomeric Aβ peptides exhibit very high conformational flexibility [6, 7], which represents one of the major challenges for this therapeutic target. The initial random coil structure shows characteristics of an α-helix and β-sheet mixture that transforms into a final structure predominantly comprising β-sheets [8]. At this stage, a nucleation phase occurs that initiates Aβ aggregation. Soluble Aβ oligomers have been shown to form by a nucleation-dependent process, wherein most neurotoxic Aβ42 becomes a seed in the aggregation process and also enhances the oligomerization of Aβ40, the most prevalent species of Aβ in the central nervous system (CNS) [9]. The conformational state of Aβ42 thus plays a critical role in the formation of oligomers, especially in the formation of initiation seeds for Aβ aggregation.
In this study, we focused on tramiprosate, 3-aminopropanesulfonic acid, a homolog of the amino acid taurine, that has been shown in preclinical studies to bind to soluble Aβ and inhibit Aβ aggregation, leading to a reduction of Aβ plaque load [10] (Fig. 1). The safety and efficacy of tramiprosate has been evaluated in the nonclinical program and in 16 clinical trials, including two phase III trials in over 2000 subjects with AD. Across these studies, tramiprosate safety and tolerability were favorable, and the main adverse events were nausea and vomiting, which are being addressed by the development of tramiprosate prodrug ALZ-801. While the phase III studies in patients with mild-to-moderate AD did not meet the primary efficacy endpoints, a pre-specified subgroup analysis revealed robust, sustained, and clinically meaningful cognitive and functional effects in patients with AD homozygous for the ε4 allele of apolipoprotein E4 (APOE4/4 homozygotes) [11]. Importantly, there was evidence of an APOE4 gene dose effect, where the order of efficacy on both cognition and function followed the number of APOE4 alleles. The APOE4/4 homozygotes showed larger (almost double) efficacy than APOE4 heterozygotes, and the APOE4 heterozygotes showed higher efficacy than non-carriers. This APOE4 gene-dose effect likely reflects the fact that APOE4 carriers have a higher burden of amyloid pathology, with APOE4/4 homozygotes thought to have the highest burden of Aβ oligomer pathology. To date, the mechanism of action (MOA) for tramiprosate and the nature of its molecular interactions with Aβ peptides, has not been elucidated.

Chemical structure of tramiprosate (left) and amino acid sequence of amyloid beta Aβ42 (right)
2 Objectives
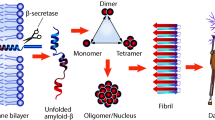
The two main objectives of this research were to (1) elucidate and characterize the molecular MOA of tramiprosate via an integrated application of three independent molecular methodologies—ion mobility spectrometry–mass spectrometry (IMS–MS), nuclear magnetic resonance (NMR), and thermodynamics analysis—and (2) present an integrated translational analysis that links the MOA, conformation of the target, stoichiometry, and pharmacokinetic dose exposure to the observed clinical outcome in APOE4/4 homozygote subjects. We hereby report the discovery and elucidation of a novel multi-ligand enveloping MOA, which mediates the Aβ anti-aggregation activity of tramiprosate and potentially underpins its clinical efficacy in patients with amyloid-bearing AD (Fig. 2). A number of intertwining molecular aspects of this MOA form a coherent understanding of the control of conformational flexibility of Aβ and its impact on clinical outcome. The integrated translational analysis shows a consistent alignment of these preclinical-to-clinical elements, describing a unique example of translational medicine.

An illustration of the impact of tramiprosate on amyloid beta Aβ42 conformation and the resultant anti-aggregation effects. Comparison of Aβ42 conformation of β-sheets in the pathological process leading to Alzheimer’s disease with the semi-cyclic conformation of Aβ42 under multi-ligand tramiprosate effect, which prevents the formation of Aβ42 oligomers. AD Alzheimer’s disease
3 Methods
3.1 Molecular Modeling and Molecular Dynamics Simulations
All molecular modeling was performed using the Schrödinger suite (2015-3; Schrödinger, LLC; New York, NY, USA; 2015). Molecular dynamics simulations were run using Desmond [12]. The simulations were run on GeForce GTX Titan Black graphics processing unit cards. The optimized potential for liquid simulations (OPLS 3.0) force field [13] was used to model all interactions, and the SPC model was used for waters. The 1IYT Aβ42 NMR structure from the Protein Data Bank (PDB) was used as a starting point for molecular dynamics simulations. This structure is primarily alpha helical and is representative of the peptide in an apolar environment. A 20-Å box of water or a mixed solvent box of 1 % tramiprosate in water was added around the peptide using Schrödinger system set-up tools. Ions were added to neutralize the charge of the entire system. Simulations were equilibrated and run under NPT conditions [constant number (N), pressure (P) and temperature (T)] with periodic boundary conditions. A Nose–Hoover Thermostat and Martina–Tobias–Klein barostat were used to control temperature and pressure, respectively. Simulations were run in replicates of three for 100 ns each, and the results were compiled for analysis. Principal component (PC) analysis was performed using ProDy [14] and plotted using custom python scripts.
3.2 Ion Mobility Spectrometry–Mass Spectrometry (IMS–MS)
The conditions used for MS, using a Waters Synapt G2-S, were as follows: positive polarity in sensitivity mode; capillary = 2.5 kV; nebulizer = 2 mbar; source temperature = 80 °C; desolvation temperature = 60 °C; sample cone setting = 35 V; source offset setting = 60 V; and mass range = 500–4000 m/z. These conditions were maintained throughout the study to ensure consistency of the data and to avoid influencing the detection of oligomers due to preferential ionization conditions.
Samples were directly infused into the mass spectrometer at a flow rate of 10 µl/min using a Protea PM-1000 Syringe Pump and Hamilton 1-ml syringe. The data acquisition of the amyloid peptide was performed using a Waters Synapt G2-S quadrupole time of flight mass spectrometer (Q-TOF MS) with traveling wave ion mobility (Waters Corp., Milford, MA, USA). The data were acquired using the systems sensitivity mode to allow for the detection of the less abundant oligomers. Samples were infused at room temperature. The IMS–MS studies were conducted at Protea, Inc. (Morgantown, WV, USA).
3.2.1 Sample Preparation
We reconstituted 1 mg of recombinant human Aβ42 peptide from BioLegend (99% purity, cat# 843801) in 200 µl of Fisher Optima LC/MS (liquid chromatography/MS) grade water (cat# W6-1) and vortexed it vigorously for 2 min to solubilize the peptide creating a 5 mg/ml solution. Samples were then diluted to a final concentration of 22 pmol/µl prior to incubation. The sample mixtures were then incubated at room temperature for 0, 4, and 24 h. After the acquisition of incubated samples was completed, the raw data were analyzed using the Waters MassLynx v2.4 suite with DriftScover v2.7 to visualize drift times for the peptide.
3.2.2 Amyloid Beta Aβ42 Species Characterization
Aβ42 species characterization using IMS–MS was performed by direct infusion at 22 pmol/µl in water. The peptide was prepared in water to maintain the native state conformation of the peptide, and ion mobility data acquisition was performed to detect and characterize the conformational changes of the native state monomer and any oligomers that may have formed during the incubation.
3.2.3 Tramiprosate IMS–MS Binding Study
The data acquisition for Aβ42 peptide was performed using a Waters Synapt G2-S Q-TOF MS with traveling wave ion mobility (Waters Corp.). The data were acquired using the systems sensitivity mode to allow for the detection of the less abundant oligomers. Samples were infused at room temperature as in the previous section.
We reconstituted 1 mg of tramiprosate in 1 ml of Fisher Optima LC/MS grade water (cat# W6-1) and vortexed it vigorously for 2 min until completely dissolved. The sample was then diluted to create 220, 2200, and 22,000 pmol/µl solutions to perform a 10-, 100-, and 1000-fold molar excess for the binding experiments with Aβ42.
We reconstituted 1 mg of recombinant human Aβ42 peptide in 200 µl of Fisher Optima LC/MS grade water and vortexed vigorously to solubilize to a 5 mg/ml solution. Samples were then diluted to their final concentrations prior to incubation. The sample mixtures were incubated at room temperature for 0, 4, and 24 h, followed by analysis as described in the previous subsections.
3.3 Nuclear Magnetic Resonance Spectroscopy
3.3.1 Aβ42 Preparation
15N-uniformly labeled Aβ42 peptide was purchased from rPeptide (Bogart, GA, USA) and used without further purification. The buffer system described by Roche et al. [15], except for NaOH, was used to acquire the NMR data of Aβ42 titrated with tramiprosate (90% H2O/10% D2O sodium phosphate buffer, pH 7.4 at 37 °C). NaOH was omitted from the sample preparation as it may interfere with tramiprosate binding. The total concentration of Aβ42 in the sample was 75 μM to limit any initial aggregation. The D2O was used to lock the NMR spectrometer.
3.3.2 NMR Experiments
NMR experiments were conducted at 800 MHz on a Bruker AVANCE II spectrometer using a 5 mm HCN cryogenic probe. The probe sample temperature was initially set to 10 °C then slowly warmed to 25 °C and to 37 °C upon insertion of the sample. Spectra were recorded at both 25 and 37 °C. A 1D 3919 Watergate [16] experiment was first conducted to optimize the water suppression and 1H spectral width for the 2D experiments. A relaxation delay of 1.5 s was used with 128 scans. The 1D Watergate experiment was optimized to suppress the largest peak (H2O) in the spectrum. The optimized parameters were then transferred to the 2D experiments. 2D1H-15N SOFAST-HMQC with 3919 Watergate were used [16, 17]. A total of 128 increments was acquired in t1 (15N) with 96 scans per increment. A J(15N-1H) coupling of 95 Hz was used. All spectra were processed using TopSpin 3.5. Assignments were taken from the literature [15, 18, 19].
3.4 Human Plasma and Brain Pharmacokinetic Analyses, Cerebrospinal Fluid (CSF) Aβ42 Levels, and Pharmacokinetic–Pharmacodynamic Translation
Plasma and cerebrospinal fluid (CSF) concentrations of tramiprosate were determined in frozen samples at 78 weeks of the completed North American phase III study using validated LC-MS/MS methods [lower limit of quantitation (LLQ) = 5 and 2.5 ng/ml in plasma and CSF, respectively]. The steady-state drug level in human brain was projected based on the brain/plasma drug exposure relationship derived from a rodent model, assuming comparable brain penetration and intra-cerebral kinetics of tramiprosate between the two species following oral administration [20, 21]. Pharmacokinetic data analyses were conducted using Winnonlin Professional v5.0.1 (Pharsight, Mountain View, CA, USA). The CSF Aβ42 concentrations were measured by enzyme-linked immunosorbent assay (ELISA) in patients with AD in the tramiprosate phase II trial as previously described [22] and were used in the present pharmacokinetic–pharmacodynamic analyses.
4 Results
4.1 Multi-Ligand Binding Mode of Tramiprosate and Effects on Aβ42 Monomer Conformation
To address the high conformational flexibility of Aβ42 and characterize its interaction with tramiprosate, we used IMS with a Q-TOF MS with traveling wave ion mobility. IMS is a powerful technique capable of separating molecular ions based on their size and conformation and can also be used to characterize the stoichiometry of ligand–protein complexes [23].
This IMS–MS analysis (Fig. 3) illustrated both the stoichiometry of the drug–protein complex and the shape of Aβ42 and also showed that multiple molecules of tramiprosate bind to a single molecule of Aβ42, in agreement with the previous studies [24]. These results indicate that tramiprosate formed a dynamic solvation envelope surrounding Aβ42 that interacted with the peptide in a dynamic manner. Figure 3 also shows that Aβ42 alone adopted many different conformations, as indicated by a long yellow zone and how the multitude of those conformations changed with each additional bound molecule of tramiprosate. Analysis of the arrival time distribution clearly showed this conformational shift. As additional tramiprosate molecules interacted with Aβ42 monomer, many conformations of Aβ42 transitioned into a more compact, presumably semi-cyclic, conformation (Sect. 4.4). The most extended conformations of Aβ42 on the right part of the yellow zone gradually disappeared with each additional bound molecule of tramiprosate, indicating the formation of more compact and stabilized conformations. With three or more bound tramiprosate molecules, only the most compact conformer populations, and none of the extended populations, were detected. This suggests that the binding of the drug to the peptide has a significant effect on the generation of more defined and stabilized populations of Aβ42 conformers. Additional bound molecules of the drug further stabilized the narrow peak of the population of Aβ42 conformers.

Ion mobility spectrometry–mass spectrometry (IMS–MS) driftscope plot of the IMS drift time versus mass/charge (m/z) of amyloid beta Aβ42-tramiprosate stoichiometry. Aβ42 alone shows long time drifts (yellow zone), indicating many different populations of conformers. With an increasing number of bound tramiprosate molecules, the drift time of the Aβ42 conformers changes, indicating the presence of fewer and more stabilized conformations. Some of the extended conformers on the right completely disappear
4.2 Tramiprosate Prevents Formation of Aβ42 Oligomers
We next evaluated whether the Aβ42 conformation-stabilizing activity of tramiprosate affects aggregation, specifically the oligomer aggregation stages from monomers through soluble decamer species. To this end, we examined the formation of soluble Aβ42 oligomers in the absence or presence of tramiprosate by IMS–MS (Figs. 4, 5; Table 1). As expected, the critical neurotoxic oligomers (i.e. dimer, trimer, tetramer, pentamer, hexamer, and decamer) [25, 26] formed following an incubation of Aβ42 monomers; the identities of the oligomer species were further characterized at multiple charge states. However, in the presence of 1000-fold molar excess tramiprosate, the formation of the corresponding oligomers was inhibited. To explore a concentration–response relationship, we incubated monomeric Aβ42 with a 100-fold or 1000-fold molar excess of tramiprosate for 24 h. At a 100-fold molar excess, tramiprosate partially reduced the number of detectable oligomers. Strikingly, at 1000-fold molar excess, tramiprosate completely abrogated the full range of Aβ42 oligomer species (Table 2). Together, the results showed a concentration-dependent effect for tramiprosate in preventing the formation of Aβ42 oligomers, with complete inhibition achieved at the highest concentration tested (i.e., 1000-fold molar excess). Importantly, these findings suggest that tramiprosate stabilizes Aβ42 in its monomeric form and prevents the initiation stage of Aβ42 aggregation.

Ion mobility spectrometry–mass spectrometry (IMS–MS) 2D arrival time data showing annotated detection of oligomers of amyloid beta Aβ42 after 24-h incubation in the absence of tramiprosate. m/z mass/charge

Ion mobility spectrometry–mass spectrometry (IMS–MS) 2D arrival time data. a Detection of no oligomers in the amyloid beta Aβ42 + 1000 × tramiprosate sample after 24-h incubation. b The corresponding mass spectrum detecting only Aβ42 monomers in different charge states. m/z mass/charge
Together, these data show that the tramiprosate-enveloping mechanism, wherein Aβ42 peptide is enveloped by a cloud of tramiprosate reminiscent of a solvation effect (Sect. 4.4), has implications for clinical activity, especially because high molar excess of the tramiprosate was required in the clinical trials [11].
4.3 NMR Experiments Identify Aβ42 Residues that Interact with Tramiprosate
Next, we used 2D heteronuclear multiple quantum correlation NMR spectroscopy (2D 1H-15N HMQC NMR) of uniformly 15N-labeled Aβ42 peptide (in 90% H2O/10% D2O sodium phosphate buffer, pH 7.4 at 37 °C) to determine how tramiprosate binds to the peptide. Based on the peak dispersion of the spectrum (Fig. 6), monomeric Aβ42 adopted a random conformation, as expected [15]. The 2D 1H-15N HMQC NMR experiments were conducted on samples containing 75 μM Aβ42 titrated with tramiprosate to produce tramiprosate to Aβ42 ratios of 10:1, 100:1, 500:1, 1000:1, 3000:1, and 5000:1. Peak assignments of Aβ42 titrated with tramiprosate were then compared with 2D 1H-15N HMQC spectra of Aβ42 alone. When a 1000-fold excess of tramiprosate was added to the peptide solution, significant chemical shift perturbations were observed. No change was observed in the Aβ42 1H-15N HMQC spectrum at a ratio of 10:1 tramiprosate to Aβ42, but minor changes were observed at a ratio of 100:1. Significant changes in the 2D 1H-15N HMQC peaks began to arise at the ratio of 500:1, which plateaued (i.e., reached a steady state) at a ratio of 1000:1 tramiprosate to Aβ42. Further increases of the ratio to 3000:1 and 5000:1 had no effect on the chemical shift perturbation.

2D 1H-15N heteronuclear multiple quantum correlation (HMQC) nuclear magnetic resonance (NMR) spectrum showing interactions of tramiprosate with amyloid beta Aβ42. a 2D 1H-15N HMQC NMR spectrum with assignments. Aβ42 alone is shown in blue, and Aβ42 with tramiprosate at a ratio of 1:1000 is overlaid in red. b An expanded view of part of a. Assignments in red indicate a significant observed chemical shift perturbation. c Example of a chemical shift perturbation of R5 residue observed when Aβ42 was incubated with tramiprosate at a ratio of 1:1000. Residue R5 is an isolated peak that clearly shows a chemical shift as the tramiprosate concentration is increased. The dotted lines illustrate the center of each peak to gauge the change in chemical shift at each concentration level. The color coding represents no tramiprosate (blue), 100:1 (gold), 500:1 (green) and 1000:1 (red) tramiprosate to Aβ42
At a 1000-fold excess of tramiprosate over Aβ42, 22 Aβ42 residues showed significant chemical shift perturbations. The most dramatic changes were observed for R5, H6, S8, G9, Y10, K16, L17, V18, F19, N27, K28, and M35. The 2D 1H-15N HMQC peaks from these residues exhibited at least a 10 Hz chemical shift change in the 1H dimension, with K16 and K28 having chemical shift perturbations of 13.5 and 16.1 Hz, respectively, indicating a substantial interaction with tramiprosate. E3, V12, H13, H14, D23, S26, G25, G33, V36, and V39 showed smaller, yet still significant, chemical shift perturbations, indicating that they also interact with tramiprosate. Taken together, these results show that tramiprosate interacts with residues that span the length of Aβ42 in a concentration-dependent mode, which supports the IMS–MS data. Importantly, the strong tramiprosate binding to K16 and K28 supports tramiprosate-mediated disruption of the Lys28-Asp23 and/or Lys28-Glu22 salt bridges and suppression of neurotoxicity and misfolding [7, 27–29], given that these two lysine residues have been previously demonstrated to play a key role in mediation of these activities [8].
4.4 Molecular and Conformational Dynamics
Given the intrinsically disordered nature of Aβ42 and a high conformational dynamics, the interaction with tramiprosate is unlikely to be described by a static structural model with a single tramiprosate molecule bound. Hence, commonly applied structure-based drug-discovery approaches such as molecular docking are unlikely to provide a complete understanding of the MOA of tramiprosate. This represents a challenge to the characterization of the secondary structures of Aβ42 peptides because of their disordered nature and high aggregation propensity.
The characterization of free energy landscapes has been successful in rationalizing the conformational and folding behavior of such disordered proteins, and it provides a concrete representation of the conformational states of such proteins. A previous study [30] described changes in conformation from an α-helical structure to a disordered state, with portions of the peptide adopting a β-sheet structure described by a molecular dynamics simulation. To characterize the structure of Aβ42 alone and with different levels of excess tramiprosate, we performed a series of all atom molecular dynamics simulations. In the absence of tramiprosate, in water alone, Aβ42 adopted a number of very different conformations and was characterized by a disordered structure (Fig. 7a), which is in agreement with other published findings [30]. However, in the presence of increasing concentrations of tramiprosate, the peptide assumed a more conformationally stable form. The observed increase in conformational stability was concentration dependent. A two orders of magnitude molar excess of tramiprosate forced the protein to adopt a semi-cyclic conformation that was stabilized further by a salt bridge formed by the Asp1 N-terminal amino group and the C-terminal carboxylate of Ala42. This semi-cyclic conformation remained stable in the presence of tramiprosate. The Aβ42 was enveloped by multiple molecules of the drug, which interacted with many transient binding sites in a very dynamic manner. Figure 7b shows a molecular dynamics screenshot with six molecules of tramiprosate binding to a semi-cyclic Aβ42 conformer.

Analysis of molecular dynamics simulations with and without 1% tramiprosate. a Representative disordered structure of amyloid beta Aβ42. b Representative Aβ42 semicyclic ordered structure with six tramiprosate molecules bound. c Principle component analysis of simulation of Aβ42 folding alone in water. d Principle component analysis of simulation of Aβ42 folding in the presence of 1% tramiprosate
To describe the large conformational changes observed in these simulations, we performed a PC analysis of the free energy surface. This analysis distills the complex motions of a flexible protein into the largest uncorrelated motions, or PCs. The first major motion (PC1) of Aβ42 can be described as a bending of the two helices towards each other like a hinge, and the second motion (PC2) can be described as a twisting of the two helices. Without tramiprosate, Aβ42 exhibited a typical trait of intrinsically disordered proteins: it lacked a narrow, well-defined energy minimum for any single folded structure (Fig. 7c). When PC1 and PC2 were mapped according to their free energy, a number of energy wells were observed (Fig. 7c), which correspond to the multiple Aβ42 conformations detected experimentally via IMS–MS. The 1% tramiprosate solution, corresponding to an Aβ42 : tramiprosate molar ratio of 1:250, stabilized the peptide in the semi-cyclic conformation; the energy surface as described by PC analysis showed stabilization of the semi-cyclic conformation as a well-defined energy well (Fig. 7d). This correlates well with the conformer stabilization detected by IMS–MS arrival time distribution (Fig. 3). The stabilization of a single conformation prevents Aβ42 from changing form and aggregating into pathogenic oligomers. Both in the stabilization of a single conformation and in the characterization of multiple transient tramiprosate binding sites, these results correlate with the IMS–MS experiments, where we detected up to 13 molecules of tramiprosate bound to Aβ42, in agreement with previous MS data [24, 31]. Interestingly, tramiprosate above 3 mM concentrations did not bind to plasma proteins from human, dogs, and rats in a standard plasma protein-binding study using an ultrafiltration technique [32], suggesting an absence of non-specific binding to plasma proteins such as albumin (data not shown).
Molecular dynamics simulations with free energy landscape analysis predicted a strong effect of tramiprosate on the intrinsically disordered conformations of Aβ42; the effect leads to a defined population of semi-cyclic conformers characterized as a stabilized energy well in the PC plot (Fig. 7d). Structurally, this conformation has a cyclic nature and retains more order than Aβ42 alone. Visual inspection of the simulations demonstrated the transient binding and unbinding of numerous tramiprosate molecules simultaneously along the Aβ42 surface. All of these interactions form a dynamic equilibrium, leading to a very tight conformer population. Taken together, these results suggest that tramiprosate stabilizes the semi-cyclic conformation of Aβ42 and prevents the formation of an initiation seed, thus preventing aggregation of the peptide (Table 3).
4.5 Translational Analyses of Human Brain Drug Exposure vs. the Target
Tramiprosate was measured in the CSF specimens of patients with AD at week 78 from the phase III study [10, 22, 33], and its average concentration at the top tramiprosate dose of 150 mg twice a day (bid) was 60.4 nM (n = 11). Furthermore, based on the brain tissue/plasma exposure ratio derived from rodents and human plasma drug exposure at week 78 after tramiprosate 150 mg bid, we projected the steady-state tramiprosate concentration in brain parenchyma to be approximately 130 nM (Table 4).
In an earlier phase II trial in patients with AD, tramiprosate produced a dose-related CSF Aβ42 reduction, suggesting target engagement [22]. In this study, the basal mean CSF concentration of Aβ42 was 179 ± 101 pg/ml (i.e., 0.04 nM, n = 46; Table 5); this concentration aligns with the reported CSF Aβ42 levels that ranged from 144 to 500 pg/ml [4, 34–36] corresponding to 0.035–0.1 nM, in patients with AD (n = 100) [34] or prodromal/early-stage AD (n = 100) [35, 36], as measured by ELISA or MS. Furthermore, brain Aβ42 measures vary in the AD literature, but the reported microdialysis studies in humans have shown that brain interstitial soluble Aβ42 are approximately equivalent to CSF Aβ42 levels [37, 38], and, therefore, the latter can be used as a suitable surrogate for brain pharmacokinetic–pharmacodynamic analyses. Thus, when comparing the ratio of brain tramiprosate : Aβ42, there is an approximately 1300- to 3700-fold excess of tramiprosate over soluble Aβ42 at the steady state based on tramiprosate measured in the brain from patients with AD (Table 4), sufficient to exert a full therapeutic effect of tramiprosate. This analysis fully aligns with the molecular stoichiometry as characterized by the the IMS–MS, NMR, and molecular dynamics approaches.
5 Discussion
In this study, we identified a novel enveloping MOA for the small-molecule Aβ-anti-aggregation agent tramiprosate. This mechanism is characterized by a multi-ligand stoichiometry, a critical excess of the ligand to target ratio, and a dose-dependent modulation of the Aβ42 conformational space, resulting in a more stabilized semi-cyclic conformation of Aβ42 and, eventually, the prevention of neurotoxic Aβ42 oligomer formation. This MOA may be responsible for the clinical cognitive and functional benefits of tramiprosate as previously observed in patients with mild-to-moderate AD [11].
Specifically, at the molecular level, we showed that tramiprosate enveloped soluble Aβ42 monomers and prevented their self-assembly into the primary monomeric misfolded Aβ42 conformation, and consequently arrested the initiation phase of Aβ42 aggregation, thus preventing the formation of neurotoxic Aβ42 oligomer species. This enveloping mechanism exerted a surprising and significant degree of control over the Aβ42 conformational landscape.
This finding is important, especially considering that the tramiprosate molecule is very small (139 Da) yet capable of controlling the structural flexibility of a large peptide/small protein such as Aβ42 under the determined conditions. This may also provide insights for a better understanding of the protein–protein and protein–peptide interaction processes in a living organism and in disease states. The challenge to modulate Aβ42 conformational dynamics has been one of the major reasons that this relatively small protein has been such an elusive target in AD drug development. We hypothesize that the enveloping occurs after a critical mass of tramiprosate (i.e. a sufficient concentration relative to Aβ42 monomer) is reached in the CNS. Because of the relatively weak nature of the transient binding of tramiprosate to Aβ42, the monomeric peptide requires a large excess of tramiprosate molecules to overcome the rapid off rates. Thus, the binding and unbinding occur rapidly enough that, only at a ratio of approximately 1:1000 of Aβ42:tramiprosate (at the ratio of 1:500, functional interaction becomes measurable), Aβ42 becomes enveloped by the drug and a full inhibition of oligomer formation is achieved.
Our molecular dynamics calculations showed a multi-ligand interaction of tramiprosate with both anions and cations of the Aβ42 side chains. Our NMR study identified the interaction of tramiprosate’s sulfonic anion with Lys16 and Lys28 as the strongest. Important roles for both amino groups have been established previously [39]. Relevant to the former, a recent study [8] showed that replacement of Lys16 with Ala abolished or dramatically reduced the neurotoxicity of Aβ40 and Aβ42, suggesting that tramiprosate binding to Lys16 may have a similar effect. The interaction of tramiprosate with Lys28 is particularly critical, because it potentially disrupts the formation of Lys28–Asp23 salt bridges (Fig. 8). Several groups [8, 40–44] have shown that the Lys28–Asp23 intramolecular salt bridge stabilizes the conformation that is important for seed formation and Lys28–Asp23 intermolecular salt bridges that form the basis of amyloid relays [45] (Fig. 9).

a Amyloid beta Aβ42 trimer with the first molecule depicted as a β-sheet in blue with Lys28–Asp23 salt bridge also in blue (PDB source 2BEG). The formation of this stabilizing Lys28–Asp23 salt bridge is disrupted by tramiprosate and, consequently, tramiprosate inhibits the formation of not only the critical seeding conformation but also the stabilizing structural element of the otherwise forming oligomers. b Aβ42 conformation adopted under excess tramiprosate conditions. Tramiprosate binds to a number of residues, most prevalently to Lys16 and Lys28 but also to Asp23, and thus prevents the formation of the Lys28–Asp23 salt bridge. Note that Lys28 as well as Asp23 point outward the conformer

Solid-state nuclear magnetic resonance (NMR) of amyloid fibril where tetramer is depicted with each individual amyloid beta Aβ42 molecule colored differently to highlight the crosslinking intermolecular salt bridge Lys28–Asp23 (source PDB 2BEG). Thus, red Lys28 forms a salt bridge with yellow Asp23. This intermolecular salt bridge stabilizes the growing superstructure
The potential impact of tramiprosate is that its anti-aggregation MOA is upstream of the Aβ oligomer formation cascade. Based on principles of biomolecular recognition and the present results, it is reasonable to suggest that, because of its interaction with Lys28, tramiprosate may prevent and block the formation of all salt bridges that are key for the salt bridge between Lys28 and Ala42, Glu22 or Asp23. This may have implications for tramiprosate and its MOA in light of recent structural characterizations of amyloid aggregates with Lys28–Ala42 salt bridges [42, 43] (Fig. 10). Thus, this action is suggested to play a critical role in the prevention of seed formation (nucleation phase) as well as growing of existing oligomers (extension phase). This interpretation is further supported by a study [45] showing that, at pH 6–8, both amino and guanidino groups are in protonated states, and the dissociation structures contain deprotonated carboxylates capable of forming salt bridges. Tramiprosate’s sulfonic acid group competing with the aspartate carboxylate (Asp23) for Lys28 amino group will preferentially form the corresponding salt bridge and thus prevent the formation of the seeding conformers.

Recently published high atomic resolution of full molecular structures of amyloid beta Aβ42 aggregates [42, 43] illustrating the salt bridge between Lys28 and C-terminal Ala42 (highlighted and annotated). The figure highlights Lys28–Ala42 as examples of salt bridges in the structures that are to be disrupted by tramiprosate binding
Another important consideration is the putative endogenous role of Aβ42 monomers in brain. To date, the physiological role of Aβ42 is not fully understood. For another aggregating protein, α-synuclein, which is implicated in Parkinson’s disease, a simple reduction of its levels is associated with synaptic failure [46], whereas whether a substantive reduction of monomeric Aβ42 levels might also result in detrimental clinical defects is unclear. Thus, therapeutic agents such as tramiprosate that preferentially prevent the formation of oligomers by an upstream action directly on Aβ42 monomers, without affecting Aβ production, unlike beta-secretase 1 (BACE1) inhibitors or γ-secretase inhibitors, may yield a new class of AD therapeutics with improved safety and efficacy. Consistent with this MOA, long-term treatment with tramiprosate (over 78 weeks) was well tolerated and devoid of vasogenic edema side effects, also referred to as ARIA (amyloid-related imaging abnormalities reported for some of the immunological therapies), in over 2000 patients with AD treated to date [11].
We also correlated the molecular mechanism results with the clinical pharmacokinetic and efficacy data [10, 11, 22, 33]. The data from our IMS–MS, NMR, and molecular dynamics experiments suggest the requirement of three orders of magnitude excess of tramiprosate relative to soluble Aβ42 to achieve a complete prevention of Aβ42 oligomer formation and aggregation. This excess ratio is in line with the projected tramiprosate concentrations in the CNS in humans based on the present translational pharmacokinetic dose-exposure analyses. The measured steady-state average concentration of tramiprosate in the brain at the dose of 150 mg tramiprosate bid from the phase III North American AD trial was 130 nM, which is 1300- to 3700-fold in excess of human CNS soluble Aβ42 levels based on the data from subjects with AD in the previous tramiprosate clinical trials, as well as the reported range in patients with AD [4, 34, 36]. Importantly, clinical cognitive and functional improvements have been demonstrated in subjects with AD in the tramiprosate phase III AD trial [11]. This suggests that the results from our current mechanism study reflect the therapeutic effect of tramiprosate in patients with AD.
While clinical efficacy of tramiprosate is suggested in a genetically defined subset of patients with AD with high amyloid burden, and its presented mechanistic understanding represents therapeutic promise, it is clear that a single-target approach to AD has not yet yielded an effective therapy. Considering the rather complex pathophysiological features of this disease, which involves multiple molecular, biochemical, and cellular pathways and systems (e.g., cholinergic function, amyloid, tau, and inflammatory components), combination therapies targeting multiple steps of amyloid cascade (e.g., tramiprosate in combination with BACE1 inhibitors, monoclonal antibodies, or insulin-degrading enzymes, etc.) or both amyloid and non-amyloid pathways (e.g., tramiprosate in combination with tau inhibitors or symptomatic agents), it is likely that future therapies will involve an approach similar to that of precision medicine, which will likely comprise the combination of more than one therapeutic modality tailored to a particular stage of the disease and/or disease phenotype. Important to this point, the clinical efficacy of tramiprosate observed in the phase III North American trial [11] was identified on top of concurrent acetylcholinesterase inhibitors (e.g., donepezil) and memantine and thus represents the first-step combination therapy approach.
6 Conclusion
Our study shows that (1) tramiprosate modulates the Aβ42 conformational landscape in a concentration-dependent manner, resulting in the stabilization of Aβ42 monomers and inhibits the formation of oligomers and subsequent aggregation and (2) the observed molecular stoichiometry is consistent with the clinical drug dose exposure versus target relationship that has been shown to achieve a robust clinically meaningful efficacy in patients with APOE4/4 homozygous AD in the previous phase III trials, suggesting that the MOA findings of tramiprosate most likely underpin its clinical outcome. The discovery of the unique enveloping MOA of tramiprosate may broaden our understanding of the control of conformationally flexible peptides/proteins, which may find potential utility in the development of disease-modifying therapies for AD and related neurodegenerative disorders caused by misfolded proteins.
References
Scheltens P, Blennow K, Breteler MMB, De Strooper B, Frisoni GB, Salloway S, et al. Alzheimer’s disease. Lancet. 2016;388:505–17.
Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med. 2016;8:595–608.
Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, et al. Diffusible, nonfibrillar ligands derived from Aβ 1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci. 1998;95:6448–53.
Lue L-F, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, et al. Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer’s disease. Am J Pathol. 1999;155:853–62.
Viola KL, Klein WL. Amyloid β oligomers in Alzheimer’s disease pathogenesis, treatment, and diagnosis. Acta Neuropathol. 2015;129:183–206.
Crescenzi O, Tomaselli S, Guerrini R, Salvadori S, D’Ursi AM, Temussi PA, et al. Solution structure of the Alzheimer amyloid β-peptide (1-42) in an apolar microenvironment. Eur J Biochem. 2002;269:5642–8.
Maji SK, Amsden JJ, Rothschild KJ, Condron MM, Teplow DB. Conformational dynamics of amyloid β-protein assembly probed using intrinsic fluorescence. Biochemistry. 2005;44:13365–76.
Sinha S, Lopes DHJ, Bitan G. A key role for lysine residues in amyloid β-protein folding, assembly, and toxicity. ACS Chem Neurosci. 2012;3:473–81.
Jarrett JT, Lansbury PTJ. Seeding “one-dimensional crystallization” of amyloid: a pathogenic mechanism in Alzheimer’s disease and scrapie? Cell. 1993;73:1055–8.
Gervais F, Paquette J, Morissette C, Krzywkowski P, Yu M, Azzi M, et al. Targeting soluble Aβ peptide with Tramiprosate for the treatment of brain amyloidosis. Neurobiol Aging. 2007;28:537–47.
Abushakra S, Porsteinsson AP, Vellas B, Cummings J, Gauthier S, Hey JA, et al. Clinical benefits of tramiprosate in Alzheimer’s disease are associated with higher number of APOE4 alleles: the “APOE4 gene-dose effect”. J Prevent Alzheimers Dis. 2016;3:1–10.
Bowers KJ, Chow E, Xu H, Dror R, Eastwood MP, Gregersen BA, et al. Scalable algorithms for molecular dynamics simulations on commodity clusters. In: SC conference, proceedings of the ACMIEEE. New York: IEEE; 2006. p. 43–56.
Shivakumar D, Harder E, Damm W, Friesner RA, Sherman W. Improving the prediction of absolute solvation free energies using the next generation OPLS force field. J Chem Theory Comput. 2012;8:2553–8.
Bakan A, Meireles LM, Bahar I. ProDy: protein dynamics inferred from theory and experiments. Bioinformatics. 2011;27:1575–7.
Roche J, Shen Y, Lee JH, Ying J, Bax A. Monomeric Aβ 1–40and Aβ 1–42 peptides in solution adopt very similar Ramachandran map distributions that closely resemble random coil. Biochemistry. 2016;55:762–75.
Sklenar V, Piotto M, Leppik R, Saudek V. Gradient-tailored water suppression for 1H-15N HSQC experiments optimized to retain full sensitivity. J Magn Reson A. 1993;102:241–5.
Gal M, Schanda P, Brutscher B, Frydman L. UltraSOFAST HMQC NMR and the repetitive acquisition of 2D protein spectra at Hz rates. J Am Chem Soc. 2007;129:1372–7.
Yan Y, Wang C. Aβ42 is more rigid than Aβ40 at the C terminus: implications for Aβ aggregation and toxicity. J Mol Biol. 2006;364:853–62.
Hou L, Shao H, Zhang Y, Li H, Menon NK, Neuhaus EB, et al. Solution NMR studies of the Aβ(1−40) and Aβ(1−42) peptides establish that the Met35 oxidation state affects the mechanism of amyloid formation. J Am Chem Soc. 2004;126:1992–2005.
Summerfield SG, Lucas AJ, Porter RA, Jeffrey P, Gunn RN, Read KR, et al. Toward an improved prediction of human in vivo brain penetration. Xenobiotica. 2008;38:1518–35.
de Lange EC. The mastermind approach to CNS drug therapy: translational prediction of human brain distribution, target site kinetics, and therapeutic effects. Fluids Barriers CNS. 2013;10:12.
Aisen PS, Saumier D, Briand R, Laurin J, Gervais F, Tremblay P, et al. A Phase II study targeting amyloid-beta with 3APS in mild-to-moderate Alzheimer disease. Neurology. 2006;67:1757–63.
Young LM, Cao P, Raleigh DP, Ashcroft AE, Radford SE. Ion mobility spectrometry–mass spectrometry defines the oligomeric intermediates in amylin amyloid formation and the mode of action of inhibitors. J Am Chem Soc. 2014;136:660–70.
Young LM, Saunders JC, Mahood RA, Revill CH, Foster RJ, Tu L-H, et al. Screening and classifying small-molecule inhibitors of amyloid formation using ion mobility spectrometry-mass spectrometry. Nat Chem. 2015;7:73–81.
Ono K, Yamada M. Low-n oligomers as therapeutic targets of Alzheimer’s disease. J Neurochem. 2011;117:19–28.
Townsend M, Shankar GM, Mehta T, Walsh DM, Selkoe DJ. Effects of secreted oligomers of amyloid beta-protein on hippocampal synaptic plasticity: a potent role for trimers. J Physiol. 2006;572:477–92.
Lazo ND, Grant MA, Condron MC, Rigby AC, Teplow DB. On the nucleation of amyloid β-protein monomer folding. Protein Sci. 2005;14:1581–96.
Sciarretta KL, Gordon DJ, Petkova AT, Tycko R, Meredith SC. Aβ40-Lactam(D23/K28) models a conformation highly favorable for nucleation of amyloid. Biochemistry. 2005;44:6003–14.
Xiao Y, Ma B, McElheny D, Parthasarathy S, Long F, Hoshi M, et al. Aβ(1–42) fibril structure illuminates self-recognition and replication of amyloid in Alzheimer’s disease. Nat Struct Mol Biol. 2015;22:499–505.
Tomaselli S, Esposito V, Vangone P, van Nuland NAJ, Bonvin AMJJ, Guerrini R, et al. The α-to-β conformational transition of Alzheimer’s Aβ-(1-42) peptide in aqueous media is reversible: a step by step conformational analysis suggests the location of β conformation seeding. Chembiochem. 2006;7:257–67.
Martineau E, Guzman JM, Rodionova L, Kong X, Mayer PM, Aman AM. Investigation of the noncovalent interactions between anti-amyloid agents and amyloid β peptides by ESI-MS. J Am Soc Mass Spectrom. 2010;21:1506–14.
Soine PJ, Blanke RV, Guzelian PS, Schwartz CC. Preferential binding of chlordecone to the protein and high density lipoprotein fractions of plasma from humans and other species. J Toxicol Environ Health A. 1982;9:107–18.
Gauthier S, Aisen PS, Ferris SH, Saumier D, Duong A, Haine D, et al. Effect of tramiprosate in patients with mild-to-moderate Alzheimer’s disease: exploratory analyses of the MRI sub-group of the Alphase study. J Nutr Health Aging. 2009;13:550–7.
Shaw LM, Vanderstichele H, Knapik-Czajka M, Clark CM, Aisen PS, Petersen RC, et al. Cerebrospinal fluid biomarker signature in Alzheimer’s disease neuroimaging initiative subjects. Ann Neurol. 2009;65:403–13.
Leinenbach A, Pannee J, Dulffer T, Huber A, Bittner T, Andreasson U, et al. Mass spectrometry-based candidate reference measurement procedure for quantification of amyloid in cerebrospinal fluid. Clin Chem. 2014;60:987–94.
Pannee J, Portelius E, Minthon L, Gobom J, Andreasson U, Zetterberg H, et al. Reference measurement procedure for CSF amyloid beta (Aβ)1-42 and the CSF Aβ1-42/Aβ1-40 ratio: a cross-validation study against amyloid PET. J Neurochem. 2016;139(4):651–8.
Brody DL, Magnoni S, Schwetye KE, Spinner ML, Esparza TJ, Stocchetti N, et al. Amyloid dynamics correlate with neurological status in the injured human brain. Science. 2008;321:1221–4.
Herukka S-K, Rummukainen J, Ihalainen J, von Und Zu Fraunberg M, Koivisto AM, Nerg O, et al. Amyloid-beta and tau dynamics in human brain interstitial fluid in patients with suspected normal pressure hydrocephalus. J Alzheimers Dis. 2015;46:261–9.
Usui K, Hulleman JD, Paulsson JF, Siegel SJ, Powers ET, Kelly JW. Site-specific modification of Alzheimer’s peptides by cholesterol oxidation products enhances aggregation energetics and neurotoxicity. Proc Natl Acad Sci. 2009;106:18563–8.
Triguero L, Singh R, Prabhakar R. Comparative molecular dynamics studies of wild-type and oxidized forms of full-length Alzheimer amyloid β-peptides Aβ(1−40) and Aβ(1−42). J Phys Chem B. 2008;112:7123–31.
Lu J-X, Qiang W, Yau W-M, Schwieters CD, Meredith SC, Tycko R. Molecular structure of β-amyloid fibrils in Alzheimer’s disease brain tissue. Cell. 2013;154:1257–68.
Colvin MT, Silvers R, Ni QZ, Can TV, Sergeyev I, Rosay M, et al. Atomic resolution structure of monomorphic Aβ 42amyloid fibrils. J Am Chem Soc. 2016;138:9663–74.
Wälti MA, Ravotti F, Arai H, Glabe CG, Wall JS, Böckmann A, et al. Atomic-resolution structure of a disease-relevant Aβ(1-42) amyloid fibril. Proc Natl Acad Sci. 2016;113:E4976–84.
Tycko R. Progress towards a molecular-level structural understanding of amyloid fibrils. Curr Opin Struct Biol. 2004;14:96–103.
Kobayashi S, Tanaka Y, Kiyono M, Chino M, Chikuma T, Hoshi K, et al. Dependence pH and proposed mechanism for aggregation of Alzheimer’s disease-related amyloid-β(1-42) protein. J Mol Struct. 2015;1094:109–17.
Burre J, Sharma M, Sudhof TC. Definition of a molecular pathway mediating alpha-synuclein neurotoxicity. J Neurosci. 2015;35:5221–32.
Acknowledgements
IMS–MS was performed by Protea Biosciences; NMR was performed by MaratechNMR. Dr. Aidan Power (Alzheon) kindly reviewed the manuscript and internal editorial comments. We thank Helena Kocis for her kind help with the preparation of graphics files.
Author Contributions
PK conceived the enveloping MOA and designed all studies, interpreted the data in collaboration with JH, and wrote the manuscript in collaboration with JH, JY, WS, SR. Molecular dynamics calculations were performed by WS and SR. Pharmacokinetic/pharmacodynamic analyses were performed by JY and JH. All co-authors reviewed and contributed to the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Alzheon Inc. sponsored the research described in this article and paid the open access fee.
Conflict of interest
PK, MT, JH, and JY are employees of Alzheon Inc. WS and SR are employees of Schrödinger, which had a scientific services agreement in place to perform some of the work in this manuscript. KB has served as a consultant or on advisory boards for Alzheon, Eli Lilly, Fujirebio Europe, IBL International, Novartis, and Roche Diagnostics, and is a co-founder of Brain Biomarker Solutions in Gothenburg AB, a GU Venture-based platform company at the University of Gothenburg. HF has no competing interests.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Kocis, P., Tolar, M., Yu, J. et al. Elucidating the Aβ42 Anti-Aggregation Mechanism of Action of Tramiprosate in Alzheimer’s Disease: Integrating Molecular Analytical Methods, Pharmacokinetic and Clinical Data. CNS Drugs 31, 495–509 (2017). https://doi.org/10.1007/s40263-017-0434-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40263-017-0434-z