
Abstract
Major depressive disorder (MDD) is a chronic and potentially life threatening illness that carries a staggering global burden. Characterized by depressed mood, MDD is often difficult to diagnose and treat owing to heterogeneity of syndrome and complex etiology. Contemporary antidepressant treatments are based on improved monoamine-based formulations from serendipitous discoveries made > 60 years ago. Novel antidepressant treatments are necessary, as roughly half of patients using available antidepressants do not see long-term remission of depressive symptoms. Current development of treatment options focuses on generating efficacious antidepressants, identifying depression-related neural substrates, and better understanding the pathophysiological mechanisms of depression. Recent insight into the brain’s mesocorticolimbic circuitry from animal models of depression underscores the importance of ionic mechanisms in neuronal homeostasis and dysregulation, and substantial evidence highlights a potential role for ion channels in mediating depression-related excitability changes. In particular, hyperpolarization-activated cyclic nucleotide-gated (HCN) channels are essential regulators of neuronal excitability. In this review, we describe seminal research on HCN channels in the prefrontal cortex and hippocampus in stress and depression-related behaviors, and highlight substantial evidence within the ventral tegmental area supporting the development of novel therapeutics targeting HCN channels in MDD. We argue that methods targeting the activity of reward-related brain areas have significant potential as superior treatments for depression.
Similar content being viewed by others

Introduction
Depression is a highly debilitating mental disorder with a heavy societal burden. The lifetime prevalence of major depressive disorder (MDD) is > 16% and the recurrence of depression episodes within 2 to 5 years is > 40% [1,2,3]. According to the Diagnostic and Statistical Manual, 5th Edition, MDD diagnosis is justified when a patient displays at least 5 of 9 possible criteria: chronic symptoms of depressed or irritable mood, anhedonia, weight change, sleep disruptions, psychomotor dysfunction, fatigue, feelings of worthlessness or guilt, diminished ability to concentrate, and recurrent suicidal ideation [4]. Core symptoms of depression include depressed mood and anhedonia. Heterogeneity of symptoms in patients meeting criteria for clinical depression has resulted in multiple studies of differential treatments for patients with varying MDD symptoms and severity, spanning efficacy of antidepressant combinations with or without adjunctive psychotherapy [5].
To date, pharmacological treatments for depression encompass first-generation antidepressants, including tricyclic antidepressants and monoamine oxidase inhibitors, and second-generation advances comprising selective serotonin reuptake inhibitors, serotonin-norepinephrine reuptake inhibitors, atypical antidepressants, and serotonin modulators. While first-generation antidepressants have mostly fallen out of favor owing to their adverse side effects, second-generation antidepressants are believed to have the same general efficacy [6]. These antidepressant medications all act to increase some or all of the monoamine neurotransmitters [7]. Currently, selective serotonin reuptake inhibitors such as fluoxetine are the most widely prescribed class of antidepressants [8]. A limitation of these antidepressant treatments is the delay of several weeks between treatment initiation and improvement of depressive symptoms [8]. Another major issue in current depression treatment is improving therapeutic options for resistant or refractory depression, a chronic condition resulting from a lack of satisfactory response to antidepressant therapies [9]. Approximately 20% to 30% of patients are resistant to treatment [2]. This patient population, in particular, highlights the pressing need for better therapeutics to combat the recurrent suffering and rising global burden of the disease.
Complexities in understanding both the pathophysiology of MDD and mechanisms of antidepressant efficacy have slowed the pipeline of new therapeutics, despite many important discoveries. Historically, the catecholamine hypothesis of depression served as the basis for development of antidepressant treatments. This hypothesis posits that decreased levels of the catecholamines serotonin, norepinephrine, and dopamine (DA) contribute to the pathophysiology of depression [10]. While antidepressants reverse this monoamine deficit within hours, their delayed behavioral action suggests downstream changes, including signaling cascades, gene changes (epigenetic, transcriptional, and translational plasticity), neurogenesis, synaptic remodeling, and altered neurotransmission [11,12,13,14,15,16,17,18]. Chronic neurotransmitter administration appears to induce plasticity mechanisms, ultimately leading to antidepressant action. Recent clinical studies have identified rapidly acting, glutamate/acetylcholine-targeting antidepressant compounds (ketamine, scopolamine), which alleviate symptoms of treatment-resistant depression and lower the risk of suicidal ideation in patients with MDD. These novel glutamatergic approaches have expanded our understanding of effective treatment strategies and raised the possibility of more direct pharmacological targets [11, 19,20,21,22,23,24,25,26,27].
In addition to elucidating mechanisms of antidepressant action, it is also important to investigate the neurobiology of depression and depression symptoms. While the complete etiology of depression is unknown, genetic and environmental stress factors have been shown to contribute to MDD onset. Notably, prolonged stress is thought to catalyze the onset and/or recurrence of depression [28]. Research into the neurobiology of stress has helped inform better animal models of depression for testing antidepressant efficacy and probing brain structures involved in MDD. Preclinical models of depression, particularly in mice, have vastly improved the ability to study mechanisms of antidepressant action, as well as identify potential targets for antidepressant treatment. Historically, antidepressants were tested for efficacy using the measure of acute antidepressant response to behavioral despair, or an animal’s active or passive response to a stressor, via the forced swim test or tail suspension test [29,30,31,32]. However, most recent preclinical studies assay additional measures of depressive-like behaviors, including anhedonia, social interaction, anxiety, memory dysfunction, and homeostatic symptoms in rodent models of depression. Furthermore, genetically derived lines of “depressed” animals have been selectively bred to model and test particular depressive behavioral phenotypes [33, 34]. Current animal models of stress-induced depression have employed chronic social stress or chronic unpredictable mild stressors to produce a constellation of depression symptoms [35]. These paradigms for the generation of depressive-like behaviors in animals are designed to ethologically recapitulate the environmental stressors that can exacerbate a vulnerability to depression in humans.
Chronic unpredictable/variable mild stress utilizes the inflexibility of animals to respond to a randomized variety of mild stressors, including tail suspension, restraint, and foot shock over weeks to months. Both female and male animals that have undergone chronic mild stress will exhibit profound anhedonia, fear, and anxiety behaviors that can be reversed by chronic but not acute antidepressant administration, suggesting an appropriate model for analyzing antidepressant plasticity mechanisms [36,37,38]. Chronic social defeat stress (CSDS) also requires repeated antidepressant administration for behavioral rescue. The ethological modeling of depression in CSDS results from a robust chronic social stressor that utilizes both the physical and psychological stress of social subordination [39]. Using retired CD-1 breeder mice that are screened for aggressive territorial behavior, smaller genetically identical C57BL/6 mice are introduced to the home cage of the CD-1 for a short physical interaction. Following social subordination, the C57BL/6 mice are further socially stressed with overnight psychosensory exposure, as they are separated from the CD-1 by a clear, perforated barrier for the remaining 24 h. This procedure is repeated for 10 days with a different CD-1 every day, producing profound and lasting anhedonia and social avoidance, in addition to abnormalities in circadian rhythm, metabolism, and hypothalamic–pituitary–adrenal axis response [39, 40]. Interestingly, a subset of mice that undergo this stress resemble control animals in their reward processing, allowing researchers to parse animals that are “susceptible” from animals that are “resilient” to social defeat stress to investigate the mechanisms of susceptibility and resilience to depression [40,41,42,43,44,45,46,47,48,49,50].
In addition to preclinical studies in animal models of depression, clinical investigations probing target brain regions of antidepressant action, brain activity in depressed patients, and the efficacy of deep brain stimulation have highlighted mesocorticolimbic circuitry as the general neural substrate for depression-related behaviors and antidepressant efficacy [11, 40,41,42,43, 46, 47, 51,52,53,54,55,56,57,58,59,60]. This neural circuit encodes reward-related behaviors and encompasses projections between brain areas including the prefrontal cortex (PFC), hippocampus, ventral tegmental area (VTA), amygdala, and nucleus accumbens (NAc). Mesocorticolimbic circuit dysfunction has been implicated in a range of psychiatric disorders, including MDD. Indeed, depressed patients exhibit increased activity in limbic regions and decreased connectivity between corticolimbic regions [61, 62]. Imbalances in activity of these brain areas may result from both extrinsic factors (changes in signaling between brain areas) and intrinsic factors (changes in neuronal excitability). Thus, dysfunctional brain activity and depression pathology may be due to changes in excitability to maintain homeostasis [63]. Hyperpolarization-activated cyclic nucleotide-gated (HCN) channels are key regulators of neuronal excitability [64,65,66]. As we will describe in this review, HCN channels have been implicated in a number of stress and depression-related behaviors, and our evolving understanding of the role of HCN channels in multiple brain areas suggests that these channels are promising targets for future therapeutics.
HCN Channels
Neuronal signaling within a region and at a circuit level is affected by properties of neuronal excitability and ion-channel distribution. Ion-channel dysfunction can lead to unbalanced excitability and subsequent neuronal dysfunction. HCN channels are present throughout the brain, and modulate neuronal excitability and activity [64]. HCN channels are known to regulate cell activity via the hyperpolarization-activated current, Ih (also known as h current), consisting of sodium and potassium cations [65]. HCN channels are activated by hyperpolarized states greater than –40 mV, increase their activation as the cell becomes more hyperpolarized, and do not display inactivation [66]. Since HCN channels are open at hyperpolarized states greater than –40 mV, Ih currents are often present during the resting state [67]. This allows HCN channels to modulate neuronal excitability by stabilizing the neuronal membrane potential against both excitatory and inhibitory inputs. HCN channels consist of 4 subtypes, HCN 1–4, that can assemble in various combinations and conformations. HCN channels can be modulated by cyclic nucleotide cyclic adenosine 3’,5’-monophosphate (cAMP) or protein kinase-mediated phosphorylation of the channel subunits [68].
HCN channels are expressed in the brain, heart, and retina [69, 70]. HCN1 appears to be the most common isoform present in the hippocampus, neocortex, and cerebellar cortex. HCN2 expression is predominant in the midbrain and thalamus, while HCN4 is the predominant isoform in the heart, thalamic nuclei, basal ganglia, and olfactory bulb [64, 71]. Ih currents in the central nervous system serve to mediate membrane properties of particular cells, including resting membrane potential, firing frequency, synaptic transmission, and synaptic input integration [72,73,74,75,76,77,78,79]. This allows HCN channels to influence the rhythmicity of neuronal networks, and to control the integration of cell signaling and firing activity. HCN channels play other important roles; besides setting pacemaker firing for the heart rhythm, HCN channels help regulate oscillatory networks, including sleep and arousal [80]. Importantly, Ih and HCN channels influence neural activity and excitability by affecting synaptic integration and plasticity [67, 81,82,83,84,85,86]. This has a profound impact on learning, memory, and encoding of behaviors.
HCN Channels and Depression
Genetic studies of HCN channels in depressed patients have not yet conclusively identified a strong association between single-nucleotide polymorphisms in HCN channel genes with depression and stress sensitivity [87, 88]. However, the generation of transgenic HCN channel null animals has allowed researchers to probe the behavioral outcomes of dysregulated neuronal activity from HCN signaling ablation. Since HCN channel variations are present throughout the peripheral nervous system [89], including in cardiomyocytes [90], researchers have developed animal models with a selective knockout of HCN1, HCN2, or tetratricopeptide repeat-containing Rab8b interacting protein (TRIP8b) for exclusive expression in the brain. TRIP8b is a brain-specific protein encoded by the Pex51 gene that is involved in HCN trafficking and appearance at dendrites [91,92,93,94]. When TRIP8b expression is eliminated, Ih currents are attenuated. Since HCN channels are important for vital functions outside of the brain, targeting the TRIP8b is another method to test HCN function exclusively in the brain.
The involvement of HCN channels in depression behaviors was first investigated using these 3 knockout lines. While anxiety-like behaviors, as tested by the elevated plus maze and dark/light boxes, were not ameliorated in global HCN knockout lines, mice with reduced Ih as a consequence of null HCN1, HCN2, and TRIP8b all displayed reduced behavioral despair [94]. In all 3 HCN channel knockout lines, Ih dysfunction was shown to be involved in depression behaviors as demonstrated by behavioral reductions in time spent immobile and latency to immobility in forced swim and tail suspension tests [94]. These findings highlight the importance of parsing the roles of HCN channel subtypes in reward circuitry, in order to determine the contribution of Ih to discrete brain regions and control of particular affective behaviors. Importantly, HCN channels are expressed in brain areas involved in reward, including the PFC, hippocampus, and VTA [69, 70]. While a number of depression studies have delineated the potential mechanism of HCN channel action in the hippocampus and PFC, recent work in the VTA has also clarified an important role for HCN channels in depression (Fig. 1).

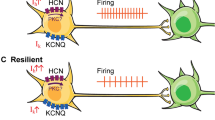
Role of hyperpolarization-activated cyclic nucleotide-gated (HCN)1 in depression behaviors. (A) Global HCN knockout (KO) in the brain produces reduced behavioral despair in forced swim test and tail suspension test for all transgenic lines (HCN1–/–, HCN2–/–, and TRIP8b–/–)[94]. (B) Selective dorsal hippocampal (HPC) reduction of HCN1 by short hairpin RNA (shRNA) HCN1 exhibited reduced behavioral despair in forced swim test, and, interestingly, anxiolytic effects in the open-field test and elevated plus maze test [108]. (C) Selective blockade of HCN channels in the prefrontal cortex (PFC) by ZD7288 or knockdown by shRNA HCN1.1 or shRNA HCN1.2 show improvements in working memory [113]. (D) Microinfusion of HCN blockers ZD7288 or DK-AH 269 into the ventral tegmental area (VTA) of susceptible/depressed mice reversed social avoidance behaviors [41]. Unexpectedly, chronic HCN channel potentiation by repeated lamotrigine administration or overexpression of HCN2 in dopamine neurons by herpes simplex virus–LS1L–HCN2 in susceptible animals induced resilience to depression behaviors [43]
HCN Channels in the Hippocampus
Chronic stress has been shown to weaken hippocampal connections [95]. Beyond the preclinical wealth of research implicating the hippocampus in depression behaviors [96], positron emission tomography imaging studies have shown that chronic antidepressants induce changes in the hippocampus, as observed by regional glucose metabolism [52, 97,98,99], and patients with severe depression show reduced posterior hippocampal volume [100]. Consequently, the role of HCN channels in mediating depression behaviors within the hippocampus is of particular interest.
HCN channels have been heavily studied in the CA1 region of the hippocampus, and decreases in excitability in the hippocampus are correlated with increases in Ih current [101]. Ih currents appear to modulate neuronal excitability, independent of synaptic plasticity changes mediated by N-methyl-D-aspartate (NMDA) receptor-dependent changes in excitability [101]. HCN channels are highly expressed along the distal dendrites of pyramidal neurons in the CA1 region where they serve to modulate network excitability for integrating incoming signals, normalizing temporal summation, and mediating the propagation of information by dampening Ca2+ signaling [102,103,104]. In particular, HCN channel density increases directly with distance from the soma to the distal apical dendrites in the hippocampal CA1 region [78]. Moreover, HCN channels are integral for mediating the theta rhythmic properties of the hippocampus, through involvement of the intrinsic resonance properties [105,106,107]. Thus, the ability of HCN channels to regulate intrinsic cellular excitability may represent a global homeostatic regulation of activity that prevents hyperexcitability by a negative-feedback mechanism [101].
To investigate the potential antidepressant role of HCN channels exclusively in the hippocampus, Kim et al. [108] utilized lentiviral short hairpin RNA (shRNA) to target the expression of HCN1 channels in the dorsal CA1 region of rats. Following knockdown of HCN1 channel proteins within this region, pyramidal neurons exhibited expected alterations in intrinsic membrane properties, including more hyperpolarized resting membrane potentials, higher input resistance, and slower membrane time constant. Pyramidal neurons also exhibited increased cellular excitability, which extended to a widespread increase of hippocampal activity. HCN1 channel knockdown produced anxiolytic effects in the open-field test and the elevated plus maze; animals spent significantly more time in both the center of the open-field box and the open arm of the elevated plus maze. HCN1 knockdown was also antidepressant: compared with control animals, HCN1 shRNA-injected rats displayed less passive activity in the forced swim test, behaving similarly to rats injected with fluoxetine or ketamine [108].
TRIP8b knockout mice show the same antidepressant-like reduction in time spent immobile in the forced swim test and tail suspension test. Behavioral modifications stemming from hippocampal HCN channel activity reduction can be bidirectionally modulated through manipulation of the brain-specific auxillary subunit TRIP8b [109]. Restoration of TRIP8b through injection of TRIP8b-overexpressing adenoassociated viral vectors into the CA1 region of TRIP8b knockout mice rescues Ih current in CA1 pyramidal neurons. This hippocampal Ih current recovery prevents rescue of the behavioral despair observed in all global HCN knockout lines (HCN1, HCN2, TRIP8b), underscoring the importance of the hippocampus in mediating depression behaviors. In both the forced swim and tail suspension tests, Han et al. [109] found that the reduction in time spent immobile of TRIP8b knockout mice was absent in TRIP8b knockout mice expressing TRIP8b-adenoassociated viral vector in the CA1 region of the hippocampus. Thus, by rescuing HCN channel expression in CA1, the antidepressant effects of HCN knockdown were ablated. These results highlight the importance of investigating region-specific actions of depression; global and hippocampal-specific knockdown of HCN channels produce antidepressant behaviors, but anxiolytic effects are observed only with hippocampal-specific ablation of Ih channels.
HCN Channels in the PFC
HCN channels also influence excitability in the PFC [110]. PFC HCN channels have important implications for stress-induced psychiatric disorders, as evidenced by the role Ih currents play in strengthening PFC recurrent connections and the consequences of HCN channel dysfunction in PFC. Chronic stress is extensively damaging to the cognitive center of the brain, affecting working memory, regulation of information processing, and error computation [111, 112]. Working memory is critically regulated by the catecholamines norepinephrine and DA, which activate α2A adrenoceptors, DA D1 receptors (D1Rs), and cAMP signaling in PFC. Chronic stress induces both high levels of norepinephrine-α2A activity and a disrupted balance of DA-D1R activity that together disturb recurrent PFC network activity and induce cognitive dysfunction in working memory [113,114,115,116,117,118].
The intrinsic mechanisms underlying working memory impairment involve excessive cAMP production, which targets HCN channels [68, 113]. Ih currents vulnerable to cAMP modulation are heteromers of HCN1 and HCN2 subunits [69, 119, 120] and are most highly expressed in cortical layers with a high concentration of α2A adrenoceptors [69, 113, 121,122,123]. Within the PFC, HCN channels are also co-localized with D1Rs on dendritic spines [115]. Increased cAMP is associated with increased conductance of Ih current and increased opening of HCN channels [113]. HCN channels are found on the distal apical dendrites of layer V and on the dendritic spines of deep layer III pyramidal cells in PFC [113, 114]. Since HCN channels are largely localized to dendrites, Ih currents serve to regulate synaptic inputs. Specifically, the dendritic Ih may serve as an electrotonic shunt to limit the spread of excitatory action potentials.
HCN channel activity disruption results in increased recurrent network interactions, as measured by both intracellular and extracellular multiple unit recordings in layer V pyramidal cell slices [113]. The effects of Ih current reduction can be tested with ZD7288, an HCN blocker. Blockade of HCN channels by ZD7288 also increases functionally connected firing activity. This suggests that HCN channels play a role in PFC network firing properties. Furthermore, HCN channel blockade by ZD7288 produces working memory improvements. Prior to high DA D1R stimulation, the working memory dysfunction induced by the D1R agonist SKF38393 can be prevented with ZD7288, likely through a cAMP–protein kinase A phosphorylation mechanism [115, 124].
Working memory perturbations can be investigated in animals using shRNA to knock down HCN1 channels or with ZD7288 to block HCN channels. Rats that received HCN1 channel knockdown or blockade demonstrated improved spatial working memory, as tested through T-maze memory performance [113]. Thus, increasing cAMP signaling suppresses prefrontal firing via opened HCN channels on dendrites of pyramidal neurons, either through α2A adrenoceptors or D1Rs. This suggests that HCN channels are well positioned to regulate PFC network dynamics by gating neuronal firing from cortico–cortico connections to influence the strength of the prefrontal microcircuit and output efficacy.
When chronic stress and consequent HCN channel dysfunction desynchronize cortical networks, the “top-down” control of other brain areas is disinhibited. This may affect prefrontal control over behavior, emotion, and cognition. While chronic cAMP is necessary for the synaptic plasticity observed in long term potentiation of hippocampal cells, cAMP typically plays a transient role in working memory signaling; high levels of cAMP from dysregulated α2A adrenoceptor or D1R-mediated signaling after chronic stress can lead to lowered PFC strength from open HCN channels [113, 115, 118]. This results in disrupted PFC network connectivity, weakened network connections, and reduced control of other brain areas, including the amygdala and striatum [111, 112]. During stress, reduced and unpatterned PFC networks will project an incorrect representation of working memory and may incidentally disinhibit affective and stress pathways in the brain.
HCN Channels in the VTA
Chronic stress may strengthen the activity of limbic circuitry, including that of the amygdala and striatum [125,126,127]. This positive feedback loop may be reinforced by disinhibition of the PFC on limbic pathways. Stress increases VTA DA activity and release in target regions [126, 128, 129]. HCN channels reduce the excitability of the PFC and hippocampus. Within the VTA, Ih currents may also serve to modulate neuronal excitability. HCN channels appear on the dendrites of VTA DA neurons [130], and regulate the amplitude and duration of the afterhyperpolarization observed after an action potential [75, 76, 131, 132]. While HCN1 is the main channel subunit within the PFC and hippocampus, HCN 2–4 channels are most expressed in midbrain DA neurons [133]. While HCN2 appears to be the most abundantly expressed channel subunit, all HCN subunits are expressed within the VTA [64, 69]. We have observed an increase in activity of the VTA and reward neurocircuitry following chronic stress; in particular, HCN channels appear to play a role in reward dysfunction and depression in VTA DA neurons.
Midbrain VTA DA neurons exhibit 2 types of firing activity: single-spike “tonic” firing or “phasic” short multispike burst firing, which are believed to maintain basal levels of DA in the extracellular space and signal reward or aversion through a transient increase of DA, respectively [134,135,136,137,138]. Pathologically, DA neuron-firing activity in response to acute and chronic stress can be incredibly complicated [42, 118, 139]. DA neurons are identified in vivo by a slow firing rate of 2 to 10 Hz, duration of unfiltered biphasic action potential waveform > 2.2 ms, and sensitivity to DA or DA receptor agonists [140]. Classically, in vitro VTA or substantia nigra DA neurons have been identified by a large Ih current [76, 141], but more recent research has conclusively shown that this criterion is insufficient [131, 142]. These researchers and others have highlighted the heterogeneity of VTA DA neuron characterization [143]. VTA DA neurons are functionally heterogeneous, and HCN channel expression within the VTA subpopulations of VTA DA projection neurons is variable. VTA DA neurons that project to the NAc exhibit a large, pronounced Ih current, while VTA DA neurons that project to the PFC have a small Ih current [43, 76, 131, 144]. The previously standard identification of DA neurons by the Ih current underlies its importance in the physiological firing activity of most VTA DA neurons and possible involvement in pathological perturbations.
In the CSDS model of depression, we have observed a similar phenomenon of VTA Ih current induction and VTA DA neuron hyperactivity that mediates behavioral susceptibility to CSDS [40]. Since Ih currents are implicated in the excitatory drive of VTA DA neurons, we investigated the Ih current as a pathological mechanism underlying VTA DA hyperactivity [40,41,42,43, 76, 132]. We found that CSDS increased the Ih current in susceptible animals, and that HCN channels play a role in the pathophysiological increase in firing rate characteristic of susceptibility [43]. VTA microinfusion of Ih inhibitors ZD7288 or DK-AH 269 rescued social avoidance in susceptible animals [41]. Moreover, following chronic administration of the antidepressant fluoxetine, susceptible animals exhibited a rescue of social avoidance behavior, as well as a return of Ih current to baseline levels [41]. These findings suggest that Ih current inhibition is a potential target for antidepressant treatment and expand upon prior findings in the PFC and hippocampus.
Unexpectedly, further investigation of the resilient subpopulation of defeated animals revealed a more complex mechanism of Ih current mediation of depression-like behaviors (Fig. 2). We found that resilience involves a dynamic induction of homeostatic plasticity that maintains adaptive behavioral responses to stress. Resilient animals exhibited VTA DA firing activity that resembled that of control animals, despite undergoing strenuous social stress [40,41,42,43]. We therefore expected the Ih current in resilient animals to be similar to that of control animals. Instead, recordings of the VTA DA Ih current of resilient animals revealed an Ih current larger than that of susceptible mice [43]. Further investigations into the excitability of VTA DA neurons following CSDS revealed intrinsic changes in both susceptible and resilient mice; the decreased excitability of resilient animals compared with control mice corresponded with the increased potassium current observed in the VTA DA of resilient mice [43, 46]. These findings suggest that social defeat stress induces an increased Ih current in resilient animals that may trigger an active, “self-tuning” mechanism that restores firing rate to control animal levels.

Ih current bidirectional modulation in the ventral tegmental area (VTA) of susceptible animals. (A, C) Microinfusion of hyperpolarization-activated cyclic nucleotide-gated (HCN) blockers ZD7288 or DK-AH269 into the VTA of susceptible animals rescues the social avoidance depression phenotype [41]. (B, D) HCN channel potentiation of susceptible animals by 5-day microinfusion of lamotrigine (LTG) induces homeostatic plasticity mechanisms in the VTA for resilience-like behavior and unsusceptible defeat stress phenotype. (E, F) Five-day LTG microinfusion produces larger Ih currents, which induces compensatory potassium currents to rescue social interaction behavior [43]
In order to probe the functional role of VTA DA Ih currents in susceptible and resilient behaviors, Friedman et al. [43, 82, 145, 146] utilized multiple techniques, including pharmacological, viral, and optogenetic manipulations. Lamotrigine (LTG) is a Food and Drugs Administration (FDA)-approved Ih current potentiator that is used as anticonvulsant and a mood stabilizer for bipolar disorder. Chronic microinfusion of LTG into the VTA of susceptible animals produced a reversal of social avoidance behavior, as measured by time spent in the interaction zone with a novel social target, and anhedonia, as measured by sucrose preference [43]. LTG infusion also increased Ih and potassium currents in test mice compared with vehicle-treated animals. However, as LTG has multiple known ion-channel targets, including sodium channels, we virally infected VTA DA neurons to overexpress HCN2 channels. HCN2 overexpression in VTA DA neurons of susceptible mice rescued social avoidance behaviors, sucrose preference deficits, behavioral despair in the forced swim test, and the corresponding increase in Ih and potassium currents [43].
Although the alterations in Ih and potassium currents rescued susceptible behaviors, we performed additional experiments to further investigate the physiological resilient phenotype. To probe the hyperactivity of VTA DA neurons resulting from social defeat stress, we induced excessive activation of VTA DA neurons using optogenetics, light-activated channels that can temporally control the firing activity of neurons [147, 148]. We infected VTA DA neurons with light-activated channelrhodopsin-2 and repeatedly increased the firing rate of VTA DA neurons in susceptible animals. Chronic, excessive hyperactivity of VTA DA neurons rescued social avoidance behavior, sucrose preference, behavioral despair in the forced swim test, and induced control firing activity and excitability with increased potassium currents [43]. Previous investigations into the functional and behavioral heterogeneity of VTA DA neurons attributed this pathological hyperactivity to VTA DA neurons projecting to the NAc (VTA–NAc pathway) but not those projecting to the mPFC in susceptible animals, suggesting that the VTA–NAc pathway mediates hyperactivity to encode susceptible behaviors [42]. This finding further supported the involvement of Ih currents in VTA–NAc DA-mediated susceptibility, as the VTA-mPFC has a smaller Ih. Further confirming the preferential role of VTA–NAc Ih currents, excessive optogenetic stimulation of the VTA–mPFC and VTA–NAc DA pathways produced induction of Ih and potassium currents specifically in the VTA–NAc DA pathway [43]. These results support the involvement of HCN channels in depression behaviors, and provide multiple therapeutic strategies for the reversal of depression or induction of resilience.
Collectively, these results support published literature finding that Ih currents are involved in reward and learning, and demonstrate that HCN channel pathology may mediate depression behaviors. Interestingly, HCN channels have been studied in the context of the rewarding aspects of ethanol. The increase in firing rate of VTA DA neurons from ethanol is blocked with the Ih inhibitor ZD7288, and chronic ethanol administration decreases the density of HCN channels in VTA DA neurons [75]. Stress may regulate Ih through a noradrenergic mechanism, as demonstrated by the decrease in Ih current amplitude when α2-adrenoreceptors are activated within the VTA [149]. The effects of this noradrenergic mechanism may be similar to those observed in PFC following cAMP inhibition [113]. Furthermore, increased VTA DA neuronal firing following corticotropin-releasing factor application is blocked by Ih current inhibition [132]. Since stress promotes the release of corticotropin-releasing factor, activating the glucocorticoid stress hormone pathway in the hypothalamic–pituitary–adrenal axis, and subsequently the mesolimbic DA system [150, 151], the relationship between depression and HCN channels in the VTA requires additional investigation.
Failure of HCN and KCNQ Homeostatic Plasticity in Depression
In order to maintain excitability at a basal level, neurons may undergo intrinsic changes to adjust or return cell activity to a target range for nonpathological firing, a concept introduced as neuronal plasticity or homeostatic plasticity [152, 153]. In the social-defeat model, resilient mice have a large Ih current and increased potassium currents in both peak and sustained components, suggesting the involvement of multiple potassium channels in regulating the excitability of VTA DA neurons for the resilience phenotype [43]. The induction of a large Ih current in susceptible mice by LTG appears to indirectly produce increased potassium currents in VTA DA neurons that bring neuronal firing back to control levels, suggesting a homeostatic plasticity mechanism and providing another therapeutic target to promote resilience or reverse susceptibility to depression [43, 154]. Previously published microarray data found an upregulation of KCNQ3 (kv7.3) channels in resilient animals, providing a more specific potassium channel target for antidepressant treatment [40, 46]. KCNQ3 channels are slow voltage-gated potassium channels, and all KCNQ channels are inhibitory potassium currents that are important for maintaining neuronal resting membrane potential, neuronal excitability, and action potential activity in concert with HCN channels [155,156,157,158,159]. KCNQ channels mediate “M” currents, since they can be closed by muscarinic receptor stimulation [160]. From a therapeutic potential, KCNQ channels have been studied in anxiety and bipolar disorders as a secondary consequence of HCN channel neuronal excitability changes [161,162,163,164].
Thus, we sought to elucidate the role of KCNQ3 in susceptibility and resilience to social-defeat stress. Overexpression of KCNQ3 channels in VTA, intra-VTA infusion of pharmacological KCNQ potassium openers flupirtine, retigabine, or BMS-204352, or intraperitoneal injection of retigabine normalized VTA neuronal activity and rescued social avoidance, anhedonia in the sucrose preference test, and behavioral despair in the forced swim test [46]. Notably, targeting KCNQ channels with retigabine (ezogabine) [165], an FDA-approved potassium channel opener for treatment of epilepsy, has antidepressant efficacy in depressed patients [166]. Based on these observations, multiple avenues can be exploited to target homeostatic plasticity within the VTA and induce antidepressant therapeutic effects: normalizing the pathological increase of Ih currents or promoting KCNQ channel function.
This interaction of KCNQ channel and HCN channel activity has also been observed in the PFC and may influence stress-induced impairments in working memory [110, 115, 156]. cAMP activity increases Ih currents in PFC and cAMP–PKA signaling increases the probability of KCNQ channel current [120, 155]. HCN and KCNQ channels may work in concert to reduce firing and stress-induced dysfunctional DA signaling within the PFC, particularly through D1Rs [115]. Since HCN and KCNQ channels also mediate the excitability of CA1 hippocampal pyramidal cells, KCNQ mechanisms may be involved in the processing of stress within the hippocampus [167]. Altogether, these results suggest another ion channel therapeutic target for antidepressant treatment.
HCN Channels: Ketamine Targets
As classical antidepressants require several weeks to produce therapeutic benefit, a major priority of the field is the identification of antidepressants that work rapidly [168]. Ketamine, an anesthetic that is a noncompetitive ionotropic glutamatergic NMDA receptor antagonist, produces long-lasting and rapid antidepressant responses in some patients with treatment-resistant MDD when administered at even a single subanesthetic dose (0.5–10 mg/kg) [25]. While the bulk of current research aims to define the glutamatergic locus of antidepressant action, interestingly the synaptic and behavioral effects of ketamine also involve HCN channels [169, 170]. In particular, ketamine interacts with HCN1 homodimers and heterodimers (HCN1–HCN2 configurations) exclusively to inhibit Ih currents [170].
In animals, the antidepressant effects of global HCN ablation appear to be linked to the effects of low-dose ketamine [169]. Ketamine induces membrane hyperpolarization, inhibits Ih currents, and reduces resonant properties of cortical pyramidal neurons in wild-type mice as measured ex vivo. HCN1 ablation also produces aberrant cortical synchrony and rhythmicity [170]. These effects are not observed in HCN1 knockout mice ex vivo, suggesting ketamine targets and inhibits HCN1 channels. When animals lacking HCN channels are administered ketamine, the behavioral actions of ketamine appear to be occluded; anhedonic measures of behavior, such as sucrose preference, can no longer be rescued by ketamine [169, 170]. Ketamine was also ineffective in rescuing depression-like behaviors in HCN knockout mice tested for novelty suppressed feeding, a measure of stress-induced anxiety and chronic antidepressant responsiveness. Interestingly, ketamine also increased behavioral despair in the forced swim test in these animals. As mentioned earlier, HCN knockout normally produces a reduction in time spent immobile in the forced swim test in mice. Ketamine is thought to promote an NMDA-mediated increase of surface abundance of HCN1 channels and an increase in HCN channel activity via NMDA receptor activation-induced calcium influx [91, 101]. This provides credence that Ih is involved in mediating an excitation–inhibition balance that likely modulates depression-related behaviors.
Antidepressant actions of ketamine involving hippocampal HCN channels have also been identified. Electrophysiological investigations of the role of ketamine in the expression of HCN channels have revealed a mainly presynaptic mechanism of HCN in ketamine’s actions [169]. To distinguish between presynaptic and postsynaptic HCN action, HCN channel blockers (ZD7288, zatebradine) that block postsynaptic HCN channels and isolate presynaptic excitatory transmission are applied to the CA1 hippocampal preparation. In this configuration, stimulation of Schaffer collateral pathways and measurement of field excitatory postsynaptic potentials indicates that ketamine still enhances presynaptic excitatory transmission. HCN channels appear to be critical to ketamine’s actions in the hippocampus antidepressant action of ketamine and hippocampal synaptic markers are eliminated by inhibition or deletion of the HCN1 channel.
Considering the findings of Ih current pathology in mesolimbic DA circuitry following social defeat stress, as well as the observed antidepressant effect of HCN knockout models, we can hypothesize that ketamine may act to block HCN channels and rescue the behavioral deficits linked to increased Ih current in susceptible animals. Thus, ketamine may recapitulate the antidepressant effects of attenuating Ih current globally in the brain, the hippocampus, the PFC, or the VTA. Use of the antidepressants fluoxetine or imipramine also reverses the susceptible phenotype, but only following chronic administration [39, 57, 171,172,173]. Chronic antidepressant treatment with fluoxetine reverses the observed pathological increases in Ih currents within the VTA of susceptible mice, mirroring the behavioral rescue of susceptible animals with Ih current blockers ZD7288 or DK-AH269 [41]. Conventional antidepressants may work to indirectly influence HCN channel activity through signaling cascades involving cAMP or cAMP-dependent kinase mediated phosphorylation of the HCN channel subunits [68, 119]. Another target for therapeutic action could be the signaling protein brain-derived neurotrophic factor (BDNF), as both antidepressants and ketamine activate BDNF signaling via cAMP response element-binding protein transcriptional regulation throughout mesocorticolimbic brain areas (including hippocampus and NAc) [12, 23, 39, 40, 174,175,176,177,178,179,180]. Notably, when HCN1 channel function is ablated with HCN1 shRNA, producing antidepressant and anxiolytic behaviors, BDNF is also increased [108]. These BDNF effects may work in concert with glutamate signaling activation, another process implicated in HCN function [91, 101, 181]. While low-dose ketamine has yielded promising clinical results, a number of issues require further examination. Beyond the risks for neurotoxicity, addiction and abuse, ketamine produces dissociative and psychotomimetic side effects and its antidepressant efficacy appears to be unsustainable with serial infusions [182, 183]. Thus, targeting HCN channels may be a promising alternative therapy to ketamine.
Therapeutic Potential of HCN Channels
Neuropsychiatric diseases, particularly depression, are characterized by a change in the intrinsic membrane excitability of neurons, a phenomenon that affects the responsiveness of neuronal populations in various brain areas to stimuli. HCN channels exert an important role in neuronal excitability, mediating rhythmic activity, resting membrane potential, neurotransmission, and synaptic plasticity within multiple reward-related brain regions. There are multiple findings attributing an important role to HCN channels in expression of depression-like behaviors. This suggests that pharmacological regulation of Ih currents offers a novel therapeutic approach for depression treatment (see Table 1 for clinically relevant therapeutics).
HCN channels have been implicated in several human diseases, namely cardiac-associated diseases and neurological diseases involving epilepsy and neuropathic pain [184,185,186,187,188,189]. HCN channel therapeutics are most well known to control cardiac activity. Blockers of HCN channel activity, such as DK-AH269, ZD-7288, zatebradine, and ivabradine, are efficacious in lowering heart rate in patients with cardiac diseases but produce side effects, including bradycardia (abnormally lowered heart rate) and phosphenes (transiently enhanced visual brightness) [190,191,192]. While all 4 blockers have been used for preclinical investigations [193,194,195,196], ivabradine is the only FDA-approved HCN-specific blocker [197]. Ivabradine is a relatively well tolerated and commonly prescribed HCN blocker for cardiac disease that is unique in its use-dependency clinical property. As Ih current increases, ivabradine is able to better access the inner pore of the HCN channels to block activity [198]. Ivabradine has shown efficacy in preclinical and clinical studies of neuropathic pain [199]. Since ivabradine does not cross the blood–brain barrier, a brain-permeable derivative will be necessary for therapeutic action within the central nervous system [200]. Regardless, DK-AH269, ZD-7288, zatebradine, and ivabradine are useful as HCN channel blockers and Ih current inhibitors for preclinical analysis of HCN in depression [193,194,195].
LTG and gabapentin are well-tolerated and FDA-approved anticonvulsant/epilepsy drugs that are known to potentiate Ih currents [184, 196, 201]. LTG and gabapentin may be useful for neuropathic pain [202], particularly gabapentin [203,204,205]. Gabapentin has also been evaluated in patients with bipolar disease. There are relatively mixed results for use of gabapentin as monotherapy to stabilize moods in bipolar disease, but it appears to be relatively well tolerated as an adjunctive therapy [206,207,208,209,210,211,212,213]. The efficacy of gabapentin in unipolar depression is also unclear [214, 215]. However, gabapentin was successful in treating anxiety, nightmares, and insomnia in patients with post-traumatic stress disorder, including those who also suffered from MDD [216, 217]. Gabapentin may be particularly useful for insomnia, as alleviation of sleep deprivation was also observed in alcohol-dependent individuals [218, 219]. For treatment of anxiety disorders, specifically social phobia and panic disorder, gabapentin may serve well as an adjunctive medication [220,221,222]. In conjunction with fluoxetine, gabapentin was effective in treating obsessive compulsive disorder, although the psychiatric symptoms rebounded after discontinuation of gabapentin [223, 224].
LTG is an approved medication for bipolar disorder, producing relief of depression symptoms and a decrease in mania [225,226,227,228,229,230,231,232,233,234,235,236]. LTG is often administered in conjunction with lithium to stabilize mood [237, 238]. In particular, chronic LTG administration decreased impulsivity, mood instability, and suicidal behaviors in patients with bipolar disease [239,240,241,242,243]. As a mood stabilizer, LTG has also been studied in patients with schizophrenia, where it had strongest efficacy as an adjunctive therapy [244,245,246,247,248,249]. Notably, LTG has been used off label to combat treatment-resistant depression, suggesting efficacy in this patient population [214, 215, 250,251,252], although its use as an antidepressant augmentation agent requires further analysis due to mixed success [253, 254]. Investigations of the effectiveness of LTG in other mood disorders, such as post-traumatic stress disorder, suggest that it can be useful to reduce symptoms including avoidance, numbing, and re-experiences of flashbacks or nightmares [255]. However, additional studies of LTG treatment in OCD are necessary to clarify its therapeutic value [256, 257]. LTG may also be useful for combating comorbidities of substance abuse with bipolar disorder for cocaine addiction [258,259,260], or cocaine dependence only [261].
LTG has shown relative efficacy in patients with alcohol dependence [262]. When LTG was administered in conjunction with clozapine as an antipsychotic medication for patients with schizophrenia and alcohol dependence, patients displayed a decrease in alcohol seeking and consumption [263]. Patients with comorbidities of bipolar disorder and alcohol dependence show improvements in depression, mania, alcohol craving, and alcohol consumption [264]. Gabapentin has also demonstrated efficacy in treating alcohol dependence [265]. The use of LTG and gabapentin to combat alcohol abuse is interesting; although the mechanism of ethanol action is not specific, acute ethanol influences HCN channels by increasing Ih currents [75, 266], and repeated ethanol and withdrawal from ethanol results in a decrease in Ih currents within the VTA in DA neurons [75, 267]. Ethanol also disrupts PFC function and affects Ih current within the hippocampus, although this phenomenon is far less studied in these 2 brain regions [268, 269]. These findings suggest that possible pathology of the reward circuitry in response to ethanol use can be remedied through potentiation of Ih currents. Thus, while LTG is useful for treatment of bipolar disorder, the benefit of gabapentin seems limited to anxiety and alcohol dependence [270]. Further research is required to conclusively define a role for gabapentin and LTG in other psychiatric disorders.
From a practical standpoint, it is important to develop more specific HCN channel drugs. HCN channel agonists, LTG and gabapentin, have multiple molecular mechanisms of action besides potentiating Ih currents, including regulation of voltage-gated sodium and calcium channels [82, 196, 205, 236]. Ih current-specific action of DK-AH269 and ZD-7288 have been elucidated particularly well in pyramidal neurons of the CA1 hippocampal region to probe the modulation of excitatory postsynaptic potentials; there is a lack of consensus regarding actions of HCN channels in hippocampal mossy fiber plasticity [271,272,273,274]. These findings suggest that HCN blockers DK-AH269 and ZD-7288 have off-target effects, particularly through putative glutamate-receptor mediated signaling [272, 274]. Guanfacine is another indirect modulator of HCN signaling that works through inhibition of cAMP–HCN signaling via activation of α2A-adrenoreceptors. Guanfacine has been investigated for efficacy in schizophrenia, and is currently prescribed for attention-deficit/hyperactivity disorder [275, 276] to improve working memory, impulse control, stress control, and emotional regulation [277,278,279]. Off-target effects of these drugs highlight the need to develop selective compounds.
A caveat of HCN channel modulation in the brain as a treatment for psychiatric disorders, in particular use of HCN channel blockers, is the essential role of Ih currents in cardiac pacemaker activity. Developing drugs to target specific HCN channel isoforms, such as HCN1 versus HCN4 specific antagonists, could alleviate negative side effects. However, since TRIP8b is a critical protein for HCN activity within the brain and is not expressed in the heart, developing TRIP8b-specific drugs could surpass these current therapeutic limitations [280]. TRIP8b knockout is antidepressant in mice [94]. Thus, antagonizing TRIP8b binding to HCN channels, particularly at the C-terminal tail, can impair trafficking of the HCN channel and reduce Ih current, producing an antidepressant effect [94, 281,282,283,284]. A fluorescence polarization screen for small molecules disrupting TRIP8b and HCN interactions demonstrated the feasibility of identifying compounds that modulate HCN channel activity and neuronal excitability through TRIP8b [280].
Currently, there are limited therapeutic options designed for Ih current modulation in the brain. Increased focus on developing novel therapeutics to target HCN channels may produce benefit for patients with depression who are underserved by the current standard of care, especially since preclinical research has highlighted the antidepressant promise of reducing HCN channel activity.
References
Kessler RC, Chiu WT, Demler O, Merikangas KR, Walters EE. Prevalence, severity, and comorbidity of 12-month DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62(6):617-27.
Fava M. Diagnosis and definition of treatment-resistant depression. Biol Psychiatry. 2003;53(8):649-59.
Solomon DA, Keller MB, Leon AC, et al. Multiple recurrences of major depressive disorder. Am J Psychiatry. 2000;157(2):229-33.
American Psychiatric Association., American Psychiatric Association. DSM-5 Task Force. Diagnostic and statistical manual of mental disorders : DSM-5. 5th ed. Washington, D.C.: American Psychiatric Association; 2013. xliv, 947 p. p.
Rush AJ, Trivedi MH, Wisniewski SR, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry. 2006;163(11):1905-17.
Gartlehner G, Thaler K, Hill S, Hansen RA. How should primary care doctors select which antidepressants to administer? Current psychiatry reports. 2012;14(4):360-9.
Stahl SM. Basic psychopharmacology of antidepressants, part 1: Antidepressants have seven distinct mechanisms of action. J Clin Psychiatry. 1998;59 Suppl 4:5-14.
Nierenberg AA, Ostacher MJ, Huffman JC, Ametrano RM, Fava M, Perlis RH. A brief review of antidepressant efficacy, effectiveness, indications, and usage for major depressive disorder. Journal of occupational and environmental medicine. 2008;50(4):428-36.
Kessler RC, Berglund P, Demler O, et al. The epidemiology of major depressive disorder: results from the National Comorbidity Survey Replication (NCS-R). JAMA. 2003;289(23):3095-105.
Schildkraut JJ. The catecholamine hypothesis of affective disorders: a review of supporting evidence. Am J Psychiatry. 1965;122(5):509-22.
Duman RS, Aghajanian GK, Sanacora G, Krystal JH. Synaptic plasticity and depression: new insights from stress and rapid-acting antidepressants. Nat Med. 2016;22(3):238-49.
Duman RS, Monteggia LM. A neurotrophic model for stress-related mood disorders. Biol Psychiatry. 2006;59(12):1116-27.
Vialou V, Feng J, Robison AJ, Nestler EJ. Epigenetic mechanisms of depression and antidepressant action. Annu Rev Pharmacol Toxicol. 2013;53:59-87.
Warner-Schmidt JL, Duman RS. Hippocampal neurogenesis: opposing effects of stress and antidepressant treatment. Hippocampus. 2006;16(3):239-49.
Christoffel DJ, Golden SA, Russo SJ. Structural and synaptic plasticity in stress-related disorders. Rev Neurosci. 2011;22(5):535-49.
Castren E, Hen R. Neuronal plasticity and antidepressant actions. Trends in neurosciences. 2013;36(5):259-67.
Willner P, Scheel-Kruger J, Belzung C. The neurobiology of depression and antidepressant action. Neurosci Biobehav Rev. 2013;37(10 Pt 1):2331-71.
Krishnan V, Nestler EJ. The molecular neurobiology of depression. Nature. 2008;455(7215):894-902.
Kavalali ET, Monteggia LM. Synaptic mechanisms underlying rapid antidepressant action of ketamine. Am J Psychiatry. 2012;169(11):1150-6.
Furey ML, Drevets WC. Antidepressant efficacy of the antimuscarinic drug scopolamine: a randomized, placebo-controlled clinical trial. Arch Gen Psychiatry. 2006;63(10):1121-9.
Drevets WC, Zarate CA, Jr., Furey ML. Antidepressant effects of the muscarinic cholinergic receptor antagonist scopolamine: a review. Biol Psychiatry. 2013;73(12):1156-63.
Voleti B, Navarria A, Liu RJ, et al. Scopolamine rapidly increases mammalian target of rapamycin complex 1 signaling, synaptogenesis, and antidepressant behavioral responses. Biol Psychiatry. 2013;74(10):742-9.
Autry AE, Adachi M, Nosyreva E, et al. NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature. 2011;475(7354):91-5.
Berman RM, Cappiello A, Anand A, et al. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry. 2000;47(4):351-4.
Zarate CA, Jr., Singh JB, Carlson PJ, et al. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry. 2006;63(8):856-64.
Wohleb ES, Gerhard D, Thomas A, Duman RS. Molecular and Cellular Mechanisms of Rapid-Acting Antidepressants Ketamine and Scopolamine. Curr Neuropharmacol. 2017;15(1):11-20.
Gerhard DM, Wohleb ES, Duman RS. Emerging treatment mechanisms for depression: focus on glutamate and synaptic plasticity. Drug Discov Today. 2016;21(3):454-64.
Kessler RC. The effects of stressful life events on depression. Annual review of psychology. 1997;48:191-214.
Lucki I. The forced swimming test as a model for core and component behavioral effects of antidepressant drugs. Behav Pharmacol. 1997;8(6-7):523-32.
Porsolt RD, Le Pichon M, Jalfre M. Depression: a new animal model sensitive to antidepressant treatments. Nature. 1977;266(5604):730-2.
Steru L, Chermat R, Thierry B, Simon P. The tail suspension test: a new method for screening antidepressants in mice. Psychopharmacology (Berl). 1985;85(3):367-70.
Cryan JF, Mombereau C, Vassout A. The tail suspension test as a model for assessing antidepressant activity: review of pharmacological and genetic studies in mice. Neurosci Biobehav Rev. 2005;29(4-5):571-625.
Henn FA, Vollmayr B. Stress models of depression: forming genetically vulnerable strains. Neurosci Biobehav Rev. 2005;29(4-5):799-804.
Li B, Piriz J, Mirrione M, et al. Synaptic potentiation onto habenula neurons in the learned helplessness model of depression. Nature. 2011;470(7335):535-9.
Nestler EJ, Hyman SE. Animal models of neuropsychiatric disorders. Nature Neuroscience. 2010;13(10):1161-9.
Willner P. Validity, reliability and utility of the chronic mild stress model of depression: a 10-year review and evaluation. Psychopharmacology (Berl). 1997;134(4):319-29.
Willner P. Chronic mild stress (CMS) revisited: consistency and behavioural-neurobiological concordance in the effects of CMS. Neuropsychobiology. 2005;52(2):90-110.
Hodes GE, Pfau ML, Purushothaman I, et al. Sex Differences in Nucleus Accumbens Transcriptome Profiles Associated with Susceptibility versus Resilience to Subchronic Variable Stress. J Neurosci. 2015;35(50):16362-76.
Berton O, McClung CA, Dileone RJ, et al. Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science. 2006;311(5762):864-8.
Krishnan V, Han MH, Graham DL, Berton O, Renthal W, Russo SJ, et al. Molecular adaptations underlying susceptibility and resistance to social defeat in brain reward regions. Cell. 2007;131(2):391-404.
Cao JL, Covington HE, 3rd, Friedman AK, et al. Mesolimbic dopamine neurons in the brain reward circuit mediate susceptibility to social defeat and antidepressant action. J Neurosci. 2010;30(49):16453-8.
Chaudhury D, Walsh JJ, Friedman AK, et al. Rapid regulation of depression-related behaviours by control of midbrain dopamine neurons. Nature. 2013;493(7433):532-6.
Friedman AK, Walsh JJ, Juarez B, et al. Enhancing depression mechanisms in midbrain dopamine neurons achieves homeostatic resilience. Science. 2014;344(6181):313-9.
Feder A, Nestler EJ, Charney DS. Psychobiology and molecular genetics of resilience. Nat Rev Neurosci. 2009;10(6):446-57.
Russo SJ, Murrough JW, Han MH, Charney DS, Nestler EJ. Neurobiology of resilience. Nat Neurosci. 2012;15(11):1475-84.
Friedman AK, Juarez B, Ku SM, et al. KCNQ channel openers reverse depressive symptoms via an active resilience mechanism. Nat Commun. 2016;7:11671.
Walsh JJ, Friedman AK, Sun H, et al. Stress and CRF gate neural activation of BDNF in the mesolimbic reward pathway. Nat Neurosci. 2014;17(1):27-9.
Friedman A. Neuroscience. Jump-starting natural resilience reverses stress susceptibility. Science. 2014;346(6209):555.
Golden SA, Covington HE, Berton O, Russo SJ. A standardized protocol for repeated social defeat stress in mice. Nature Protocols. 2011;6(8):1183-91.
Pfau ML, Russo SJ. Peripheral and Central Mechanisms of Stress Resilience. Neurobiol Stress. 2015;1:66-79.
Mayberg HS. Limbic-cortical dysregulation: a proposed model of depression. J Neuropsychiatry Clin Neurosci. 1997;9(3):471-81.
Mayberg HS, Brannan SK, Tekell JL, et al. Regional metabolic effects of fluoxetine in major depression: serial changes and relationship to clinical response. Biol Psychiatry. 2000;48(8):830-43.
Mayberg HS, Lozano AM, Voon V, et al. Deep brain stimulation for treatment-resistant depression. Neuron. 2005;45(5):651-60.
Schlaepfer TE, Cohen MX, Frick C, et al. Deep brain stimulation to reward circuitry alleviates anhedonia in refractory major depression. Neuropsychopharmacology. 2008;33(2):368-77.
Krishnan V, Han MH, Mazei-Robison M, et al. AKT signaling within the ventral tegmental area regulates cellular and behavioral responses to stressful stimuli. Biol Psychiatry. 2008;64(8):691-700.
Covington HE, 3rd, Lobo MK, Maze I, et al. Antidepressant effect of optogenetic stimulation of the medial prefrontal cortex. J Neurosci. 2010;30(48):16082-90.
Vialou V, Robison AJ, Laplant QC, et al. DeltaFosB in brain reward circuits mediates resilience to stress and antidepressant responses. Nat Neurosci. 2010;13(6):745-52.
Christoffel DJ, Golden SA, Dumitriu D, et al. IkappaB kinase regulates social defeat stress-induced synaptic and behavioral plasticity. J Neurosci. 2011;31(1):314-21.
Bagot RC, Parise EM, Pena CJ, et al. Ventral hippocampal afferents to the nucleus accumbens regulate susceptibility to depression. Nat Commun. 2015;6:7062.
Nestler EJ, Carlezon WA, Jr. The mesolimbic dopamine reward circuit in depression. Biol Psychiatry. 2006;59(12):1151-9.
Delvecchio G, Fossati P, Boyer P, et al. Common and distinct neural correlates of emotional processing in Bipolar Disorder and Major Depressive Disorder: a voxel-based meta-analysis of functional magnetic resonance imaging studies. Eur Neuropsychopharmacol. 2012;22(2):100-13.
Anand A, Li Y, Wang Y, Gardner K, Lowe MJ. Reciprocal effects of antidepressant treatment on activity and connectivity of the mood regulating circuit: an FMRI study. J Neuropsychiatry Clin Neurosci. 2007;19(3):274-82.
Fitzgerald PB, Laird AR, Maller J, Daskalakis ZJ. A meta-analytic study of changes in brain activation in depression. Hum Brain Mapp. 2008;29(6):683-95.
Monteggia LM, Eisch AJ, Tang MD, Kaczmarek LK, Nestler EJ. Cloning and localization of the hyperpolarization-activated cyclic nucleotide-gated channel family in rat brain. Brain research Molecular brain research. 2000;81(1-2):129-39.
Ludwig A, Zong X, Jeglitsch M, Hofmann F, Biel M. A family of hyperpolarization-activated mammalian cation channels. Nature. 1998;393(6685):587-91.
Robinson RB, Siegelbaum SA. Hyperpolarization-activated cation currents: from molecules to physiological function. Annu Rev Physiol. 2003;65:453-80.
Nolan MF, Malleret G, Dudman JT, et al. A behavioral role for dendritic integration: HCN1 channels constrain spatial memory and plasticity at inputs to distal dendrites of CA1 pyramidal neurons. Cell. 2004;119(5):719-32.
Wahl-Schott C, Biel M. HCN channels: structure, cellular regulation and physiological function. Cell Mol Life Sci. 2009;66(3):470-94.
Notomi T, Shigemoto R. Immunohistochemical localization of Ih channel subunits, HCN1-4, in the rat brain. J Comp Neurol. 2004;471(3):241-76.
Moosmang S, Stieber J, Zong X, Biel M, Hofmann F, Ludwig A. Cellular expression and functional characterization of four hyperpolarization-activated pacemaker channels in cardiac and neuronal tissues. European journal of biochemistry. 2001;268(6):1646-52.
Brewster AL, Chen Y, Bender RA, Yeh A, Shigemoto R, Baram TZ. Quantitative analysis and subcellular distribution of mRNA and protein expression of the hyperpolarization-activated cyclic nucleotide-gated channels throughout development in rat hippocampus. Cereb Cortex. 2007;17(3):702-12.
Surges R, Freiman TM, Feuerstein TJ. Input resistance is voltage dependent due to activation of Ih channels in rat CA1 pyramidal cells. J Neurosci Res. 2004;76(4):475-80.
Lamas JA. A hyperpolarization-activated cation current (Ih) contributes to resting membrane potential in rat superior cervical sympathetic neurones. Pflugers Arch. 1998;436(3):429-35.
Nolan MF, Dudman JT, Dodson PD, Santoro B. HCN1 channels control resting and active integrative properties of stellate cells from layer II of the entorhinal cortex. J Neurosci. 2007;27(46):12440-51.
Okamoto T, Harnett MT, Morikawa H. Hyperpolarization-activated cation current (Ih) is an ethanol target in midbrain dopamine neurons of mice. J Neurophysiol. 2006;95(2):619-26.
Neuhoff H, Neu A, Liss B, Roeper J. I(h) channels contribute to the different functional properties of identified dopaminergic subpopulations in the midbrain. J Neurosci. 2002;22(4):1290-302.
Magee JC. Dendritic lh normalizes temporal summation in hippocampal CA1 neurons. Nat Neurosci. 1999;2(6):508-14.
Lorincz A, Notomi T, Tamas G, Shigemoto R, Nusser Z. Polarized and compartment-dependent distribution of HCN1 in pyramidal cell dendrites. Nat Neurosci. 2002;5(11):1185-93.
Beaumont V, Zucker RS. Enhancement of synaptic transmission by cyclic AMP modulation of presynaptic Ih channels. Nat Neurosci. 2000;3(2):133-41.
He C, Chen F, Li B, Hu Z. Neurophysiology of HCN channels: from cellular functions to multiple regulations. Prog Neurobiol. 2014;112:1-23.
Bender RA, Kirschstein T, Kretz O, et al. Localization of HCN1 channels to presynaptic compartments: novel plasticity that may contribute to hippocampal maturation. J Neurosci. 2007;27(17):4697-706.
Poolos NP, Migliore M, Johnston D. Pharmacological upregulation of h-channels reduces the excitability of pyramidal neuron dendrites. Nat Neurosci. 2002;5(8):767-74.
Huang Z, Lujan R, Martinez-Hernandez J, Lewis AS, Chetkovich DM, Shah MM. TRIP8b-independent trafficking and plasticity of adult cortical presynaptic HCN1 channels. J Neurosci. 2012;32(42):14835-48.
Thuault SJ, Malleret G, Constantinople CM, et al. Prefrontal cortex HCN1 channels enable intrinsic persistent neural firing and executive memory function. J Neurosci. 2013;33(34):13583-99.
Brager DH, Lewis AS, Chetkovich DM, Johnston D. Short- and long-term plasticity in CA1 neurons from mice lacking h-channel auxiliary subunit TRIP8b. J Neurophysiol. 2013;110(10):2350-7.
He W, Xu X, Lv Q, Guo L. Low dose ZD7288 attenuates the ischemia/reperfusion-induced impairment of long-term potentiation induction at hippocampal Schaffer collateral-CA1 synapses. Cell Mol Neurobiol. 2014;34(4):611-7.
Kelmendi B, Holsbach-Beltrame M, McIntosh AM, et al. Association of polymorphisms in HCN4 with mood disorders and obsessive compulsive disorder. Neurosci Lett. 2011;496(3):195-9.
McIntosh AM, Simen AA, Evans KL, et al. Genetic variation in Hyperpolarization-activated cyclic nucleotide-gated channels and its relationship with neuroticism, cognition and risk of depression. Front Genet. 2012;3:116.
Dunlop J, Vasilyev D, Lu P, Cummons T, Bowlby MR. Hyperpolarization-activated cyclic nucleotide-gated (HCN) channels and pain. Curr Pharm Des. 2009;15(15):1767-72.
Baruscotti M, Bucchi A, Difrancesco D. Physiology and pharmacology of the cardiac pacemaker ("funny") current. Pharmacol Ther. 2005;107(1):59-79.
Noam Y, Zha Q, Phan L, et al. Trafficking and surface expression of hyperpolarization-activated cyclic nucleotide-gated channels in hippocampal neurons. J Biol Chem. 2010;285(19):14724-36.
Santoro B, Piskorowski RA, Pian P, Hu L, Liu H, Siegelbaum SA. TRIP8b splice variants form a family of auxiliary subunits that regulate gating and trafficking of HCN channels in the brain. Neuron. 2009;62(6):802-13.
Lewis AS, Schwartz E, Chan CS, Noam Y, Shin M, Wadman WJ, et al. Alternatively spliced isoforms of TRIP8b differentially control h channel trafficking and function. J Neurosci. 2009;29(19):6250-65.
Lewis AS, Vaidya SP, Blaiss CA, et al. Deletion of the hyperpolarization-activated cyclic nucleotide-gated channel auxiliary subunit TRIP8b impairs hippocampal Ih localization and function and promotes antidepressant behavior in mice. J Neurosci. 2011;31(20):7424-40.
Kim JJ, Yoon KS. Stress: metaplastic effects in the hippocampus. Trends in neurosciences. 1998;21(12):505-9.
Sapolsky RM. Depression, antidepressants, and the shrinking hippocampus. Proc Natl Acad Sci U S A. 2001;98(22):12320-2.
Kennedy SH, Evans KR, Kruger S, et al. Changes in regional brain glucose metabolism measured with positron emission tomography after paroxetine treatment of major depression. Am J Psychiatry. 2001;158(6):899-905.
Campbell S, MacQueen G. An update on regional brain volume differences associated with mood disorders. Curr Opin Psychiatry. 2006;19(1):25-33.
Campbell S, Macqueen G. The role of the hippocampus in the pathophysiology of major depression. J Psychiatry Neurosci. 2004;29(6):417-26.
Campbell S, Marriott M, Nahmias C, MacQueen GM. Lower hippocampal volume in patients suffering from depression: a meta-analysis. Am J Psychiatry. 2004;161(4):598-607.
Fan Y, Fricker D, Brager DH, et al. Activity-dependent decrease of excitability in rat hippocampal neurons through increases in I(h). Nat Neurosci. 2005;8(11):1542-51.
Magee JC. Dendritic hyperpolarization-activated currents modify the integrative properties of hippocampal CA1 pyramidal neurons. J Neurosci. 1998;18(19):7613-24.
Tsay D, Dudman JT, Siegelbaum SA. HCN1 channels constrain synaptically evoked Ca2+ spikes in distal dendrites of CA1 pyramidal neurons. Neuron. 2007;56(6):1076-89.
Magee JC. Dendritic integration of excitatory synaptic input. Nat Rev Neurosci. 2000;1(3):181-90.
Hu H, Vervaeke K, Storm JF. Two forms of electrical resonance at theta frequencies, generated by M-current, h-current and persistent Na+ current in rat hippocampal pyramidal cells. J Physiol. 2002;545(Pt 3):783-805.
Narayanan R, Johnston D. The h channel mediates location dependence and plasticity of intrinsic phase response in rat hippocampal neurons. J Neurosci. 2008;28(22):5846-60.
Narayanan R, Johnston D. Long-term potentiation in rat hippocampal neurons is accompanied by spatially widespread changes in intrinsic oscillatory dynamics and excitability. Neuron. 2007;56(6):1061-75.
Kim CS, Chang PY, Johnston D. Enhancement of dorsal hippocampal activity by knockdown of HCN1 channels leads to anxiolytic- and antidepressant-like behaviors. Neuron. 2012;75(3):503-16.
Han Y, Heuermann RJ, Lyman KA, Fisher D, Ismail QA, Chetkovich DM. HCN-channel dendritic targeting requires bipartite interaction with TRIP8b and regulates antidepressant-like behavioral effects. Mol Psychiatry. 2016.
Day M, Carr DB, Ulrich S, Ilijic E, Tkatch T, Surmeier DJ. Dendritic excitability of mouse frontal cortex pyramidal neurons is shaped by the interaction among HCN, Kir2, and Kleak channels. J Neurosci. 2005;25(38):8776-87.
Arnsten AF. Stress weakens prefrontal networks: molecular insults to higher cognition. Nat Neurosci. 2015;18(10):1376-85.
Arnsten AF. Stress signalling pathways that impair prefrontal cortex structure and function. Nat Rev Neurosci. 2009;10(6):410-22.
Wang M, Ramos BP, Paspalas CD, et al. Alpha2A-adrenoceptors strengthen working memory networks by inhibiting cAMP-HCN channel signaling in prefrontal cortex. Cell. 2007;129(2):397-410.
Paspalas CD, Wang M, Arnsten AF. Constellation of HCN channels and cAMP regulating proteins in dendritic spines of the primate prefrontal cortex: potential substrate for working memory deficits in schizophrenia. Cereb Cortex. 2013;23(7):1643-54.
Gamo NJ, Lur G, Higley MJ, et al. Stress Impairs Prefrontal Cortical Function via D1 Dopamine Receptor Interactions With Hyperpolarization-Activated Cyclic Nucleotide-Gated Channels. Biol Psychiatry. 2015;78(12):860-70.
Arnsten AF. Catecholamine and second messenger influences on prefrontal cortical networks of "representational knowledge": a rational bridge between genetics and the symptoms of mental illness. Cereb Cortex. 2007;17 Suppl 1:i6-15.
Birnbaum S, Gobeske KT, Auerbach J, Taylor JR, Arnsten AF. A role for norepinephrine in stress-induced cognitive deficits: alpha-1-adrenoceptor mediation in the prefrontal cortex. Biol Psychiatry. 1999;46(9):1266-74.
Vijayraghavan S, Wang M, Birnbaum SG, Williams GV, Arnsten AF. Inverted-U dopamine D1 receptor actions on prefrontal neurons engaged in working memory. Nat Neurosci. 2007;10(3):376-84.
Chen S, Wang J, Siegelbaum SA. Properties of hyperpolarization-activated pacemaker current defined by coassembly of HCN1 and HCN2 subunits and basal modulation by cyclic nucleotide. J Gen Physiol. 2001;117(5):491-504.
Ulens C, Tytgat J. Functional heteromerization of HCN1 and HCN2 pacemaker channels. J Biol Chem. 2001;276(9):6069-72.
Aoki C, Venkatesan C, Kurose H. Noradrenergic modulation of the prefrontal cortex as revealed by electron microscopic immunocytochemistry. Adv Pharmacol. 1998;42:777-80.
Aoki C, Venkatesan C, Go CG, Forman R, Kurose H. Cellular and subcellular sites for noradrenergic action in the monkey dorsolateral prefrontal cortex as revealed by the immunocytochemical localization of noradrenergic receptors and axons. Cereb Cortex. 1998;8(3):269-77.
Goldman-Rakic PS, Lidow MS, Gallager DW. Overlap of dopaminergic, adrenergic, and serotoninergic receptors and complementarity of their subtypes in primate prefrontal cortex. J Neurosci. 1990;10(7):2125-38.
Vargas G, Lucero MT. Modulation by PKA of the hyperpolarization-activated current (Ih) in cultured rat olfactory receptor neurons. J Membr Biol. 2002;188(2):115-25.
Packard MG, Teather LA. Amygdala modulation of multiple memory systems: hippocampus and caudate-putamen. Neurobiol Learn Mem. 1998;69(2):163-203.
Thierry AM, Tassin JP, Blanc G, Glowinski J. Selective activation of mesocortical DA system by stress. Nature. 1976;263(5574):242-4.
Inglis FM, Moghaddam B. Dopaminergic innervation of the amygdala is highly responsive to stress. J Neurochem. 1999;72(3):1088-94.
Abercrombie ED, Keefe KA, DiFrischia DS, Zigmond MJ. Differential effect of stress on in vivo dopamine release in striatum, nucleus accumbens, and medial frontal cortex. J Neurochem. 1989;52(5):1655-8.
Tidey JW, Miczek KA. Social defeat stress selectively alters mesocorticolimbic dopamine release: an in vivo microdialysis study. Brain Res. 1996;721(1-2):140-9.
Arencibia-Albite F, Paladini C, Williams JT, Jimenez-Rivera CA. Noradrenergic modulation of the hyperpolarization-activated cation current (Ih) in dopamine neurons of the ventral tegmental area. Neuroscience. 2007;149(2):303-14.
Lammel S, Hetzel A, Hackel O, Jones I, Liss B, Roeper J. Unique properties of mesoprefrontal neurons within a dual mesocorticolimbic dopamine system. Neuron. 2008;57(5):760-73.
Wanat MJ, Hopf FW, Stuber GD, Phillips PE, Bonci A. Corticotropin-releasing factor increases mouse ventral tegmental area dopamine neuron firing through a protein kinase C-dependent enhancement of Ih. J Physiol. 2008;586(8):2157-70.
Franz O, Liss B, Neu A, Roeper J. Single-cell mRNA expression of HCN1 correlates with a fast gating phenotype of hyperpolarization-activated cyclic nucleotide-gated ion channels (Ih) in central neurons. Eur J Neurosci. 2000;12(8):2685-93.
Schultz W, Dayan P, Montague PR. A neural substrate of prediction and reward. Science. 1997;275(5306):1593-9.
Matsumoto M, Hikosaka O. Two types of dopamine neuron distinctly convey positive and negative motivational signals. Nature. 2009;459(7248):837-41.
Brischoux F, Chakraborty S, Brierley DI, Ungless MA. Phasic excitation of dopamine neurons in ventral VTA by noxious stimuli. Proc Natl Acad Sci U S A. 2009;106(12):4894-9.
Grace AA, Floresco SB, Goto Y, Lodge DJ. Regulation of firing of dopaminergic neurons and control of goal-directed behaviors. Trends in neurosciences. 2007;30(5):220-7.
Schultz W. Behavioral dopamine signals. Trends in neurosciences. 2007;30(5):203-10.
Tye KM, Mirzabekov JJ, Warden MR, et al. Dopamine neurons modulate neural encoding and expression of depression-related behaviour. Nature. 2013;493(7433):537-41.
Ungless MA, Grace AA. Are you or aren't you? Challenges associated with physiologically identifying dopamine neurons. Trends in neurosciences. 2012;35(7):422-30.
Lacey MG, Mercuri NB, North RA. Two cell types in rat substantia nigra zona compacta distinguished by membrane properties and the actions of dopamine and opioids. J Neurosci. 1989;9(4):1233-41.
Margolis EB, Lock H, Hjelmstad GO, Fields HL. The ventral tegmental area revisited: is there an electrophysiological marker for dopaminergic neurons? J Physiol. 2006;577(Pt 3):907-24.
Walsh JJ, Han MH. The heterogeneity of ventral tegmental area neurons: Projection functions in a mood-related context. Neuroscience. 2014;282:101-8.
Juarez B, Han MH. Diversity of Dopaminergic Neural Circuits in Response to Drug Exposure. Neuropsychopharmacology. 2016;41(10):2424-46.
Frye MA. Clinical practice. Bipolar disorder--a focus on depression. N Engl J Med. 2011;364(1):51-9.
Poolos NP, Warner LN, Humphreys SZ, Williams S. Comparative efficacy of combination drug therapy in refractory epilepsy. Neurology. 2012;78(1):62-8.
Boyden ES, Zhang F, Bamberg E, Nagel G, Deisseroth K. Millisecond-timescale, genetically targeted optical control of neural activity. Nat Neurosci. 2005;8(9):1263-8.
Zhang F, Wang LP, Boyden ES, Deisseroth K. Channelrhodopsin-2 and optical control of excitable cells. Nat Methods. 2006;3(10):785-92.
Inyushin MU, Arencibia-Albite F, Vazquez-Torres R, Velez-Hernandez ME, Jimenez-Rivera CA. Alpha-2 noradrenergic receptor activation inhibits the hyperpolarization-activated cation current (Ih) in neurons of the ventral tegmental area. Neuroscience. 2010;167(2):287-97.
Sarnyai Z, Shaham Y, Heinrichs SC. The role of corticotropin-releasing factor in drug addiction. Pharmacol Rev. 2001;53(2):209-43.
Hauger RL, Risbrough V, Brauns O, Dautzenberg FM. Corticotropin releasing factor (CRF) receptor signaling in the central nervous system: new molecular targets. CNS Neurol Disord Drug Targets. 2006;5(4):453-79.
Nestler EJ, Hope BT, Widnell KL. Drug addiction: a model for the molecular basis of neural plasticity. Neuron. 1993;11(6):995-1006.
Turrigiano G. Too many cooks? Intrinsic and synaptic homeostatic mechanisms in cortical circuit refinement. Annu Rev Neurosci. 2011;34:89-103.
Han MH, Nestler EJ. Neural Substrates of Depression and Resilience. Neurotherapeutics. 2017.
Delmas P, Brown DA. Pathways modulating neural KCNQ/M (Kv7) potassium channels. Nat Rev Neurosci. 2005;6(11):850-62.
George MS, Abbott LF, Siegelbaum SA. HCN hyperpolarization-activated cation channels inhibit EPSPs by interactions with M-type K(+) channels. Nat Neurosci. 2009;12(5):577-84.
Wang HS, Pan Z, Shi W, et al. KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science. 1998;282(5395):1890-3.
Wulff H, Castle NA, Pardo LA. Voltage-gated potassium channels as therapeutic targets. Nat Rev Drug Discov. 2009;8(12):982-1001.
Hernandez CC, Zaika O, Tolstykh GP, Shapiro MS. Regulation of neural KCNQ channels: signalling pathways, structural motifs and functional implications. J Physiol. 2008;586(7):1811-21.
Tatulian L, Delmas P, Abogadie FC, Brown DA. Activation of expressed KCNQ potassium currents and native neuronal M-type potassium currents by the anti-convulsant drug retigabine. J Neurosci. 2001;21(15):5535-45.
Korsgaard MP, Hartz BP, Brown WD, Ahring PK, Strobaek D, Mirza NR. Anxiolytic effects of Maxipost (BMS-204352) and retigabine via activation of neuronal Kv7 channels. J Pharmacol Exp Ther. 2005;314(1):282-92.
Sotty F, Damgaard T, Montezinho LP, et al. Antipsychotic-like effect of retigabine [N-(2-Amino-4-(fluorobenzylamino)-phenyl)carbamic acid ester], a KCNQ potassium channel opener, via modulation of mesolimbic dopaminergic neurotransmission. J Pharmacol Exp Ther. 2009;328(3):951-62.
Dencker D, Husum H. Antimanic efficacy of retigabine in a proposed mouse model of bipolar disorder. Behav Brain Res. 2010;207(1):78-83.
Wang M, Gamo NJ, Yang Y, et al. Neuronal basis of age-related working memory decline. Nature. 2011;476(7359):210-3.
Stafstrom CE, Grippon S, Kirkpatrick P. Ezogabine (retigabine). Nat Rev Drug Discov. 2011;10(10):729-30.
Nicholas T, Van Dam MK, Friedman AK, et al. Targeting KCNQ channels with retigabine (also called ezogabine) has antidepressant efficacy in depressed patients. Annual Meeting of Society of Biological Psychiatry; 2016; Atlanta, Georgia: SOBP Abstracts In Press.
Gu N, Vervaeke K, Hu H, Storm JF. Kv7/KCNQ/M and HCN/h, but not KCa2/SK channels, contribute to the somatic medium after-hyperpolarization and excitability control in CA1 hippocampal pyramidal cells. J Physiol. 2005;566(Pt 3):689-715.
Duman RS, Li N, Liu RJ, Duric V, Aghajanian G. Signaling pathways underlying the rapid antidepressant actions of ketamine. Neuropharmacology. 2012;62(1):35-41.
Zhang K, Xu T, Yuan Z, et al. Essential roles of AMPA receptor GluA1 phosphorylation and presynaptic HCN channels in fast-acting antidepressant responses of ketamine. Sci Signal. 2016;9(458):ra123.
Chen X, Shu S, Bayliss DA. HCN1 channel subunits are a molecular substrate for hypnotic actions of ketamine. J Neurosci. 2009;29(3):600-9.
Bagot RC, Cates HM, Purushothaman I, et al. Ketamine and Imipramine Reverse Transcriptional Signatures of Susceptibility and Induce Resilience-Specific Gene Expression Profiles. Biol Psychiatry. 2017;81(4):285-95.
Robison AJ, Vialou V, Sun HS, et al. Fluoxetine epigenetically alters the CaMKIIalpha promoter in nucleus accumbens to regulate DeltaFosB binding and antidepressant effects. Neuropsychopharmacology. 2014;39(5):1178-86.
Wilkinson MB, Xiao G, Kumar A, et al. Imipramine treatment and resiliency exhibit similar chromatin regulation in the mouse nucleus accumbens in depression models. J Neurosci. 2009;29(24):7820-32.
Chen B, Dowlatshahi D, MacQueen GM, Wang JF, Young LT. Increased hippocampal BDNF immunoreactivity in subjects treated with antidepressant medication. Biol Psychiatry. 2001;50(4):260-5.
Adachi M, Barrot M, Autry AE, Theobald D, Monteggia LM. Selective loss of brain-derived neurotrophic factor in the dentate gyrus attenuates antidepressant efficacy. Biol Psychiatry. 2008;63(7):642-9.
Monteggia LM, Barrot M, Powell CM, et al. Essential role of brain-derived neurotrophic factor in adult hippocampal function. Proc Natl Acad Sci U S A. 2004;101(29):10827-32.
Monteggia LM, Luikart B, Barrot M, et al. Brain-derived neurotrophic factor conditional knockouts show gender differences in depression-related behaviors. Biol Psychiatry. 2007;61(2):187-97.
Taliaz D, Loya A, Gersner R, Haramati S, Chen A, Zangen A. Resilience to chronic stress is mediated by hippocampal brain-derived neurotrophic factor. J Neurosci. 2011;31(12):4475-83.
Eisch AJ, Bolanos CA, de Wit J, et al. Brain-derived neurotrophic factor in the ventral midbrain-nucleus accumbens pathway: a role in depression. Biol Psychiatry. 2003;54(10):994-1005.
Manji HK, Drevets WC, Charney DS. The cellular neurobiology of depression. Nat Med. 2001;7(5):541-7.
Shah MM. HCN1 channels: a new therapeutic target for depressive disorders? Sci Signal. 2012;5(244):pe44.
Murrough JW, Abdallah CG, Mathew SJ. Targeting glutamate signalling in depression: progress and prospects. Nat Rev Drug Discov. 2017.
Newport DJ, Carpenter LL, McDonald WM, Potash JB, Tohen M, Nemeroff CB. Ketamine and Other NMDA Antagonists: Early Clinical Trials and Possible Mechanisms in Depression. Am J Psychiatry. 2015;172(10):950-66.
Postea O, Biel M. Exploring HCN channels as novel drug targets. Nat Rev Drug Discov. 2011;10(12):903-14.
Baruscotti M, Bottelli G, Milanesi R, DiFrancesco JC, DiFrancesco D. HCN-related channelopathies. Pflugers Arch. 2010;460(2):405-15.
Poolos NP. The Yin and Yang of the H-Channel and Its Role in Epilepsy. Epilepsy Curr. 2004;4(1):3-6.
Jiang YQ, Sun Q, Tu HY, Wan Y. Characteristics of HCN channels and their participation in neuropathic pain. Neurochemical research. 2008;33(10):1979-89.
Noam Y, Bernard C, Baram TZ. Towards an integrated view of HCN channel role in epilepsy. Curr Opin Neurobiol. 2011;21(6):873-9.
Emery EC, Young GT, Berrocoso EM, Chen L, McNaughton PA. HCN2 ion channels play a central role in inflammatory and neuropathic pain. Science. 2011;333(6048):1462-6.
Thorup L, Simonsen U, Grimm D, Hedegaard ER. Ivabradine: Current and Future Treatment of Heart Failure. Basic Clin Pharmacol Toxicol. 2017.
Guha K, Allen CJ, Hartley A, Sharma R. Ivabradine: A Current Overview. Curr Clin Pharmacol. 2016;11(4):241-9.
Bois P, Guinamard R, Chemaly AE, Faivre JF, Bescond J. Molecular regulation and pharmacology of pacemaker channels. Curr Pharm Des. 2007;13(23):2338-49.
Riccioni G, Vitulano N, D'Orazio N. Ivabradine: beyond heart rate control. Adv Ther. 2009;26(1):12-24.
Pape HC. Specific bradycardic agents block the hyperpolarization-activated cation current in central neurons. Neuroscience. 1994;59(2):363-73.
Harris NC, Constanti A. Mechanism of block by ZD 7288 of the hyperpolarization-activated inward rectifying current in guinea pig substantia nigra neurons in vitro. J Neurophysiol. 1995;74(6):2366-78.
Surges R, Freiman TM, Feuerstein TJ. Gabapentin increases the hyperpolarization-activated cation current Ih in rat CA1 pyramidal cells. Epilepsia. 2003;44(2):150-6.
Food and Drug Administration FDA approves Corlanor to treat heart failure. Apr 15, 2015 [Available from: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm442978.htm].
Bucchi A, Tognati A, Milanesi R, Baruscotti M, DiFrancesco D. Properties of ivabradine-induced block of HCN1 and HCN4 pacemaker channels. J Physiol. 2006;572(Pt 2):335-46.
Tibbs GR, Posson DJ, Goldstein PA. Voltage-Gated Ion Channels in the PNS: Novel Therapies for Neuropathic Pain? Trends Pharmacol Sci. 2016;37(7):522-42.
Savelieva I, Camm AJ. I f inhibition with ivabradine : electrophysiological effects and safety. Drug safety. 2008;31(2):95-107.
Jacob S, Nair AB. An Updated Overview on Therapeutic Drug Monitoring of Recent Antiepileptic Drugs. Drugs R D. 2016;16(4):303-16.
Coderre TJ, Kumar N, Lefebvre CD, Yu JS. A comparison of the glutamate release inhibition and anti-allodynic effects of gabapentin, lamotrigine, and riluzole in a model of neuropathic pain. J Neurochem. 2007;100(5):1289-99.
Atalay H, Solak Y, Biyik Z, Gaipov A, Guney F, Turk S. Cross-over, open-label trial of the effects of gabapentin versus pregabalin on painful peripheral neuropathy and health-related quality of life in haemodialysis patients. Clin Drug Investig. 2013;33(6):401-8.
Maneuf YP, Luo ZD, Lee K. alpha2delta and the mechanism of action of gabapentin in the treatment of pain. Semin Cell Dev Biol. 2006;17(5):565-70.
Kremer M, Salvat E, Muller A, Yalcin I, Barrot M. Antidepressants and gabapentinoids in neuropathic pain: Mechanistic insights. Neuroscience. 2016;338:183-206.
Pande AC, Crockatt JG, Janney CA, Werth JL, Tsaroucha G. Gabapentin in bipolar disorder: a placebo-controlled trial of adjunctive therapy. Gabapentin Bipolar Disorder Study Group. Bipolar Disord. 2000;2(3 Pt 2):249-55.
Carta MG, Hardoy MC, Hardoy MJ, Grunze H, Carpiniello B. The clinical use of gabapentin in bipolar spectrum disorders. J Affect Disord. 2003;75(1):83-91.
Stanton SP, Keck PE, Jr., McElroy SL. Treatment of acute mania with gabapentin. Am J Psychiatry. 1997;154(2):287.
McElroy SL, Soutullo CA, Keck PE, Jr., Kmetz GF. A pilot trial of adjunctive gabapentin in the treatment of bipolar disorder. Ann Clin Psychiatry. 1997;9(2):99-103.
Ghaemi SN, Katzow JJ, Desai SP, Goodwin FK. Gabapentin treatment of mood disorders: a preliminary study. J Clin Psychiatry. 1998;59(8):426-9.
Erfurth A, Kammerer C, Grunze H, Normann C, Walden J. An open label study of gabapentin in the treatment of acute mania. J Psychiatr Res. 1998;32(5):261-4.
Young LT, Robb JC, Patelis-Siotis I, MacDonald C, Joffe RT. Acute treatment of bipolar depression with gabapentin. Biol Psychiatry. 1997;42(9):851-3.
Houghton KT, Forrest A, Awad A, et al. Biological rationale and potential clinical use of gabapentin and pregabalin in bipolar disorder, insomnia and anxiety: protocol for a systematic review and meta-analysis. BMJ Open. 2017;7(3):e013433.
Frye MA, Ketter TA, Kimbrell TA, et al. A placebo-controlled study of lamotrigine and gabapentin monotherapy in refractory mood disorders. J Clin Psychopharmacol. 2000;20(6):607-14.
Vigo DV, Baldessarini RJ. Anticonvulsants in the treatment of major depressive disorder: an overview. Harv Rev Psychiatry. 2009;17(4):231-41.
Brannon N, Labbate L, Huber M. Gabapentin treatment for posttraumatic stress disorder. Can J Psychiatry. 2000;45(1):84.
Hamner MB, Brodrick PS, Labbate LA. Gabapentin in PTSD: a retrospective, clinical series of adjunctive therapy. Ann Clin Psychiatry. 2001;13(3):141-6.
Karam-Hage M, Brower KJ. Open pilot study of gabapentin versus trazodone to treat insomnia in alcoholic outpatients. Psychiatry Clin Neurosci. 2003;57(5):542-4.
Karam-Hage M, Brower KJ. Gabapentin treatment for insomnia associated with alcohol dependence. Am J Psychiatry. 2000;157(1):151.
Pande AC, Pollack MH, Crockatt J, et al. Placebo-controlled study of gabapentin treatment of panic disorder. J Clin Psychopharmacol. 2000;20(4):467-71.
Pande AC, Davidson JR, Jefferson JW, et al. Treatment of social phobia with gabapentin: a placebo-controlled study. J Clin Psychopharmacol. 1999;19(4):341-8.
Pollack MH, Matthews J, Scott EL. Gabapentin as a potential treatment for anxiety disorders. Am J Psychiatry. 1998;155(7):992-3.
Cora-Locatelli G, Greenberg BD, Martin J, Murphy DL. Gabapentin augmentation for fluoxetine-treated patients with obsessive-compulsive disorder. J Clin Psychiatry. 1998;59(9):480-1.
Cora-Locatelli G, Greenberg BD, Martin JD, Murphy DL. Rebound psychiatric and physical symptoms after gabapentin discontinuation. J Clin Psychiatry. 1998;59(3):131.
Bowden CL, Calabrese JR, McElroy SL, et al. The efficacy of lamotrigine in rapid cycling and non-rapid cycling patients with bipolar disorder. Biol Psychiatry. 1999;45(8):953-8.
Calabrese JR, Bowden CL, McElroy SL, et al. Spectrum of activity of lamotrigine in treatment-refractory bipolar disorder. Am J Psychiatry. 1999;156(7):1019-23.
Calabrese JR, Bowden CL, Sachs GS, Ascher JA, Monaghan E, Rudd GD. A double-blind placebo-controlled study of lamotrigine monotherapy in outpatients with bipolar I depression. Lamictal 602 Study Group. J Clin Psychiatry. 1999;60(2):79-88.
Pichler EM, Hattwich G, Grunze H, Muehlbacher M. Safety and tolerability of anticonvulsant medication in bipolar disorder. Expert Opin Drug Saf. 2015;14(11):1703-24.
Geddes JR, Calabrese JR, Goodwin GM. Lamotrigine for treatment of bipolar depression: independent meta-analysis and meta-regression of individual patient data from five randomised trials. Br J Psychiatry. 2009;194(1):4-9.
Walden J, Hesslinger B, van Calker D, Berger M. Addition of lamotrigine to valproate may enhance efficacy in the treatment of bipolar affective disorder. Pharmacopsychiatry. 1996;29(5):193-5.
Suppes T, Brown ES, McElroy SL, et al. Lamotrigine for the treatment of bipolar disorder: a clinical case series. J Affect Disord. 1999;53(1):95-8.
Fogelson DL, Sternbach H. Lamotrigine treatment of refractory bipolar disorder. J Clin Psychiatry. 1997;58(6):271-3.
Sporn J, Sachs G. The anticonvulsant lamotrigine in treatment-resistant manic-depressive illness. J Clin Psychopharmacol. 1997;17(3):185-9.
Kusumakar V, Yatham LN. An open study of lamotrigine in refractory bipolar depression. Psychiatry Res. 1997;72(2):145-8.
Labbate LA, Rubey RN. Lamotrigine for treatment-refractory bipolar disorder. Am J Psychiatry. 1997;154(9):1317.
Ketter TA, Manji HK, Post RM. Potential mechanisms of action of lamotrigine in the treatment of bipolar disorders. J Clin Psychopharmacol. 2003;23(5):484-95.
van der Loos ML, Mulder P, Hartong EG, et al. Long-term outcome of bipolar depressed patients receiving lamotrigine as add-on to lithium with the possibility of the addition of paroxetine in nonresponders: a randomized, placebo-controlled trial with a novel design. Bipolar Disord. 2011;13(1):111-7.
van der Loos ML, Mulder PG, Hartong EG, et al. Efficacy and safety of lamotrigine as add-on treatment to lithium in bipolar depression: a multicenter, double-blind, placebo-controlled trial. J Clin Psychiatry. 2009;70(2):223-31.
Pinto OC, Akiskal HS. Lamotrigine as a promising approach to borderline personality: an open case series without concurrent DSM-IV major mood disorder. J Affect Disord. 1998;51(3):333-43.
Preston GA, Marchant BK, Reimherr FW, Strong RE, Hedges DW. Borderline personality disorder in patients with bipolar disorder and response to lamotrigine. J Affect Disord. 2004;79(1-3):297-303.
Leiberich P, Nickel MK, Tritt K, Pedrosa Gil F. Lamotrigine treatment of aggression in female borderline patients, Part II: an 18-month follow-up. J Psychopharmacol. 2008;22(7):805-8.
Tritt K, Nickel C, Lahmann C, et al. Lamotrigine treatment of aggression in female borderline-patients: a randomized, double-blind, placebo-controlled study. J Psychopharmacol. 2005;19(3):287-91.
Hahn CG, Gyulai L, Baldassano CF, Lenox RH. The current understanding of lamotrigine as a mood stabilizer. J Clin Psychiatry. 2004;65(6):791-804.
Stahl SM. Anticonvulsants as mood stabilizers and adjuncts to antipsychotics: valproate, lamotrigine, carbamazepine, and oxcarbazepine and actions at voltage-gated sodium channels. J Clin Psychiatry. 2004;65(6):738-9.
Kolivakis TT, Beauclair L, Margolese HC, Chouinard G. Long-term lamotrigine adjunctive to antipsychotic monotherapy in schizophrenia: further evidence. Can J Psychiatry. 2004;49(4):280.
Tiihonen J, Hallikainen T, Ryynanen OP, et al. Lamotrigine in treatment-resistant schizophrenia: a randomized placebo-controlled crossover trial. Biol Psychiatry. 2003;54(11):1241-8.
Premkumar TS, Pick J. Lamotrigine for schizophrenia. The Cochrane database of systematic reviews. 2006(4):CD005962.
Reid JG, Gitlin MJ, Altshuler LL. Lamotrigine in psychiatric disorders. J Clin Psychiatry. 2013;74(7):675-84.
Tiihonen J, Wahlbeck K, Kiviniemi V. The efficacy of lamotrigine in clozapine-resistant schizophrenia: a systematic review and meta-analysis. Schizophr Res. 2009;109(1-3):10-4.
Mihara K, Nakamura A, Nemoto K, et al. Lamotrigine augmentation therapy in a case with treatment-resistant unipolar depression that showed insufficient response to electroconvulsive therapy. Psychiatry Clin Neurosci. 2016;70(2):126.
Nakamura A, Mihara K, Nagai G, et al. Prediction of an Optimal Dose of Lamotrigine for Augmentation Therapy in Treatment-Resistant Depressive Disorder From Plasma Lamotrigine Concentration at Week 2. Ther Drug Monit. 2016;38(3):379-82.
Ivkovic M, Damjanovic A, Jovanovic A, Cvetic T, Jasovic-Gasic M. Lamotrigine versus lithium augmentation of antidepressant therapy in treatment-resistant depression: efficacy and tolerability. Psychiatr Danub. 2009;21(2):187-93.
Barbee JG, Thompson TR, Jamhour NJ, et al. A double-blind placebo-controlled trial of lamotrigine as an antidepressant augmentation agent in treatment-refractory unipolar depression. J Clin Psychiatry. 2011;72(10):1405-12.
Barbee JG, Jamhour NJ. Lamotrigine as an augmentation agent in treatment-resistant depression. J Clin Psychiatry. 2002;63(8):737-41.
Hertzberg MA, Butterfield MI, Feldman ME, et al. A preliminary study of lamotrigine for the treatment of posttraumatic stress disorder. Biol Psychiatry. 1999;45(9):1226-9.
Kumar TC, Khanna S. Lamotrigine augmentation of serotonin re-uptake inhibitors in obsessive-compulsive disorder. Aust N Z J Psychiatry. 2000;34(3):527-8.
Bruno A, Mico U, Pandolfo G, et al. Lamotrigine augmentation of serotonin reuptake inhibitors in treatment-resistant obsessive-compulsive disorder: a double-blind, placebo-controlled study. J Psychopharmacol. 2012;26(11):1456-62.
Brown ES, Nejtek VA, Perantie DC, Orsulak PJ, Bobadilla L. Lamotrigine in patients with bipolar disorder and cocaine dependence. J Clin Psychiatry. 2003;64(2):197-201.
Brown ES, Perantie DC, Dhanani N, Beard L, Orsulak P, Rush AJ. Lamotrigine for bipolar disorder and comorbid cocaine dependence: a replication and extension study. J Affect Disord. 2006;93(1-3):219-22.
Brown ES, Sunderajan P, Hu LT, Sowell SM, Carmody TJ. A randomized, double-blind, placebo-controlled, trial of lamotrigine therapy in bipolar disorder, depressed or mixed phase and cocaine dependence. Neuropsychopharmacology. 2012;37(11):2347-54.
Margolin A, Avants SK, DePhilippis D, Kosten TR. A preliminary investigation of lamotrigine for cocaine abuse in HIV-seropositive patients. Am J Drug Alcohol Abuse. 1998;24(1):85-101.
Krupitsky EM, Rudenko AA, Burakov AM, et al. Antiglutamatergic strategies for ethanol detoxification: comparison with placebo and diazepam. Alcohol Clin Exp Res. 2007;31(4):604-11.
Kalyoncu A, Mirsal H, Pektas O, Unsalan N, Tan D, Beyazyurek M. Use of lamotrigine to augment clozapine in patients with resistant schizophrenia and comorbid alcohol dependence: a potent anti-craving effect? J Psychopharmacol. 2005;19(3):301-5.
Rubio G, Lopez-Munoz F, Alamo C. Effects of lamotrigine in patients with bipolar disorder and alcohol dependence. Bipolar Disord. 2006;8(3):289-93.
Brower KJ, Myra Kim H, Strobbe S, Karam-Hage MA, Consens F, Zucker RA. A randomized double-blind pilot trial of gabapentin versus placebo to treat alcohol dependence and comorbid insomnia. Alcohol Clin Exp Res. 2008;32(8):1429-38.
Brodie MS, Pesold C, Appel SB. Ethanol directly excites dopaminergic ventral tegmental area reward neurons. Alcohol Clin Exp Res. 1999;23(11):1848-52.
Hopf FW, Martin M, Chen BT, Bowers MS, Mohamedi MM, Bonci A. Withdrawal from intermittent ethanol exposure increases probability of burst firing in VTA neurons in vitro. J Neurophysiol. 2007;98(4):2297-310.
Tu Y, Kroener S, Abernathy K et al. Ethanol inhibits persistent activity in prefrontal cortical neurons. J Neurosci. 2007;27(17):4765-75.
Yan H, Li Q, Fleming R, Madison RD, Wilson WA, Swartzwelder HS. Developmental sensitivity of hippocampal interneurons to ethanol: involvement of the hyperpolarization-activated current, Ih. J Neurophysiol. 2009;101(1):67-83.
Berlin RK, Butler PM, Perloff MD. Gabapentin Therapy in Psychiatric Disorders: A Systematic Review. Prim Care Companion CNS Disord. 2015;17(5).
Williams AD, Jung S, Poolos NP. Protein kinase C bidirectionally modulates Ih and hyperpolarization-activated cyclic nucleotide-gated (HCN) channel surface expression in hippocampal pyramidal neurons. J Physiol. 2015;593(13):2779-92.
Chevaleyre V, Castillo PE. Assessing the role of Ih channels in synaptic transmission and mossy fiber LTP. Proc Natl Acad Sci U S A. 2002;99(14):9538-43.
Mellor J, Nicoll RA, Schmitz D. Mediation of hippocampal mossy fiber long-term potentiation by presynaptic Ih channels. Science. 2002;295(5552):143-7.
Chen C. ZD7288 inhibits postsynaptic glutamate receptor-mediated responses at hippocampal perforant path-granule cell synapses. Eur J Neurosci. 2004;19(3):643-9.
Biederman J, Melmed RD, Patel A, et al. A randomized, double-blind, placebo-controlled study of guanfacine extended release in children and adolescents with attention-deficit/hyperactivity disorder. Pediatrics. 2008;121(1):e73-84.
Sallee FR, McGough J, Wigal T, Donahue J, Lyne A, Biederman J. Guanfacine extended release in children and adolescents with attention-deficit/hyperactivity disorder: a placebo-controlled trial. J Am Acad Child Adolesc Psychiatry. 2009;48(2):155-65.
Arnsten AF, Wang M. Targeting Prefrontal Cortical Systems for Drug Development: Potential Therapies for Cognitive Disorders. Annu Rev Pharmacol Toxicol. 2016;56:339-60.
Connor DF, Arnsten AF, Pearson GS, Greco GF. Guanfacine extended release for the treatment of attention-deficit/hyperactivity disorder in children and adolescents. Expert Opin Pharmacother. 2014;15(11):1601-10.
Arnsten AF, Jin LE. Guanfacine for the treatment of cognitive disorders: a century of discoveries at Yale. Yale J Biol Med. 2012;85(1):45-58.
Han Y, Lyman K, Clutter M et al. Identification of Small-Molecule Inhibitors of Hyperpolarization-Activated Cyclic Nucleotide-Gated Channels. J Biomol Screen. 2015;20(9):1124-31.
Santoro B, Hu L, Liu H, et al. TRIP8b regulates HCN1 channel trafficking and gating through two distinct C-terminal interaction sites. J Neurosci. 2011;31(11):4074-86.
Bankston JR, Camp SS, DiMaio F, Lewis AS, Chetkovich DM, Zagotta WN. Structure and stoichiometry of an accessory subunit TRIP8b interaction with hyperpolarization-activated cyclic nucleotide-gated channels. Proc Natl Acad Sci U S A. 2012;109(20):7899-904.
Han Y, Lyman KA, Clutter M et al. Method for Identifying Small Molecule Inhibitors of the Protein-protein Interaction Between HCN1 and TRIP8b. J Vis Exp. 2016(117).
Han Y, Noam Y, Lewis AS et al. Trafficking and gating of hyperpolarization-activated cyclic nucleotide-gated channels are regulated by interaction with tetratricopeptide repeat-containing Rab8b-interacting protein (TRIP8b) and cyclic AMP at distinct sites. J Biol Chem. 2011;286(23):20823-34.
Acknowledgments
This work was supported by grants from the National Institute of Mental Health (M.H.H., R21MH112081; S.M.K., F31MH108326), and the Brain and Behavior Research Foundation (M.H.H., NARSAD). We thank Dr. Madeline L. Pfau for critical reading of the review.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 1224 kb)
Rights and permissions
About this article

Cite this article
Ku, S.M., Han, MH. HCN Channel Targets for Novel Antidepressant Treatment. Neurotherapeutics 14, 698–715 (2017). https://doi.org/10.1007/s13311-017-0538-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-017-0538-7