
Abstract
Axonal excitability testing provides in vivo assessment of axonal ion channel function and membrane potential. Excitability techniques have provided insights into the pathophysiological mechanisms underlying the development of neurodegeneration and clinical features of amyotrophic lateral sclerosis (ALS) and related neuromuscular disorders. Specifically, abnormalities of Na+ and K+ conductances contribute to development of membrane hyperexcitability in ALS, thereby leading to symptom generation of muscle cramps and fasciculations, in addition to promoting a neurodegenerative cascade via Ca2+-mediated processes. Modulation of axonal ion channel function in ALS has resulted in significant symptomatic improvement that has been accompanied by stabilization of axonal excitability parameters. Separately, axonal ion channel dysfunction evolves with disease progression and correlates with survival, thereby serving as a potential therapeutic biomarker in ALS. The present review provides an overview of axonal excitability techniques and the physiological mechanisms underlying membrane excitability, with a focus on the role of axonal ion channel dysfunction in motor neuron disease and related neuromuscular diseases.
Similar content being viewed by others
Introduction
Axonal excitability techniques provide in vivo assessment of axonal membrane and ion channel function, yielding complementary information to conventional nerve conduction studies [1, 2]. Excitability techniques have been utilized across the spectrum of neurologic disorders, providing invaluable insights into the patholophysiologic processes underlying a host of neuromuscular disorders [3]. Specifically, in motor neuron disease, also termed amyotrophic lateral sclerosis (ALS), axonal excitability techniques have provided vital information on axonal ion channel dysfunction, which has been of pathophysiologic significance [4]. Importantly, abnormalities of axonal Na+ and K+ conductances have been consistently identified in ALS [5–10], resulting in symptom generation such as muscle cramps and fasciculations, as well as serving as potential therapeutic targets [11]. Similarly, axonal excitability parameters have yielded critical pathophysiological insights in ALS-mimicking disorders [3, 12–15] Table 1.
Importantly, the potential use of excitability techniques as a biomarker to provide information about axonal function relevant to the development of disease, disease progression, and treatment response may prove to be a useful endpoint in clinical trials, particularly in monitoring the effects of therapeutic agents. In this review article, we provide an overview of the physiological mechanisms underlying axonal excitability, and focus on the importance of axonal ion channel dysfunction as a pathogenic and prognostic biomarker in ALS and related neuromuscular diseases.
The Clinical Assessment of Axonal Excitability
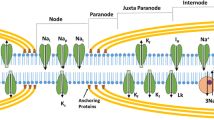
The availability and utility of axonal excitability techniques have progressed markedly over the last 20 years, with the advent of commercialization of equipment and specialized software. In contrast to monitoring maximal compound amplitudes, as occurs with conventional nerve conduction studies, excitability studies examine the function of constituent axonal conductances, assessing changes in current required to produce a constant compound muscle action potential (CMAP) response. Accordingly, excitability studies provide information about axonal function, membrane potential, and ion channel properties (Fig. 1). In clinically applied protocols, the constant response method of assessing excitability is typically utilized [2]. Threshold tracking relies on the definition of “threshold” as the stimulus strength required to produce a CMAP response of a specified target amplitude. In threshold tracking protocols, the current required to produce the target response (threshold) is tracked online, with the stimulus current altered proportionally to the discrepancy between the target and actual response [9]. Software enabling semiautomated threshold tracking has been utilized in axonal excitability protocols via the QTracS program (Institute of Neurology, UK). The modern TROND protocol was developed by Kiernan, Burke, and Bostock [16] during the 1999 Nordic course in clinical axonal electrophysiology held in Trondheim, and provides a package of excitability techniques for assessing motor nerve excitability, while sensory nerve excitability protocols were subsequently developed [17]. Interpretation of axonal excitability studies can be complex, owing to the multifactorial nature of changes in ionic conductances, as well as passive and active membrane properties. Accordingly, the development of a mathematical model of the excitability properties of human axons has assisted in interpretation of axonal excitability changes [18–20].

Schematic of axon structure and ion channel distribution, demonstrating the node of Ranvier with high density of voltage-gated Na+ channels [both transient (Nat) and persistent (Nap)] and slow K+ channels (Ks). The internode under the myelin sheath is depicted with fast K+ channels (Kf) located adjacent to the node, the hyperpolarization activated cation conductance (Ih), slow K+ channels, and voltage-independent leak conductances (Lk) along with the energy dependent Na+/K+ pump and the Na+/Ca2+ exchanger
The key axonal excitability parameters include threshold, strength-duration time constant, rheobase, threshold electrotonus, recovery cycle, and current/threshold relationship (Fig. 2). These protocols rely on an accurate stimulus response curve in which the current is progressively increased from zero until the CMAP amplitude is supramaximal, producing a sigmoid curve.

a Strength-duration time constant (τsd) plotted as the negative intercept on the x-axis of the linear relationship between stimulus width and threshold charge. b Recovery cycle plotted with the relative refractory period, superexcitability, and subexcitability depicted on the figure. c Threshold electrotonus in response to polarizing currents set to ± 40% of threshold. The initial response to polarization (F phase) is followed by a slow change in threshold (S1 phase), with threshold increase in the hyperpolarizing direction (plotted downwards) and threshold decrease in the depolarizing direction (plotted upwards), followed by accommodation to depolarization (S2 phase). d Current threshold (I/V) relationship plotted with response to depolarizing current in the upper right quadrant and response to hyperpolarizing current in the lower left quadrant
Threshold
reflects the amount of stimulus current required to activate an axon and produce a CMAP of a specific amplitude, usually 40% of the maximal CMAP amplitude [1, 2]. The threshold is related to membrane potential, as membrane hyperpolarization increases and depolarization decreases the threshold. Additional factors such as nerve ischemia may also impact on threshold, and, consequently, assessment of axonal excitability parameters (discussed later) is required to provide a clear depiction of membrane excitability [21–25].
The strength-duration time constant (τ SD ), also termed chronaxie, is a membrane time constant, assessing the relationship between stimulus strength and duration [26–28]. τSD measures the rate at which the threshold current declines as the stimulus duration is increased and can be calculated using the ratio between two different stimulus durations according to Weiss’ law (Fig. 2a) [29]. Rheobase is defined as the minimum current strength (mA) required to produce a response for a stimulus of infinite duration [1, 7]. Both τSD and rheobase are nodal properties dependent on passive membrane properties and persistent Na+ channel conductances (INaP) [30].
The persistent Na+ current represents a fraction of the total Na+ current (approximately 1–2% of total current) and does not inactivate, therefore influencing membrane excitability [30–32]. Voltage-gated Na+ channels are composed of 1 alpha (α) and 4 beta (β1–4) subunits [33, 34], with the α subunits organized in 4 homologous domains (I–IV), consisting of 6 transmembrane α helices (S1–S6) and a selectivity filter located between S5 and S6 segments. Na+ channel inactivation is mediated by an intracellular loop connecting domains III and IV, which blocks the pore from inside during continuous membrane depolarization. A total of 10 Na+ channel isoforms have been identified (Nav 1.1–Nav 1.9 and Nax [33–35]), and the Nav1.6 isoform expressed at the nodes of Ranvier is thought to produce INaP [33–36].
Changes in the τSD and rheobase may be influenced by multiple factors—with changes in resting membrane potential, nerve geometry, and structure, such as axonal loss or demyelination, and discrete changes in nodal Na+ conductances producing modification of membrane excitability [37–39].
The recovery cycle of excitability (Fig. 2b) determines the profile of excitability changes following an action potential [1]. Utilizing paired pulses, the interstimulus interval is progressively altered between 2 ms and 200 ms, revealing a characteristic pattern of excitability changes [40]. When the interstimulus interval is short (<4 ms), the current required to produce a subsequent action potential is increased [40]. The axon is completely refractory for a period of 0.5 to 1 ms during which no action potentials can be generated, owing to the inactivation of transient voltage-gated Na+ channels, termed the absolute refractory period [21]. Following this absolute refractory period, there is development of the relative refractory period, lasting up to 4 ms (Fig. 2b), during which time action potential generation is possible but more difficult as Na+ channels recover from inactivation [41]. The extent of the relative refractory period can be measured as an increase in current required to generate a CMAP (termed refractoriness) [41, 42].
Transient Na+ channel kinetics, and, accordingly, refractoriness, may be influenced by changes in membrane potential, whereby membrane depolarization increases the extent of Na+ channel inactivation and refractoriness, and hyperpolarization reduces refractoriness by reducing the degree of inactivated Na+ channels [1]. Na+ channel inactivation and refractoriness are also sensitive to temperature changes, and reduction in limb temperature markedly increases refractoriness [43, 44].
The next phase of the recovery cycle has been termed superexcitability (lasting 15 ms and peaking between 5 and 7 ms; Fig. 2b), reflecting enhanced membrane excitability due to depolarizing afterpotential formation [1, 40, 45, 46]. Superexcitability appears mediated by re-excitation of the nodal membrane produced by the depolarizing afterpotential, leading to discharge of current stored on the internodal membrane following an action potential (akin to capacitive charging of the internode). The amplitude and time course of the afterpotential is limited by activation of fast paranodal K+ channels [47]. Changes in membrane potential influence superexcitability, with membrane depolarization reducing superexcitability through activation of fast K+ channels [45], and hyperpolarization increasing superexcitability [48].
The final phase of the recovery cycle has been termed subexcitability, a period of reduced axonal excitability lasting approximately 100 ms (Fig. 2b). Subexcitability is due to reduced membrane excitability due to the slow activation of K+ channels [46]. Membrane depolarization increases subexcitability, via changes to the electrochemical gradient for K+. However, conversely, membrane depolarization secondary to increases in extracellular K+ concentration, as occurs with ischemia or renal failure, results in subexcitability reduction [48–50].
Threshold Electrotonus (TE)
describes the changes in threshold produced by long-lasting subthreshold currents, providing insight into both nodal and internodal membrane conductances (Fig. 2c) [1, 2, 51]. TE utilizes a 100-ms subthreshold current pulse with sequential depolarizing and hyperpolarizing currents set to +20% or 40% and −20 or 40% of the threshold current [4]. The changes in the threshold current can be measured at varying time intervals to reveal the excitability profile in response to subthreshold conditioning currents [16, 17].
In response to depolarizing currents, threshold is immediately reduced until mitigated by the activity of slow K+ channels which accommodate the effect of depolarization [51]. The initial fast response (F phase; Fig. 3c) reflects rapid changes in membrane threshold at the node. This is followed by slower changes in threshold over tens of milliseconds with depolarizing current, termed the S1 phase, reflecting the spread of current to the internodal membrane and peaking 20 ms after the onset of the current pulse. After this threshold begins to return to baseline, termed the S2 phase, reflecting activation of nodal and internodal slow K+ channels [2, 51].

a The strength-duration time constant reflects nodal persistent Na+ channel conductances and was significantly increased in patients with amyotrophic lateral sclerosis (ALS; filled circles) compared to controls (clear squares). b Mean SDTC was significantly increased in both familial ALS and sporadic ALS compared with controls (*p < 0.05; **p < 0.01). c Recovery cycle waveforms in patients with motor neuron disease (MND; filled circles) compared with controls (clear squares) demonstrating increased superexcitability in patients with ALS. d Threshold electrotonus waveforms in patients with ALS (filled circles) compared with controls (clear squares) demonstrating increased threshold change in threshold electrotonus depolarizing direction (TEd) 90–100 ms in patients with ALS
With hyperpolarizing current pulses, threshold is increased proportional to the current pulse (F phase). Subsequent closure of K+ channels with hyperpolarization further increases threshold [2]. During the S1 phase, further hyperpolarization occurs, peaking at 100 to 150 ms after the onset of the subthreshold conditioning current and returning to baseline. After prolonged hyperpolarization, the S3 phase commences as a result of activation of the hyperpolarizing-activated cation conductance (Ih) [1–3, 52]. Activation of Ih following prolonged hyperpolarization attenuates the increase in threshold [52] and limits the extent of hyperpolarization in response to high-frequency activity [46]. As high-frequency activity can induce conduction failure in axons with a reduced safety factor of transmission, the Ih current may be critical in preventing conduction failure [53]. Termination of subthreshold conditioning currents results in overshoot of depolarizing and hyperpolarizing thresholds, mediated by deactivation of slow K+ channels, and the slow kinetics of Ih [1].
As with other axonal excitability parameters, TE is influenced by changes in membrane potential. Specifically, membrane depolarization activates K+ channels, resulting in reduction of the S1 phase in both depolarizing and hyperpolarizing directions, causing a “fanning in” appearance of TE [1, 2, 48]. Conversely, hyperpolarization closes K+ channels, increasing both S1 and S2 phases and producing a “fanning out” appearance of TE [1, 2, 48].
The current threshold relationship (I/V relationship) utilizes a range of depolarizing and hyperpolarizing current strengths over long-duration subthreshold 200-ms pulses [48]. Currents are applied from +50% (depolarizing) to −100% (hyperpolarizing) of the control threshold in 10% steps (Fig. 2d) [16, 17]. The I/V relationship estimates rectifying properties of both nodal and internodal axonal segments [2]. Accommodation to depolarization occurs via fast and slow K+ channel activation, termed outward rectification [54]. While slow K+ channels are located at higher density at the node and fast K+ channels are located at the juxtaparanode adjacent to the node, both act broadly to reduce excitability following impulse conduction [55]. With hyperpolarizing currents, inward rectification occurs owing to the activation of Ih [52].
Axonal Excitability Changes in ALS
Disturbances in membrane excitability and axonal ion-channel function have been identified from the earliest application of axonal excitability techniques in patients with ALS [5]. Subsequently, numerous studies have reported axonal dysfunction in patients with ALS characterized by prolonged τSD, increased superexcitability, and abnormalities of threshold electrotonus [6–8, 56–58]. These changes have been specifically attributed to aberrant ionic conductances, particularly increased Na+ and decreased axonal K+ conductances, a profile that may underlie neurodegeneration and contribute to symptoms, such as fasciculations and muscle cramps [59].
Prolongation of τSD has been consistently identified in sporadic ALS and linked to neurodegeneration (Fig. 3a) [6, 7, 56, 60–63].In addition, prolongation of τSD has been identified in atypical ALS phenotypes, such as the flail arm variant [64], as well as familial forms of ALS linked to mutations in SOD1 and c9orf72 (Fig. 3b) [9, 56, 65]. The increase of τSD was most prominent in patients with ALS with a moderate degree of lower motor neuron dysfunction, suggesting that axonal hyperexcitability is an early feature in ALS [8]. Interestingly, the split-hand phenomenon [66–68], a specific sign in ALS characterized by preferential wasting of the abductor pollicis brevis muscle, was reported to be associated with greater increases in the τSD in median motor nerves [63, 69].
Upregulation of persistent Na+ conductances appears to underlie the prolongation of τSD in ALS [60, 70]. In addition, a reduction in fast and slow K+ channel conductances has also been proposed as a contributing mechanism in prolongation of τSD by inducing membrane depolarization [8]. These changes in axonal excitability have been linked to axonal degeneration and development of symptoms such as fasciculations and muscle cramps [8, 10, 56]. Recent studies have suggested that the increase in axonal excitability was an adaptive and potentially neuroprotective mechanisms [71]. This seems unlikely given that treatment with Na+ blocking agents, flecainide and mexiletine (discussed below), was not associated with a more rapid progression of the disease [11, 72, 73]. Importantly, τSD appears to be a robust biomarker of survival in ALS [74], whereby a prolonged τSD was associated with a significantly shorter survival, further underscoring the pathogenic basis of axonal hyperexcitability in ALS [80]. Similarly, rapid functional decline in motor performance has also been linked to prolonged τSD [75], further suggesting a link between excitability and clinical progression.
In concert with prolonged τSD, changes in threshold electrotonus have been identified in sporadic ALS, although the pattern of changes appears heterogeneous [5, 6, 8, 76, 77]. Specifically, a “fanned out” appearance of TE, whereby greater threshold changes were evident with depolarizing and hyperpolarizing subthreshold currents, has been documented in ALS [6, 77]. In contrast, others have documented greater changes only in response to subthreshold depolarizing currents (Fig. 3d) [5, 6]. A sudden reduction of membrane excitability in conjunction with an abrupt increase in threshold has also been reported in ALS [5], although such changes appear to be an infrequent finding [4].
TE changes in ALS evolve with disease progression, as evidenced by an increase in depolarizing TE with longitudinal follow-up over 3 months [58]. The longitudinal changes in TE were accompanied by axonal degeneration and functional decline, thereby implying a pathogenic role for TE changes in ALS. In addition, “pseudonormalization” of TE changes has also been reported with disease progression and associated with axonal degeneration [4], with the most hyperexcitable axons degenerating with advanced disease. Underscoring this notion is the greater variability of axonal excitability at different target amplitudes, with preferential hyperexcitability evident in low-threshold axons [78]. Importantly, these longitudinal changes in TE could account for the variability reported in previous axonal excitability studies in ALS [5, 78], and suggest that targeting the “hyperexcitable” axons early in the disease process could prove therapeutically useful. Separately, the TE changes in ALS appear to be more prominent in distal axonal segments [79], suggesting a propensity for axonal hyperexcitability to develop distally, and potentially explaining the notion that fasciculations are generated distally in the axon [59, 80]. A similar pattern of TE changes were reported in SOD1 and c9orf72 familial ALS, and were linked to neurodegeneration [9, 56]. In contrast, no significant TE changes were evident in asymptomatic mutation (SOD1 and c9orf72) carriers, suggesting that factors other than inheritance of genetic mutations are required for the development of such excitability changes in ALS.
In addition to changes in TE, significant abnormalities of the recovery cycle of excitability have also been reported in sporadic ALS. Specifically, an increase in superexcitability has accompanied changes in TE (Fig. 3c) [6], although this has not been a consistent finding [9], further underscoring the heterogeneity of the disease process. In part, this heterogeneity may develop in the context of dying and degenerating axons. Importantly, the increase in superexcitability appears more prominent with disease progression and is also linked to functional decline [58]. Interestingly, increased superexcitability was reported in SOD1- but not c9orf72-associated familial ALS [9, 10, 56]. As with TE, asymptomatic mutation carriers exhibited a normal level of superexcitability, underscoring the notion that axonal ion channel dysfunction develops following disease onset in familial ALS.
Mathematical modeling studies have suggested that reduction of nodal and internodal slow K+ conductances underlies the abnormalities of TE in sporadic and familial ALS cohorts [8]. Of further relevance, techniques utilizing double conditioning pulses during the recovery cycle have provided supporting evidence of impairment of slow K+ conductances in ALS [81]. Separately, increases of superexcitability in ALS reflect dysfunction of paranodal fast K+ channels [4]. Support for such an explanation has been provided by gene expression studies documenting reduced mRNA expression of K+ channel genes, both paranodal fast K+ channel genes (KCNA1 and KCNA2) and the nodal K+ channel gene (KCNQ2), in spinal motor neurons of patients with ALS [82]. Of further relevance, immunohistochemical studies have disclosed reduced axonal expression of K+ channels in patients with sporadic ALS [83].
Pathophysiological Basis of Excitability Changes in ALS
Given that fasciculations and muscle cramps are prominent features of ALS that reflect ectopic activity in motor axons [59, 80, 84], abnormalities of K+ and Na+ ion channel function in ALS is likely to underlie the generation of such symptoms. The reduction of axonal K+ currents would decrease the tendency to membrane hyperpolarization, while upregulation of persistent Na+ conductances would increase the depolarizing drive, leading to membrane hyperexcitability [85]. Importantly, reduction of K+ currents has been associated with the generation of fasciculations in ALS [5], while axonal hyperexcitability was induced by presynaptic blockade of K+ channels [86]. In addition, blockade of persistent Na+ conductances with mexiletine led to a dose-dependent reduction in muscle cramp frequency and severity [11], further underscoring the clinical importance of the axonal excitability changes in ALS.
Of further relevance, the changes in axonal ion channel function are associated with neurodegeneration in ALS [4, 87]. Although the precise mechanisms by which the axonal excitability changes lead to neurodegeneration remain to be elucidated, it has been postulated that an influx of Na+ ions results in reverse operation of the Na+/Ca2+ exchanger, with the resultant accumulation of intra-axonal accumulation of Ca2+ ions and, ultimately, activation of Ca2+-dependent enzyme pathways leading to motor neuron degeneration [88–91].Support for such a mechanism was provided by transgenic ALS mouse model studies disclosing a global reduction of Na+/K+-ATPase activity [92], leading to an increase in intracellular Na+ ion concentration [89], and thereby resulting in Ca2+-mediated neurodegeneration. Importantly, expression of the functional Na+/K+ ATPase appears to be neuroprotective in transgenic mouse models, while reduced expression was documented in the spinal cord of patients with sporadic and familial ALS [92]. Of further relevance, axons in the SOD1G127X mouse model appear to be less resistant to activity-induced changes in intracellular ion concentrations and degeneration [93], underscoring the pathogenic importance of Na+/K+ ATPase dysfunction in ALS. In addition, impairment of Na+/K+ ATPase activity along with altered expression was reported in the spinal cords of SOD1G93A mice and patients with ALS [94], and was associated with accelerated neuronal degeneration in the mouse model [94]. In contrast, the function of Na+/K+ ATPase was reportedly normal in patients with ALS, when measured from peripheral motor axons, and was associated with the development of fatigue [95]. This apparent discordance could be explained by disease heterogeneity, with the surviving axons demonstrating either normal pump function or a stronger reserve capacity of the Na+/K+ ATPase pump.
In addition to aberrant Na+/K+ ATPase activity, there is substantial evidence relating to the pathogenic role of hyperexcitability and aberrant persistent Na+ conductances in ALS from transgenic mouse models [87]. Upregulation of persistent Na+ currents was reported in cortical and spinal motoneurons in SOD1G93A mouse models [60, 96, 97], and appeared to be an early feature inhibited by riluzole [60], the only effective neuroprotective agent in ALS [98, 99]. Importantly, altered expression of specific voltage-gated Na+ channel subunits, namely the β3 subunit, could underlie the upregulation of persistent Na+ currents in ALS [100]. Of further relevance, increased persistent sodium currents and hyperexcitability were evident in brainstem motor neurons from SOD1G93A mice prior to onset of motor neuron degeneration [101]. In addition, noncell-autonomous toxicity, an important pathogenic process in ALS [87], appears to be mediated by astrocyte induced upregulation of persistent Na+ conductances leading to increased hyperexcitability and intracellular Ca2+ ion concentration [102]. Importantly, reduction of persistent Na+ conductances by either specific (mexiletine) or nonspecific (spermidine and riluzole) agents inhibited hyperexcitability, normalized Ca2+ influx and prevented motoneuron degeneration [102], thereby suggesting a potential therapeutic role for Na+ channel blocking agents in ALS.
Separately, pluripotent stem-cell techniques have provided additional support for the importance of axonal ion-channel dysfunction in ALS pathogenesis [103, 104]. Specifically, reduction in K+ currents was reported in induced pluripotent stem cell-derived motor neurons from patients with ALS harboring SOD1, C9orf72, and FUS mutations [103]. The reduction in K+ currents was associated with membrane hyperexcitability across the 3 genetic mutations, suggesting that hyperexcitability is a ubiquitous feature in ALS. Importantly, activation of voltage-gated K+ channels by retigabine reduced hyperexcitability and improved in vitro survival [103]. Of further relevance, similar techniques have confirmed that neuronal hyperexcitability is an early feature in patients with ALS with C9orf72 and TARDBP mutations, with hypoexcitability evident in advanced disease underpinned by a reduction in Na+ and K+ currents over time [104]. Taken together, these findings suggest that targeting the axonal ion channel function, especially in early stages of ALS, could be of therapeutic benefit.
A contrasting view suggested that neuronal hyperexcitability exerted neuroprotective effects in ALS [71, 105]. Specifically, enhancement of neuronal excitability in the transgenic SOD1 mouse models, through activation of metabotropic cholinergic signaling and mammalian target of rapamycin pathways, appeared neuroprotective [71], while early neuronal hyperexcitability failed to induce neurodegeneration in a separate superoxide dismutase 1 mouse model [105]. Separately, neuronal hypoexcitability has been reported in motor neuronal cell lines derived from patients with ALS, and an increase in excitability by K+ channel blockade appeared neuroprotective [106]. Separately, others have argued that neuronal excitability is not related to intrinsic axonal properties of motor neurons, but rather related to extrinsic factors [107]. Importantly, the suggestion the axonal excitability is neuroprotective has only been documented in transgenic mouse models, which are poor models of human ALS [108].
From a clinical trials perspective, axonal excitability studies have been utilized as potential biomarkers of therapeutic efficacy in ALS. Specifically, riluzole therapy was associated with partial normalization of superexcitability, while no significant changes were evident in TE or τSD [109]. In addition, excitability studies have also been utilized in clinical trials assessing the effects of Na+ channel-blocking agents. Specifically, low-dose mexiletine (300 mg) did not exert any modulating effects on axonal excitability parameters, including τSD, potentially accounting for the absence of clinical effectiveness [73]. Separately, axonal excitability studies disclosed stabilization of axonal ion channel function in patients with ALS treated with the Na+ channel-blocking agent flecainide [72], associated with a reduced rate of lower motor neuron dysfunction. These clinical studies underscore the potential of utilizing axonal excitability parameters as biomarkers of therapeutic effectiveness in a drug trial setting. Currently two phase II trials assessing efficacy of retigabine (Clinicaltrials.gov ID: NCT02450552) and high-dose mexiletine (Clinicaltrials.gov ID: NCT01811355) in ALS are in progress where axonal excitability parameters are being utilized as secondary outcome measures.
Axonal Excitability in ALS Mimic Disorders
In terms of differential diagnoses of ALS, Kennedy’s disease, also known as spinobulbomuscular atrophy, is often raised. Kennedy’s disease is a slowly progressive inherited neurodegenerative disorder of motor and sensory neurons secondary to increased expansion of the polymorphic cytosine–adenine–guanine (CAG) repeat sequence (coding glutamine) in the androgen receptor gene on the X chromosome, at Xq11-12 [110, 111]. Clinically, Kennedy’s disease is characterized by presence of generalized fasciculations, muscle cramps, fatigue, and postural hand tremor, followed by progressive muscle weakness, dysarthria, and dysphagia [110, 112].Facial fasciculations with pursing of the lips, called contraction fasciculations, are a specific feature of Kennedy’s disease and may be evident in female heterozygote or homozygous mutation carriers [112, 113].
Conventional neurophysiologic studies in Kennedy’s disease are characterized by generalized and persistent fasciculations [114]. Unlike ALS, the fasciculation potentials in Kennedy’s disease are simple with lower frequency when compared with ALS [114]. In addition, the contraction fasciculations that are characteristic of Kennedy’s disease, are electrically characterized by either myokymia-like discharges that are induced by facial movement or 20 to 40-Hz discharges of individual motor unit action potentials of 0.1 to several seconds’ duration [115].
Axonal excitability studies in Kennedy’s disease have disclosed marked abnormalities as indicated by prolonged τSD, greater changes in response to a subthreshold depolarizing currents and increased hyperpolarizing current/threshold gradient [116]. Importantly, the increase in the τSD was an early feature in Kennedy’s disease and correlated with the fasciculation frequency, suggesting that upregulation of persistent Na+ conductances was responsible for generation of symptoms, particularly fasciculations, and contributed to the neurodegeneration. The pathophysiological mechanisms by which the mutated androgen receptor induces axonal ion-channel dysfunction in Kennedy’s disease remains to be elucidated, although transcriptional dysregulation is a likely mechanism [117]. Future studies assessing the effects of activating neuroprotective cellular processes, such as molecular chaperones, ubiquitin–proteasome system, and autophagy, on axonal excitability could provide critical pathogenic insights in Kennedy’s disease.
Spinal muscular atrophy (SMA) is a pediatric disorder of the spinal motor neurons clinically characterized by development of muscle weakness and atrophy [12]. Mutations in the SMN1 underlie the development of SMA with reduction in survival motor neuron protein levels forming the pathogenic basis of motor neuron degeneration in SMA [118]. While SMN1 mutations may induce abnormalities in axonal and membrane transports, as well as axonal myelination [119], transgenic mouse model studies have identified membrane hyperexcitability of diseased motor neurons [120].
Axonal excitability studies in ambulatory patients with SMA identified reduction in motor amplitudes, without significant changes in axonal excitability parameters [12]. In contrast, prolongation of τSD, along with greater depolarizing TE changes and steeper changes in the early phase of hyperpolarizing TE, were reported in severely affected patients with SMA and correlated with disease severity. Mathematical modeling suggested that a combination of neuronal degeneration and regeneration, resulting in shorter internodal length, reduction of internodal fast K+ currents, and an increase in nodal K+ conductances, best accounted for the axonal excitability changes in SMA [12]. From a clinical perspective, changes in axonal excitability parameters, particularly in more advanced SMA, could serve as potential biomarkers of therapeutic effectiveness in future trials.
Acquired neuromyotonia (aNMT) refers to a group of autoimmune diseases characterized by continuous ectopic nerve activity, clinically manifesting with muscle cramps, fasciculations, and stiffness [121]. The neuromuscular symptoms may be accompanied by hyperhidrosis, sensory abnormalities, and central nervous system features [121], and voltage-gated K+ channel-associated antibodies are evident in approximately 40% of patients [121, 122]. Conventional neurophysiologic studies in aNMT have identified the presence of spontaneous activity, including ongoing changes (positive sharp waves and fibrillation potentials), fasciculations, myokymia, multiple discharges, muscle cramps, and repetitive after-discharges after voluntary contraction [15, 122, 123]. Importantly, the fasciculation firing rate and frequency of doublet discharges were reportedly less frequent in aNMT when compared with ALS [124].
The site of generation of ectopic activity in aNMT remains to be elucidated, and some have suggested that the ectopic focus arises at the distal nerve terminal [15, 121, 123, 125–128]. In contrast, an ectopic focus arising at the level of the anterior horn cell or more centrally [129–131], has been proposed, with the latter hypothesis supported by the presence central nervous system symptoms and inflammatory changes, including the presence of oligoclonal bands [121, 132, 133]. Separately, aNMT may resemble ALS, particularly in early disease stages [87], and rarely an “apparent” case of aNMT may progress to ALS [134]. Axonal excitability has provided critical pathophysiological insights in aNMT, disclosing largely normal axonal ion channel function [15, 135], and thereby suggesting that the ectopic focus originates at the motor nerve terminal. Importantly, despite the presence of ectopic activity, the axonal excitability findings were markedly different between aNMT and ALS, thereby further supporting the importance of axonal ion-channel dysfunction in ALS pathogenesis.
Further, benign-cramp fasciculation syndrome is a related condition characterized by fasciculations in the absence of other symptoms or disorders [124]. Patients with benign cramp-fasciculation syndrome demonstrated increased accommodation to extensive hyperpolarization, potentially due to greater Ih conductance [136]. This was not evident with conventional axonal excitability protocols, but only with stronger hyperpolarization (−70% and −100% of thresehold). In a small cohort of patients, a different pattern was identified with reduced accommodation in TE (hyperpolarizing direction; TEh) and TE (depolarizing direction; TEd), suggestive of reduced slow K+ channel function [137], and indicating that there may be subgroups of patients with different membrane excitability.
Turning attention to inflammatory neuropathies, multifocal motor neuropathy (MMN) presents with limb-onset muscle atrophy, and is characterized by conduction block, fasciculations, and muscle cramps [138]. Axonal excitability studies in patients with MMN have demonstrated prominent changes in axonal membrane function, with a substantially different excitability profile when compared with ALS. Overall, the excitability changes were suggestive of membrane hyperpolarization distal to the site of conduction block [14, 139]. Specifically, the excitability profile was characterized by increased threshold change in depolarizing and hyperpolarizing TE, along with an increase in superexcitability. It was argued that the membrane hyperpolarization in MMN was due to excessive activation of the Na+/K+-ATPase in response to focal depolarization at the site of conduction block. Interestingly, reduction of τSD and an increase in rheobase were reported in MMN after intravenous immunoglobulin infusion (IVIg), a finding related to inhibition of persistent Na+ conductances [140], and postulated as the mechanism of the neuroprotective effects of IVIg in MMN [141].
Significant abnormalities of axonal excitability have also been reported in chronic inflammatory demyelinating polyneuropathy (CIDP), an autoimmune inflammatory disorder of the sensory and motor nerves [140, 142, 143]. Specifically, CIDP can be associated with reduction of τSD, refractoriness, supernormality, and subexcitability, along with an increase in rheobase, findings attributed to the underlying pathology [140, 142, 144, 145]. However, variability in excitability between patients may reflect heterogenous clinical phenotypes. Importantly, IVIg therapy resulted in both short- and long-term partial normalization of axonal excitability, characterized by an increase in τSD and improvements in TE [144]. Consequently, axonal excitability testing may serve as a useful therapeutic biomarker of immunotherapy responsiveness in autoimmune neuropathies, and could guide more tailored treatment strategies for individual patients suffering with MMN and CIDP.
In conclusion, axonal excitability techniques have provided insights regarding nodal and internodal nerve function in ALS and related neuromuscular disorders. Importantly, axonal ion channel dysfunction, characterized by abnormalities of Na+ and K+ conductances, appear to contribute to the underlying pathogenesis resulting in symptom generation, as well as neurodegeneration. Therapeutic strategies aimed at modulating axonal ion-channel dysfunction may prove therapeutically useful, particularly in ALS, with axonal excitability parameters utilized as primary or secondary biomarkers in future clinical trials.
References
Burke D, Kiernan MC, Bostock H. Excitability of human axons. Clin Neurophysiol 2001;112:1575–1585.
Bostock H, Cikurel K, Burke D. Threshold tracking techniques in the study of human peripheral nerve. Muscle Nerve 1998;21:137–158.
Krishnan AV, Lin CS, Park SB, Kiernan MC. Axonal ion channels from bench to bedside: a translational neuroscience perspective. Prog Neurobiol 2009;89:288–313.
Kiernan MC, Burke, D. Threshold electronus and the assessment of nerve excitability in amyotrophic lateral sclerosis. In: Eisen A, editor. Clinical Neurophysiology of Motor Neuron Diseases. Amsterdam: Elsevier; 2004. p. 359–366.
Bostock H, Sharief MK, Reid G, Murray NMF. Axonal ion channel dysfunction in amyotrophic lateral sclerosis. Brain 1995;118:217–225.
Vucic S, Kiernan MC. Axonal excitability properties in amyotrophic lateral sclerosis. Clin Neurophysiol 2006;117:1458–1466.
Mogyoros I, Kiernan M, Burke D, Bostock H. Strength-duration properties of sensory and motor axons in amyotrophic lateral sclerosis. Brain 1998;121:851–859.
Kanai K, Kuwabara S, Misawa S, et al. Altered axonal excitability properties in amyotrophic lateral sclerosis: impaired potassium channel function related to disease stage. Brain 2006;129:953–962.
Geevasinga N, Menon P, Howells J, Nicholson GA, Kiernan MC, Vucic S. Axonal ion channel dysfunction in c9orf72 familial amyotrophic lateral sclerosis. JAMA Neurol 2015;72:49–57.
Tamura N, Kuwabara S, Misawa S, et al. Increased nodal persistent Na+ currents in human neuropathy and motor neuron disease estimated by latent addition. Clin Neurophysiol 2006;117:2451–2458.
Weiss MD, Macklin EA, Simmons Z, et al. A randomized trial of mexiletine in ALS: safety and effects on muscle cramps and progression. Neurology 2016;86:1474–1481.
Farrar MA, Vucic S, Johnston HM, du Sart D, Kiernan MC. Pathophysiological insights derived by natural history and motor function of spinal muscular atrophy. J Pediatr 2013;162:155–159.
Vucic S, Nicholson GA, Kiernan MC. Cortical excitability in hereditary motor neuronopathy with pyramidal signs: comparison with ALS. J Neurol Neurosurg Psychiatry 2010;81:97–100.
Kiernan MC, Guglielmi JM, Kaji R, Murray NM, Bostock H. Evidence for axonal membrane hyperpolarization in multifocal motor neuropathy with conduction block. Brain 2002;125:664–675.
Kiernan MC, Hart IK, Bostock H. Excitability properties of motor axons in patients with spontaneous motor unit activity. J Neurol Neurosurg Psychiatry 2001;70:56–64.
Kiernan MC, Burke D, Andersen KV, Bostock H. Multiple measures of axonal excitability: a new approach in clinical testing. Muscle Nerve 2000;23:399–409.
Kiernan MC, Lin CS, Andersen KV, Murray NM, Bostock H. Clinical evaluation of excitability measures in sensory nerve. Muscle Nerve 2001;24:883–892.
Bostock H, Baker M, Grafe P, Reid G. Changes in excitability and accommodation of human motor axons following brief periods of ischaemia. J Physiol (Lond) 1991;441:513–535.
Kiernan MC, Isbister GK, Lin CS, Burke D, Bostock H. Acute tetrodotoxin-induced neurotoxicity after ingestion of puffer fish. Ann Neurol 2005;57:339–348.
Howells J, Trevillion L, Bostock H, Burke D. The voltage dependence of I(h) in human myelinated axons. J Physiol 2012;590:1625–1640.
Baker M, Bostock H. Depolarization changes the mechanism of accommodation in rat and human motor axons. J Physiol (Lond) 1989;411:545–561.
Bostock H, Burke D, Hales JP. Differences in behaviour of sensory and motor axons following release of ischaemia. Brain 1994;117:225–234.
Mogyoros I, Kiernan MC, Burke D, Bostock H. Excitability changes in human sensory and motor axons during hyperventilation and ischaemia. Brain 1997;120:317–325.
Grosskreutz J, Lin C, Mogyoros I, Burke D. Changes in excitability indices of cutaneous afferents produced by ischaemia in human subjects. J Physiol (Lond) 1999;518:301–314.
Grosskreutz J, Lin CS, Mogyoros I, Burke D. Ischaemic changes in refractoriness of human cutaneous afferents under threshold-clamp conditions. J Physiol (Lond) 2000;523:807–815.
Bostock H. The strength-duration relationship for excitation of myelinated nerve: computed dependence on membrane parameters. J Physiol (Lond) 1983;341:59–74.
Mogyoros I, Kiernan MC, Burke D. Strength-duration properties of human peripheral nerve. Brain 1996;119:439–447.
Mogyoros I, Lin C, Dowla S, Grosskreutz J, Burke D. Strength-duration properties and their voltage dependence at different sites along the median nerve. Clin Neurophysiol 1999;110:1618–1624.
Weiss G. Sur la possibilité de rendre comparables entre eux les appareils servant l'excitation électrique. Arch Ital Biol 1901;35:413–446.
Bostock H, Rothwell JC. Latent addition in motor and sensory fibres of human peripheral nerve. J Physiol (Lond) 1997;498:277–294.
French CR, Sah P, Buckett KJ, Gage PW. A voltage-dependent persistent sodium current in mammalian hippocampal neurons. J Gen Physiol 1990;95:1139–1157.
Crill WE. Persistent sodium current in mammalian central neurons. Annu Rev Physiol 1996;58:349–362.
Catterall WA. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron 2000;26:13–25.
Catterall WA, Goldin AL, Waxman SG. International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol Rev 2005;57:397–409.
Catterall WA. Voltage-gated sodium channels at 60: structure, function and pathophysiology. J Physiol 2012;590:2577–2589.
Goldin AL. Resurgence of sodium channel research. Annu Rev Physiol 2001;63:871–894.
Brismar T. Electrical properties of isolated demyelinated rat nerve fibres. Acta Physiol Scand 1981;113:161–166.
Mogyoros I, Kiernan MC, Gracies JM, Burke D. The effect of stimulus duration on the latency of submaximal nerve volleys. Muscle Nerve 1996;19:1354–1356.
Kiernan MC, Krishnan AV, Lin CS, Burke D, Berkovic SF. Mutation in the Na + channel subunit SCN1B produces paradoxical changes in peripheral nerve excitability. Brain 2005;128:1841–1846.
Kiernan MC, Mogyoros I, Burke D. Changes in excitability and impulse transmission following prolonged repetitive activity in normal subjects and patients with a focal nerve lesion. Brain 1996;119:2029–2037.
Hodgkin A, Huxley A. A quantitative descirption of membrane current and its application to conduction and excitation in nerve. J Physiol (Lond) 1952:500–544.
Scholz A, Reid G, Vogel W, Bostock H. Ion channels in human axons. J Neurophysiol 1993;70:1274–1279.
Burke D, Kiernan M, Mogyoros I, Bostock H. Susceptibility to conduction block: differences in the biophysical properties of cutaneous afferents and motor axons. Physiology of ALS and Related Diseases 1997:43–53.
Kiernan MC, Cikurel K, Bostock H. Effects of temperature on the excitability properties of human motor axons. Brain 2001;124:816–825.
Barrett EF, Barrett JN. Intracellular recording from vertebrate myelinated axons: mechanism of the depolarizing afterpotential. J Physiol (Lond) 1982;323:117–144.
Baker M, Bostock H, Grafe P, Martius P. Function and distribution of three types of rectifying channel in rat spinal root myelinated axons. J Physiol (Lond) 1987;383:45–67.
McIntyre CC, Richardson AG, Grill WM. Modeling the excitability of mammalian nerve fibers: influence of afterpotentials on the recovery cycle. J Neurophysiol 2002;87:995–1006.
Kiernan MC, Bostock H. Effects of membrane polarization and ischaemia on the excitability properties of human motor axons. Brain 2000;123:2542–2551.
Krishnan AV, Phoon RK, Pussell BA, Charlesworth JA, Bostock H, Kiernan MC. Altered motor nerve excitability in end-stage kidney disease. Brain 2005;128:2164–2174.
Kiernan MC, Walters RJ, Andersen KV, Taube D, Murray NM, Bostock H. Nerve excitability changes in chronic renal failure indicate membrane depolarization due to hyperkalaemia. Brain 2002;125:1366–1378.
Bostock H, Baker M. Evidence for two types of potassium channel in human motor axons in vivo. Brain Res 1988;462:354–358.
Pape HC. Queer current and pacemaker: the hyperpolarization-activated cation current in neurons. Annu Rev Physiol 1996;58:299–327.
Vagg R, Mogyoros I, Kiernan MC, Burke D. Activity-dependent hyperpolarization of human motor axons produced by natural activity. J Physiol 1998;507:919–925.
Lin CS, Kiernan MC, Burke D, Bostock H. Assessment of nerve excitability properties in peripheral nerve disease. In: Kimura J, editor. Handbook of Clinical Neurophysioloy. 7. Amsterdam: Elsevier; 2006. p. 381–403.
Roper J, Schwarz JR. Heterogeneous distribution of fast and slow potassium channels in myelinated rat nerve fibres. J Physiol (Lond) 1989;416:93–110.
Vucic S, Kiernan MC. Upregulation of persistent sodium conductances in familial ALS. J Neurol Neurosurg Psychiatry 2010;81:222–227.
Nakata M, Baba H, Kanai K, et al. Changes in Na(+) channel expression and nodal persistent Na(+) currents associated with peripheral nerve regeneration in mice. Muscle Nerve 2008;37:721–730.
Cheah BC, Lin CSY, Park SB, Vucic S, Krishnan AV, Kiernan MC. Progressive axonal dysfunction and clinical impairment in amyotrophic lateral sclerosis. Clin Neurophysiol 2012;123:2460–2467.
Layzer RB. The origin of muscle fasciculations and cramps. Muscle Nerve 1994;17:1243–1249.
Kuo JJ, Siddique T, Fu R, Heckman CJ. Increased persistent Na(+) current and its effect on excitability in motoneurones cultured from mutant SOD1 mice. J Physiol (Lond) 2005;563:843–854.
Kanai K, Kuwabara S, Arai K, Sung JY, Ogawara K, Hattori T. Muscle cramp in Machado-Joseph disease: altered motor axonal excitability properties and mexiletine treatment. Brain 2003;126:965–973.
Vucic S, Kiernan MC. Novel threshold tracking techniques suggest that cortical hyperexcitability is an early feature of motor neuron disease. Brain 2006;129:2436–2446.
Shibuya K, Misawa S, Nasu S, et al. Split hand syndrome in amyotrophic lateral sclerosis: different excitability changes in the thenar and hypothenar motor axons. J Neurol Neurosurg Psychiatry 2013;84:969–972.
Vucic S, Kiernan MC. Abnormalities in cortical and peripheral excitability in flail arm variant amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 2007;78:849–852.
Vucic S, Nicholson GA, Kiernan MC. Cortical hyperexcitability may precede the onset of familial amyotrophic lateral sclerosis. Brain 2008;131:1540–1550.
Wilbourn AJ. The “split hand syndrome”. Muscle Nerve 2000;23:138.
Kuwabara S, Sonoo M, Komori T, et al. Dissociated small hand muscle atrophy in amyotrophic lateral sclerosis: frequency, extent, and specificity. Muscle Nerve 2008;37:426–430.
Menon P, Kiernan MC, Yiannikas C, Stroud J, Vucic S. Split-hand index for the diagnosis of amyotrophic lateral sclerosis. Clin Neurophysiol 2013;124:410–416.
Menon P, Kiernan MC, Vucic S. ALS pathophysiology: Insights form the split-hand phenomenon. Clin Neurophysiol 2014;125:186–193.
Urbani A, Belluzzi O. Riluzole inhibits the persistent sodium current in mammalian CNS neurons. Eur J Neurosci 2000;12:3567–3574.
Saxena S, Roselli F, Singh K, et al. Neuroprotection through excitability and mTOR required in ALS motoneurons to delay disease and extend survival. Neuron 2013;80:80–96.
Park SB, Vucic S, Cheah BC, et al. Flecainide in Amyotrophic Lateral Sclerosis as a Neuroprotective Strategy (FANS): a randomized placebo-controlled trial. EBioMedicine 2015;2:1916–1922.
Shibuya K, Misawa S, Kimura H, et al. A single blind randomized controlled clinical trial of mexiletine in amyotrophic lateral sclerosis: efficacy and safety of sodium channel blocker phase II trial. Amyotroph Lateral Scler Frontotemporal Degener 2015;16:353–358.
Kanai K, Shibuya K, Sato Y, et al. Motor axonal excitability properties are strong predictors for survival in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 2012;83:734–738.
Shibuya K, Misawa S, Kimura H, et al. Increased motor axonal persistent sodium currents predict rapid functional declines in amyotrophic lateral sclerosis. Neurol Clin Neurosci 2016; 4:108–111.
Noto Y, Kanai K, Misawa S, et al. Distal motor axonal dysfunction in amyotrophic lateral sclerosis. J Neurol Sci 2011;302:58–62.
Horn S, Quasthoff S, Grafe P, Bostock H, Renner R, Schrank B. Abnormal axonal inward rectification in diabetic neuropathy. Muscle Nerve 1996;19:1268–1275.
Shibuta Y, Shimatani Y, Nodera H, Izumi Y, Kaji R. Increased variability of axonal excitability in amyotrophic lateral sclerosis. Clin Neurophysiol 2013;124:2046–2053.
Nakata M, Kuwabara S, Kanai K, et al. Distal excitability changes in motor axons in amyotrophic lateral sclerosis. Clin Neurophysiol 2006;117:1444–1448.
Roth G. The origin of fasciculations. Ann Neurol 1982;12:542–547.
Shibuta Y, Nodera H, Nodera A, et al. Utility of recovery cycle with two conditioning pulses for detection of impaired axonal slow potassium current in ALS. Clin Neurophysiol 2010;121:2117–2120.
Jiang Y, Yamamoto M, Kobayashi Y, et al. Gene expression profile of spinal motor neurons in sporadic amyotrophic lateral sclerosis. Ann Neurol 2005:236–251.
Shibuya K, Misawa S, Arai K, et al. Markedly reduced axonal potassium channel expression in human sporadic amyotrophic lateral sclerosis: an immunohistochemical study. Exp Neurol 2011;232:149–153.
Miller TM, Layzer RB. Muscle cramps. Muscle Nerve 2005;32:431–442.
Kiernan MC, Vucic S, Cheah BC, et al. Amyotrophic lateral sclerosis. Lancet 2011;377:942–955.
Dodson P, Billups B, Rusznak Z, Szucs G, Barker MC, Forsythe ID. Presynaptic rat Kv1.2 channels suppress synaptic terminal hyperexcitability following action potential invasion. J Physiol 2003:550:27–33.
Vucic S, Rothstein JD, Kiernan MC. Advances in treating amyotrophic lateral sclerosis: insights from pathophysiological studies. Trends Neurosci 2014;37:433–442.
Stys PK. Anoxic and ischemic injury of myelinated axons in CNS white matter: from mechanistic concepts to therapeutics. J Cereb Blood Flow Metab 1998;18:2–25.
Stys PK. General mechanisms of axonal damage and its prevention. J Neurol Sci 2005;233:3–13.
Stys PK. Sodium channel blockers as neuroprotectants in neuroinflammatory disease: a double-edged sword. Ann Neurol 2007;62:3–5.
Waxman SG. Axonal conduction and injury in multiple sclerosis: the role of sodium channels. Nat Rev Neurosci 2006;7:932–941.
Ellis DZ, Rabe J, Sweadner KJ. Global loss of Na,K-ATPase and its nitric oxide-mediated regulation in a transgenic mouse model of amyotrophic lateral sclerosis. J Neurosci 2003;23:43–51.
Alvarez S, Calin A, Graffmo KS, Moldovan M, Krarup C. Peripheral motor axons of SOD1(G127X) mutant mice are susceptible to activity-dependent degeneration. Neuroscience 2013;241:239–249.
Ruegsegger C, Maharjan N, Goswami A, et al. Aberrant association of misfolded SOD1 with Na(+)/K(+)ATPase-alpha3 impairs its activity and contributes to motor neuron vulnerability in ALS. Acta Neuropathol 2016;131:427–451.
Vucic S, Krishnan AV, Kiernan MC. Fatigue and activity dependent changes in axonal excitability in amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 2007;78:1202–1208.
Pieri M, Carunchio I, Curcio L, Mercuri NB, Zona C. Increased persistent sodium current determines cortical hyperexcitability in a genetic model of amyotrophic lateral sclerosis. Exp Neurol 2009;215:368–379.
Quinlan KA. Links between electrophysiological and molecular pathology of amyotrophic lateral sclerosis. Integ Comp Biol 2011;51:913–925.
Bensimon G, Lacomblez L, Meininger V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med 1994;330:585–591.
Lacomblez L, Bensimon G, Leigh PN, Guillet P, Meininger V. Dose-ranging study of riluzole in amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis/Riluzole Study Group II. Lancet 1996;347:1425–1431.
Nutini M, Spalloni A, Florenzano F, et al. Increased expression of the beta3 subunit of voltage-gated Na + channels in the spinal cord of the SOD1G93A mouse. Mol Cell Neurosci 2011;47:108–118.
van Zundert B, Peuscher MH, Hynynen M, et al. Neonatal neuronal circuitry shows hyperexcitable disturbance in a mouse model of the adult-onset neurodegenerative disease amyotrophic lateral sclerosis. J Neurosci 2008;28:10864–10874.
Fritz E, Izaurieta P, Weiss A, et al. Mutant SOD1-expressing astrocytes release toxic factors that trigger motoneuron death by inducing hyperexcitability. J Neurophysiol 2013;109:2803–2814.
Wainger BJ, Kiskinis E, Mellin C, et al. Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell Rep 2014;7:1–11.
Devlin A-C, Burr K, Borooah S, et al. Human iPSC-derived motoneurons harbouring TARDBP or C9ORF72 ALS mutations are dysfunctional despite maintaining viability. Nat Commun 2015;12:6.
Leroy F, Lamotte d'Incamps B, Imhoff-Manuel RD, Zytnicki D. Early intrinsic hyperexcitability does not contribute to motoneuron degeneration in amyotrophic lateral sclerosis. eLife 2014;3:e04046.
Naujock M, Stanslowsky N, Bufler S, et al. 4-Aminopyridine induced activity rescues hypoexcitable motor neurons from ALS patient-derived induced pluripotent stem cells. Stem Cells 2016;34:1563–1575.
Delestree N, Manuel M, Iglesias C, Elbasiouny SM, Heckman CJ, Zytnicki D. Adult spinal motoneurones are not hyperexcitable in a mouse model of inherited amyotrophic lateral sclerosis. J Physiol 2014;592:1687–1703.
Turner MR, Hardiman O, Benatar M, et al. Controversies and priorities in amyotrophic lateral sclerosis. Lancet Neurol 2013;12:310–322.
Vucic S, Lin CS-Y, Cheah BC, et al. Riluzole exerts central and peripheral modulating effects in amyotrophic lateral sclerosis. Brain 2013;136:1361–1370.
Kennedy WR, Alter M, Sung JH. Progressive proximal spinal and bulbar muscular atrophy of late onset. A sex-linked recessive trait. Neurology 1968;18:671–680.
La Spada AR, Wilson EM, Lubahn DB, Harding AE, Fischbeck KH. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature 1991;352:77–79.
Gallo JM. Spinobulbar muscular atrophy (Kennedy's disease). In: A. E, editor. Clinical Neurophysiology of Motor Neuron Diseases Hanbook of Clinical Neurophysiology. 4. Amsterdam: Elsevier B.V; 2004. p. 209–214.
Schmidt BJ, Greenberg CR, Allingham-Hawkins DJ, Spriggs EL. Expression of X-linked bulbospinal muscular atrophy (Kennedy disease) in two homozygous women. Neurology 2002;59:770–772.
Hirota N, Eisen A, Weber M. Complex fasciculations and their origin in amyotrophic lateral sclerosis and Kennedy's disease. Muscle Nerve 2000;23:1872–1875.
Olney RK, Aminoff MJ, So YT. Clinical and electrodiagnostic features of X-linked recessive bulbospinal neuronopathy. Neurology 1991;41:823–828.
Vucic S, Kiernan MC. Pathophysiological insights into motor axonal function in Kennedy's disease. Neurology 2007;69:1828–1835.
Katsuno M, Tanaka F, Adachi H, et al. Pathogenesis and therapy of spinal and bulbar muscular atrophy (SBMA). Prog Neurobiol 2012;99:246–256.
Lefebvre S, Burglen L, Reboullet S, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995;80:155–165.
Zhang Z, Lotti F, Dittmar K, et al. SMN deficiency causes tissue-specific perturbations in the repertoire of snRNAs and widespread defects in splicing. Cell 2008;133:585–600.
Mentis GZ, Blivis D, Liu W, et al. Early functional impairment of sensory-motor connectivity in a mouse model of spinal muscular atrophy. Neuron 2011;69:453–467.
Hart IK, Maddison P, Newsom-Davis J, Vincent A, Mills KR. Phenotypic variants of autoimmune peripheral nerve hyperexcitability. Brain 2002;125:1887–1895.
Hart IK, Waters C, Vincent A, et al. Autoantibodies detected to expressed K+ channels are implicated in neuromyotonia. Ann Neurol 1997;41:238–246.
Warmolts JR, Mendell JR. Neurotonia: impulse-induced repetitive discharges in motor nerves in peripheral neuropathy. Ann Neurol 1980;7:245–250.
Mills KR. Characteristics of fasciculations in amyotrophic lateral sclerosis and the benign fasciculation syndrome. Brain 2010;133:3458–3469.
Maddison P, Newsom-Davis J, Mills KR. Strength-duration properties of peripheral nerve in acquired neuromyotonia. Muscle Nerve 1999;22:823–830.
Deymeer F, Oge AE, Serdaroglu P, Yazici J, Ozdemir C, Baslo A. The use of botulinum toxin in localizing neuromyotonia to the terminal branches of the peripheral nerve. Muscle Nerve 1998;21:643–646.
Isaacs H. A syndrome of continuous muscle-fibre activity. J Neurol Neurosurg Psychiatry 1961;24:319–325.
Lance JW, Burke D, Pollard J. Hyperexcitability of motor and sensory neurons in neuromyotonia. Ann Neurol 1979;5:523–532.
Irani PF, Purohit AV, Wadia NH. The syndrome of continuous muscle fiber activity. Evidence to suggest proximal neurogenic causation. Acta Neurol Scand 1977;55:273–288.
Hosokawa S, Shinoda H, Sakai T, Kato M, Kuroiwa Y. Electrophysiological study on limb myokymia in three women. J Neurol Neurosurg Psychiatry 1987;50:877–881.
Hart IK, Waters C, Newsom-Davis J. Cerebrospinal fluid and serum from acquired neuromyotonia patients seropositive for anti-potassium channel antibodies label dentate nucleus neurons. Ann Neurol 1996;40:554–555.
Liguori R, Vincent A, Clover L, et al. Morvan's syndrome: peripheral and central nervous system and cardiac involvement with antibodies to voltage-gated potassium channels. Brain 2001;124:2417–2426.
Newsom-Davis J, Mills KR. Immunological associations of acquired neuromyotonia (Isaacs’ syndrome). Report of five cases and literature review. Brain 1993;116 453–469.
de Carvalho M, Swash M. Cramps, muscle pain, and fasciculations: not always benign? Neurology 2004;63:721–723.
Park SB, Lin CS, Krishnan AV, et al. Axaonl dysfunction with voltage gated potassium channel complex antibodies. Exp Neurol 2014;261:337–342.
Czesnik D, Howells J, Negro F, et al. Increased HCN channel driven inward rectification in benign cramp fasciculation syndrome. Brain 2010;138:3168–3179.
Shimatani Y, Nodera H, Shibuta Y, et al. Abnormal gating of axonal slow potassium current in cramp-fasciculation syndrome. Clin Neurophysiol 2015;126:1246–1254.
Arcila-Londono X, Lewis RA. Multifocal motor neuropathy. Handb Clin Neurol 2013;115:429–442.
Cappelen-Smith C, Kuwabara S, Lin CS, Burke D. Abnormalities of axonal excitability are not generalized in early multifocal motor neuropathy. Muscle Nerve 2002;26:769–776.
Boerio D, Creange A, Hogrel JY, Gueguen A, Bertrand D, Lefaucheur JP. Nerve excitability changes after intravenous immunoglobulin infusions in multifocal motor neuropathy and chronic inflammatory demyelinating neuropathy. J Neurol Sci 2010;292:63–71.
Vucic S, Black KR, Chong PS, Cros D. Multifocal motor neuropathy: decrease in conduction blocks and reinnervation with long-term IVIg. Neurology 2004;63:1264–1269.
Cappelen-Smith C, Kuwabara S, Lin CS, Mogyoros I, Burke D. Membrane properties in chronic inflammatory demyelinating polyneuropathy. Brain 2001;124:2439–2447.
Mathey EK, Park SB, Hughes RA, et al. Chronic inflammatory demyelinating polyradiculoneuropathy: from pathology to phenotype. J Neurol Neurosurg Psychiatry 2015;86:973–985.
Lin CS, Krishnan AV, Park SB, Kiernan MC. Modulatory effects on axonal function after intravenous immunoglobulin therapy in chronic inflammatory demyelinating polyneuropathy. Arch Neurol 2011;68:862–869.
Sung JY, Kuwabara S, Kaji R, et al. Threshold electrotonus in chronic inflammatory demyelinating polyneuropathy: correlation with clinical profiles. Muscle Nerve 2004;29:28–37.
Acknowledgments
This work was supported by funding to Forefront, a collaborative research group dedicated to the study of motor neuron disease, from the National Health and Medical Research Council of Australia program grant (#1037746).
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 1224 kb)
Rights and permissions
About this article

Cite this article
Park, S.B., Kiernan, M.C. & Vucic, S. Axonal Excitability in Amyotrophic Lateral Sclerosis. Neurotherapeutics 14, 78–90 (2017). https://doi.org/10.1007/s13311-016-0492-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-016-0492-9