
Abstract
After decades since the discovery of natural killer (NK) cells as potential effector cells fighting malignantly transformed and virally infected cells, little progress has been made in their clinical application. This yet unrealized therapeutic effect is presumably, at least in part, due to low numbers of functional NK cells that could be obtained from the peripheral blood relative to tumor burden. Our group hypothesized that a relatively small NK cell number to targeted malignant cells is the cause of a lack of clinical effect. We pursued obtaining large numbers of NK cells via ex vivo expansion using feeder cells that express membrane-bound IL-21. Early clinical studies demonstrate safety of administration of ex vivo expanded NK cells after transplantation using this method and suggest a therapeutic benefit in terms on decreasing relapse rate and possible control of viral infections post-transplant can be achieved. Successful application of NK cells after hematopoietic stem cell transplantation opens the possibility to effectively enhance the anti-tumor effect and decrease relapse rate post-transplant. Moreover, high doses of NK cells could prove more efficacious in enhancing anti-tumor effects, not only in hematological malignancies, with our without transplantation, but also in solid tumor oncology.
Similar content being viewed by others
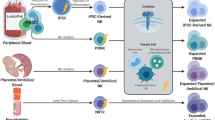

Overview of NK cells
NK cells are the most efficient effector cells of the innate immune system and are able to kill tumor cells without the need for prior sensitization [1, 2]. Cytotoxicity of NK cells is presumably based on a delicate balance between inhibitory and activating signals from the cell surface [3]. Activation of NK cell cytotoxicity is mediated by killer immunoglobulin-like receptors (KIRs) and their ligands (human leukocyte antigen (HLA) class I molecules; HLA-A3/11, HLA-Bw4-motifs, HLA-C group 1 and HLA-C group 2 alleles), which are expressed on all cells depending on the individual HLA profile. Upon engagement of these inhibitory KIRs with MHC class I molecules on a target cell, inhibitory signals are transmitted, prohibiting NK cells to kill the targeted cell. However, if the inhibitory KIRs cannot bind their MHC class I ligand (missing self), they become licensed to kill the target [4]. Killing of these MHC class I missing target cells can only be initiated after simultaneous detection of activating signals, mostly stress signals, on the surface of tumor cells. Expression of numerous activating receptors can also induce NK cell killing even in the presence of MHC class I surface expression (induced self) (Fig. 1). This intrinsic NK cell killing capacity has been utilized in therapeutic anti-tumor approaches using allogeneic as well as autologous NK cells.

NK cell function. NK cells are activated by a delicate balance of activating and inhibiting signals. By interacting with normal tissue cells that express MHC class I and stress ligands, NK cells do not become activated because the inhibitory signal is stronger than the activating signal. Middle: By interacting with tumor cells that upregulated the amount of stress molecules, NK cells become activated because the activating signals overrule in inhibitory signal (induced self). Lower: Interaction with tumor cells that have downregulated their expression of MHC class I will lead, in combination with, to NK cell activation (missing self)
NK cells in hematopoietic stem cell transplantation
Allogeneic hematopoietic stem cell transplantation is a potentially curative treatment option for different hematological malignancies. Historically, patients have been preferably transplanted with an HLA-matched donor for HLA-A, HLA-B, HLA-C, - DR ± DQ loci. The anti-tumor effect after transplant depends mainly on allogeneic T cell-mediated anti-tumor responses. Transplanting over HLA barriers has shown, in case of KIR–ligand mismatch, to induce NK cell activation. This concept was first described in 2002 by Ruggeri et al. who showed that patients with acute myeloid leukemia (AML) receiving a KIR-mismatched haploidentical stem cell transplantation showed drastic increase in five-year relapse-free survival compared to patients without a KIR mismatch (100% vs. 25%) [5]. After this report, haploidentical transplantation has been exploited in different hematological diseases, using different conditioning regimens, GvHD prophylaxis and stem cell sources. Results from these studies have been conflicting, and high incidence of GvHD as well as opportunistic infections has been a major concern [6, 7]. Optimization of the haploidentical transplantation protocols, recently by application of post-transplantation cyclophosphamide (PTCy) as the most widely used method [8, 9], has led to improved transplant outcomes in terms of safety and decreased transplant-related mortality (TRM) with outcomes similar to HLA-matched transplants. As these major drawbacks are at least partially resolved, the focus has shifted to disease relapse, which has emerged the most common cause of treatment failure, not only after HLA-matched transplants but also after haploidentical transplantation.
First results from studies investigating the effect of KIR–ligand mismatch versus non-KIR-mismatched in the PTCy setting are disagreeing. While Symons and colleagues showed that recipients of an inhibitory KIR–gene-mismatched graft had lower relapse rate and improved survival and that patients with KIR Bx donor for recipients with homozygous A haplotype had improved non-relapse mortality and survival [10], Hosokai et al. reported that KIR Bx donors for recipients with A/A haplotype had higher incidence of acute GVHD with no impact on non-relapse mortality, relapse or survival [11]. Low NK cell numbers and function early after transplantation have been observed related to the PTCy administration [12, 13], while higher numbers of NK cells have been associated with better transplant outcomes [8,9,10,11]. This association is true for both allogeneic and autologous transplantation [14,15,16,17]. Although the NK cells are about the first cells that can be detected in the peripheral blood after transplantation, their phenotype and function are immature [13]. It takes at least 3–6 months until these cells gain KIR expression and become functionally active to induce optimal anti-leukemia responses. To warrant early post-transplant graft-versus-leukemia effects—when tumor load is low—it seems essential to improve NK cell repopulation, which can be accomplished by post-transplant NK cell transfer.
NK cell expansion
Different sources of NK cells are used for NK cell therapy. Autologous as well as allogeneic NK cells can be purified from peripheral blood apheresis products or obtained from umbilical cord blood (UCB) [18, 19]. Additionally, NK cells can be developed from CD34+ UCB stem cells or from induced pluripotent stem cells [20, 21]. However, differences among NK cells obtained from these sources have not been adequately described.
Since progression-free survival after hematopoietic stem cell transplantation is positively correlated with the number of peripheral blood NK cells, it seems of importance to infuse high numbers of NK cells for NK cell therapy [22, 23]. NK cells make up only approximately 10% of mononucleated cells in the blood and even less in umbilical cord blood, while the number of NK cells that can be obtained via apheresis procedure on average 1–3 × 107/kg of an adult individual, enough in general for 1 transfer dose only [24, 25] which makes NK cell expansion necessary. Different methods have been developed for NK expansion, both in vivo and ex vivo [25].
Ex vivo expansion methods included single cytokines or cytokine cocktails containing IL-2, IL-15, IL-12 or IL-18 [18, 26]. To potentiate proliferation, anti-CD3 antibodies were added to cultures [27]. Also feeder cells are often used for NK cell proliferation. EBV-transfected cell lines, peripheral blood mononuclear cells, Wilms tumor-derived cell lines are used for this purpose [28, 29]. However, the most widely used feeder cell line is the chronic myeloid leukemia-derived cell line, K562. This cell line is either used in combination with different cytokines or transduced to express membrane-bound IL-15, IL-21 or 4-1BB (CD137) [30, 31]. Probably the best method of expansion so far, yielding high NK cell numbers, which uses K562 cells stable transfected with CD64, CD86, truncated CD19, 4-1BB and mbIL-21, has been developed by our group [32]. With this method, NK cell proliferation can continue for 8–10 weeks before NK cells go into senescence, with NK cells displaying longer telomeres and the capacity of continuous proliferating capacity. For UCB-derived NK cells, proliferation protocols are similar to those used for peripheral blood NK cell expansion and also consist of cytokine combinations in the presence or absence of different feeder cells expressing NK cell-activating ligands (membrane-bound cytokines, 4-1BB) and anti-CD3 antibodies [18, 33].
In addition to ex vivo NK cell expansion, in vivo NK cell expansion has been investigated extensively. Expansion protocols depend mainly on cytokine infusion, of which IL-2 has been the most frequently used cytokine [34]. Increase in peripheral blood NK cells depends on both mature NK cell proliferation and induced NK cell development from bone marrow-derived stem cells [35]. Drawbacks of in vivo NK cell expansion protocols are side effects due to T cell activation, which are also sensitive to most cytokines used for NK cell activation. This makes the use of in vivo NK cell expansion after stem cell transplantation less appealing not only due to systemic adverse effects but also due to the possible risk of T cell-mediated GvHD.
Post-transplantation NK cell therapy
NK cell therapy has been first utilized in a non-transplant setting in hematological malignancies and solid tumors [36]. Although successes were limited to a minority of patients, NK cell infusion was demonstrated safe and feasible [34]. These results have led to exploit post-transplantation NK cell transfer in hematological malignancies to warrant early NK cell repopulation.
NK cell infusion after hematopoietic stem cell transplantation has been utilized in autologous and allogeneic transplantation studies. In a small study in AML patients with impending relapse, Uharek et al. showed favorable two-year survival (40% vs. 11%) when treated with a haploidentical transplantation in combination with post-transplant NK cell infusion, as compared to haploidentical transplantation alone. Also in multiple myeloma patients, NK cell were infused after both autologous and allogeneic transplantation [37, 38] with no safety concerns.
We recently reported on a phase I clinical study in high-risk myeloid leukemia patients treated with haploidentical transplants and multiple donor-derived, ex vivo expanded NK cell infusions using feeder cells expressing mbIL-21 [13]. In this dose escalation study, three doses of NK cells infused on Day-2, + 7 and + 28 (up to Day + 90), up to 1 × 108/kg were safely infused without reaching a maximum tolerated dose and with no induction of grade 3 or 4 acute GvHD or higher TRM. In addition, a low incidence of CMV and BK virus reactivation was noted along with a lower than expected relapse rate for the high-risk patients treated on this study. Products were a highly pure collection of NK cells (average 99%). Infused NK cells had mature phenotype and were functionally active, holding the capacity to mediate anti-leukemia cytotoxic responses, and high IFN-γ and TNF alpha production. Out of 13 treated patients, 11 patients showed sustained remission, only one patient relapsed and another patient succumbed to infectious disease [13]. The patient who relapsed had very high risk of leukemia relapse (FLT3 + AML patient in CR2 with detectable minimal residual disease) and received the lowest dose of NK cells (1 × 105/kg). These results encouraged us to further pursue this approach in a randomized study to confirm the ability of ex vivo expanded NK cells to decrease relapse rate post-transplant, as well as explore NK cell therapy in a non-transplant setting for both hematological malignancies and possibly solid tumors. A second generation, potentially safer NK cell product with similar characteristics, produced without K562 feeder cells, is currently being explored.
Future directions
Results from current studies with mbIL-21 ex vivo expanded NK cell infusions post-transplant are very promising and hold the exciting possibility for improvement in relapse rate post-transplant, which is urgently needed especially for patients with very high-risk or advanced hematological malignancies, for which relapse rate may exceed 50%. In addition, NK cell therapy could be utilized in patients with advanced or refractory leukemia with the goal of achieving remission and offer the chance for these patients to undergo transplantation. Moreover, due to the broad anti-tumor effect and safety profile as compared for antigen-specific therapy (antibody therapy, CAR-T cells, etc.), this therapy could potentially expand for a broad variety of malignancies and extend the therapeutic armamentarium for a large number of patients.
Expansion protocols yielding the highest NK cell numbers depend, as for now, on the use of feeder cells. Although the culture conditions have been standardized according to good manufacturing practices, the use of cancer cell lines for NK cell production and clinical applications is still controversial. Therefore, new feeder cell-free methods are under investigation and one very successful method, among others, is the use of particles prepared from plasma membrane of K562-MB21-41BBL cells that are derived from the previously described K562 cell line expressing 4-1BB and membrane-bound IL-21 (PM21 particles) [39, 40], which yields high numbers of cytotoxic NK cells that home to different organ sites after infusion in NSG mice.
Since NK cell infusions seem to be safe and feasible, NK cell therapy could also be exploited in hematological malignancies other than myeloid leukemia and solid tumors. Although the number of treated patients with other malignancies is very small, solid tumors seem to be less susceptible to NK cell therapy; however, treatment successes have been reported. Tumor microenvironment plays a major role in damping the therapeutic effect in these diseases, warranting development of novel NK cell strategies that either boost NK cell activation or target the tumor microenvironment in these patients.
One of these strategies that boost NK cell activation is the use of antibody therapy. By expression of CD16 (Fc gamma RIII receptor) on the cell surface, NK cells are able to induce antibody-dependent cellular cytotoxicity (ADCC). In the last decade, many new tumor antigen-specific antibodies have been developed for immunotherapy. Examples are anti-Her2 for breast cancer, anti-EGFR for different epithelial tumors and anti-CD20 for lymphoma [41,42,43]. Since many of these enhance NK cell-mediated tumor cell killing, it can be envisioned to combine antibody treatment with NK cell therapy. As the field of antibody therapy is developing rapidly, bi-specific NK cell-activating antibodies are also being developed [44]. These antibodies recognize NK cell-activating receptors as well as tumor-specific antigens, which brings the NK cell in close physical proximity with cancer cells, to possibly enhance anti-tumor effects in this setting.
Another approach to increase NK cell activation is the use of anti-KIR antibodies that block inhibitory KIRs on the surface of NK cells, thereby mediating an NK cell-activating signal. Although the NK cell inhibitory signal to normal self is blocked, NK cells do not attack healthy cells, since these cell lack expression of activating stress molecules. Anti-KIR antibodies are often used in combination with other immunotherapeutic strategies and have been proved save. Preclinical studies in lymphoma have shown induction of NK cell-mediated lymphoma cytotoxicity when the anti-KIR antibody lirilumab is combined with anti-CD20 antibody treatment [45], and clinical studies are now being carried out (NCT01714739, NCT02481297).
The NK cell-mediated anti-tumor response could potentially be enhanced by transducing NK cells with a chimeric antigen receptor (CAR-NK), which recognizes tumor-specific antigens [46, 47]. The CAR is an artificially modified fusion protein that consists of an extracellular antigen recognition domain fused to an intracellular signaling domain. T cells transduced with CAR have been studied extensively, and the first CAR is now FDA approved for pediatric patients with CD19+ hematological malignancies. The potential advantage of CAR-NK cells over CAR-T cells is a more favorable safety profile, as their intrinsic mechanism of action prevents them for attacking non-cancerous cells, thereby preventing collateral damage. Moreover, CAR-NK cells could yield higher initial cytotoxicity and can be generated from different donors. However, they display low transfection efficiency and do not expand ex vivo [47]. Preclinical studies in hematological malignancies as well as in solid tumors have shown anti-tumor responses with either transduction of primary NK cells or of the NK-92 cell line [48,49,50]. Until now, only few phase I/II clinical studies are being carried out with CAR-NK cells (MUC1+ solid tumors NCT02839954, CD33+ acute myeloid leukemia NCT02944162, B-acute lymphoblastic leukemia NCT01974479) and results are eagerly awaited.
Methods for altering the tumor microenvironment are numerous and the use of checkpoint inhibitor is one of them. There is an increasing interest in combining NK cell therapy with checkpoint inhibitors. NK cells not only express receptors that ligate inhibiting molecules on the cancer cells but also have a role in paving the path for T cell tumor infiltration. The innate response is the initiator of chemokine and cytokine release responsible for T cell infiltration into the tumor microenvironment. Responsiveness to checkpoint inhibitors has been shown to be dependent on these infiltrates. Most interesting is combining NK cells with PD-1/PDL-1 inhibitors, which already has been shown effective in myeloma and lymphoma [51, 52]. Effects partially depend on inhibition of NK cell blocking [53].
Conclusions
Due to optimized haploidentical transplantation protocols, haploidentical stem cell transplantation for hematological malignancies has become a very attractive therapeutic option with outcomes similar to HLA-matched donors. As NK cell reconstitution post-transplant is suboptimal, it is likely that it contributes to suboptimal graft-versus-leukemia responses. As such, post-transplant NK cell administration has been of great interest. Administration of ex vivo expanded NK cells in the early post-transplant period not only has been shown to be safe and feasible, but also a very encouraging is the low relapse rate that has been noted, warranting a randomized study to confirm a possible therapeutic benefit. To enhance NK cell-mediated cytotoxicity with or without transplantation, a multitude of different approaches are of great interest for the foreseeable future, like combination with antibody therapy, checkpoint inhibition or CAR-NK cells, all of which hold the promise to make NK cell therapy a viable therapeutic approach for cancer therapy.
References
Kiessling R, Klein E, Pross H, Wigzell H. “Natural” killer cells in the mouse. II. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Characteristics of the killer cell. Eur J Immunol. 1975;5:117–21.
Kiessling R, Klein E, Wigzell H. “Natural” killer cells in the mouse. I. Cytotoxic cells with specificity for mouse Moloney leukemia cells. specificity and distribution according to genotype. Eur J Immunol. 1975;5:112–7.
Raulet DH, Held W. Natural killer cell receptors: the offs and ons of NK cell recognition. Cell. 1995;82:697–700.
Raulet DH, Vance RE, McMahon CW. Regulation of the natural killer cell receptor repertoire. Annu Rev Immunol. 2001;19:291–330.
Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science. 2002;295:2097–100.
Ciurea SO, Mulanovich V, Jiang Y, Bassett R, Rondon G, et al. Lymphocyte recovery predicts outcomes in cord blood and T cell-depleted haploidentical stem cell transplantation. Biol Blood Marrow Transplant. 2011;17:1169–75.
Ciurea SO, Saliba RM, Rondon G, Patah PA, Aung F, et al. Outcomes of patients with myeloid malignancies treated with allogeneic hematopoietic stem cell transplantation from matched unrelated donors compared with one human leukocyte antigen mismatched related donors using HLA typing at 10 loci. Biol Blood Marrow Transplant. 2011;17:923–9.
Mehta RS, Saliba RM, Chen J, Rondon G, Hammerstrom AE, et al. Post-transplantation cyclophosphamide versus conventional graft-versus-host disease prophylaxis in mismatched unrelated donor haematopoietic cell transplantation. Br J Haematol. 2016;173:444–55.
Luznik L, O’Donnell PV, Fuchs EJ. Post-transplantation cyclophosphamide for tolerance induction in HLA-haploidentical bone marrow transplantation. Semin Oncol. 2012;39:683–93.
Symons HJ, Leffell MS, Rossiter ND, Zahurak M, Jones RJ, et al. Improved survival with inhibitory killer immunoglobulin receptor (KIR) gene mismatches and KIR haplotype B donors after nonmyeloablative, HLA-haploidentical bone marrow transplantation. Biol Blood Marrow Transplant. 2010;16:533–42.
Hosokai R, Masuko M, Shibasaki Y, Saitoh A, Furukawa T, et al. Donor killer immunoglobulin-like receptor haplotype b/x induces severe acute graft-versus-host disease in the presence of human leukocyte antigen mismatch in t cell-replete hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2017;23:606–11.
Russo A, Oliveira G, Berglund S, Greco R, Gambacorta V, et al. (2017) NK cell recovery after haploidentical HSCT with post-transplant cyclophosphamide: dynamics and clinical implications. Blood.
Ciurea SO, Schafer JR, Bassett R, Denman CJ, Cao K, et al. Phase 1 clinical trial using mbIL21 ex vivo-expanded donor-derived NK cells after haploidentical transplantation. Blood. 2017;130:1857–68.
Clausen J, Wolf D, Petzer AL, Gunsilius E, Schumacher P, et al. Impact of natural killer cell dose and donor killer-cell immunoglobulin-like receptor (KIR) genotype on outcome following human leucocyte antigen-identical haematopoietic stem cell transplantation. Clin Exp Immunol. 2007;148:520–8.
Savani BN, Mielke S, Rezvani K, Montero A, Yong AS, et al. Absolute lymphocyte count on day 30 is a surrogate for robust hematopoietic recovery and strongly predicts outcome after T cell-depleted allogeneic stem cell transplantation. Biol Blood Marrow Transplant. 2007;13:1216–23.
Rueff J, Medinger M, Heim D, Passweg J, Stern M. Lymphocyte subset recovery and outcome after autologous hematopoietic stem cell transplantation for plasma cell myeloma. Biol Blood Marrow Transplant. 2014;20:896–9.
Porrata LF, Inwards DJ, Micallef IN, Johnston PB, Ansell SM, et al. Interleukin-15 affects patient survival through natural killer cell recovery after autologous hematopoietic stem cell transplantation for non-Hodgkin lymphomas. Clin Dev Immunol. 2010;2010:914945.
Tomchuck SL, Leung WH, Dallas MH. Enhanced cytotoxic function of natural killer and CD3+ CD56+ cells in cord blood after culture. Biol Blood Marrow Transplant. 2015;21:39–49.
Iyengar R, Handgretinger R, Babarin-Dorner A, Leimig T, Otto M, et al. Purification of human natural killer cells using a clinical-scale immunomagnetic method. Cytotherapy. 2003;5:479–84.
Dolstra H, Roeven MWH, Spanholtz J, Hangalapura BN, Tordoir M, et al. Successful transfer of umbilical cord blood CD34+ hematopoietic stem and progenitor-derived NK cells in older acute myeloid leukemia patients. Clin Cancer Res. 2017;23:4107–18.
Hermanson DL, Bendzick L, Pribyl L, McCullar V, Vogel RI, et al. Induced pluripotent stem cell-derived natural killer cells for treatment of ovarian cancer. Stem Cells. 2016;34:93–101.
Savani BN, Mielke S, Adams S, Uribe M, Rezvani K, et al. Rapid natural killer cell recovery determines outcome after T-cell-depleted HLA-identical stem cell transplantation in patients with myeloid leukemias but not with acute lymphoblastic leukemia. Leukemia. 2007;21:2145–52.
Dunbar EM, Buzzeo MP, Levine JB, Schold JD, Meier-Kriesche HU, et al. The relationship between circulating natural killer cells after reduced intensity conditioning hematopoietic stem cell transplantation and relapse-free survival and graft-versus-host disease. Haematologica. 2008;93:1852–8.
Meyer-Monard S, Passweg J, Siegler U, Kalberer C, Koehl U, et al. Clinical-grade purification of natural killer cells in haploidentical hematopoietic stem cell transplantation. Transfusion. 2009;49:362–71.
Koepsell SA, Miller JS, McKenna DH Jr. Natural killer cells: a review of manufacturing and clinical utility. Transfusion. 2013;53:404–10.
London L, Perussia B, Trinchieri G. Induction of proliferation in vitro of resting human natural killer cells: iL 2 induces into cell cycle most peripheral blood NK cells, but only a minor subset of low density T cells. J Immunol. 1986;137:3845–54.
Alici E, Sutlu T, Bjorkstrand B, Gilljam M, Stellan B, et al. Autologous antitumor activity by NK cells expanded from myeloma patients using GMP-compliant components. Blood. 2008;111:3155–62.
Berg M, Lundqvist A, McCoy P Jr, Samsel L, Fan Y, et al. Clinical-grade ex vivo-expanded human natural killer cells up-regulate activating receptors and death receptor ligands and have enhanced cytolytic activity against tumor cells. Cytotherapy. 2009;11:341–55.
Harada H, Watanabe S, Saijo K, Ishiwata I, Ohno T. A Wilms tumor cell line, HFWT, can greatly stimulate proliferation of CD56 + human natural killer cells and their novel precursors in blood mononuclear cells. Exp Hematol. 2004;32:614–21.
Melero I, Johnston JV, Shufford WW, Mittler RS, Chen L. NK1.1 cells express 4-1BB (CDw137) costimulatory molecule and are required for tumor immunity elicited by anti-4-1BB monoclonal antibodies. Cell Immunol. 1998;190:167–72.
Phillips JH, Lanier LL. A model for the differentiation of human natural killer cells. Studies on the in vitro activation of Leu-11 + granular lymphocytes with a natural killer-sensitive tumor cell, K562. J Exp Med. 1985;161:1464–82.
Denman CJ, Senyukov VV, Somanchi SS, Phatarpekar PV, Kopp LM, et al. Membrane-bound IL-21 promotes sustained ex vivo proliferation of human natural killer cells. PLoS ONE. 2012;7:e30264.
Satwani P, van de Ven C, Ayello J, Cairo D, Simpson LL, et al. Interleukin (IL)-15 in combination with IL-2, fms-like tyrosine kinase-3 ligand and anti-CD3 significantly enhances umbilical cord blood natural killer (NK) cell and NK-cell subset expansion and NK function. Cytotherapy. 2011;13:730–8.
Miller JS, Soignier Y, Panoskaltsis-Mortari A, McNearney SA, Yun GH, et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood. 2005;105:3051–7.
Fehniger TA, Bluman EM, Porter MM, Mrozek E, Cooper MA, et al. Potential mechanisms of human natural killer cell expansion in vivo during low-dose IL-2 therapy. J Clin Invest. 2000;106:117–24.
Rosenberg SA, Lotze MT, Muul LM, Chang AE, Avis FP, et al. A progress report on the treatment of 157 patients with advanced cancer using lymphokine-activated killer cells and interleukin-2 or high-dose interleukin-2 alone. N Engl J Med. 1987;316:889–97.
Shaffer BC, Le Luduec JB, Forlenza C, Jakubowski AA, Perales MA, et al. Phase II study of haploidentical natural killer cell infusion for treatment of relapsed or persistent myeloid malignancies following allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2016;22:705–9.
Shah N, Li L, McCarty J, Kaur I, Yvon E, et al. Phase I study of cord blood-derived natural killer cells combined with autologous stem cell transplantation in multiple myeloma. Br J Haematol. 2017;177:457–66.
Oyer JL, Pandey V, Igarashi RY, Somanchi SS, Zakari A, et al. Natural killer cells stimulated with PM21 particles expand and biodistribute in vivo: clinical implications for cancer treatment. Cytotherapy. 2016;18:653–63.
Oyer JL, Igarashi RY, Kulikowski AR, Colosimo DA, Solh MM, et al. Generation of highly cytotoxic natural killer cells for treatment of acute myelogenous leukemia using a feeder-free, particle-based approach. Biol Blood Marrow Transplant. 2015;21:632–9.
Junttila TT, Parsons K, Olsson C, Lu Y, Xin Y, et al. Superior in vivo efficacy of afucosylated trastuzumab in the treatment of HER2-amplified breast cancer. Cancer Res. 2010;70:4481–9.
Lopez-Albaitero A, Lee SC, Morgan S, Grandis JR, Gooding WE, et al. Role of polymorphic Fc gamma receptor IIIa and EGFR expression level in cetuximab mediated, NK cell dependent in vitro cytotoxicity of head and neck squamous cell carcinoma cells. Cancer Immunol Immunother. 2009;58:1853–64.
Cartron G, Dacheux L, Salles G, Solal-Celigny P, Bardos P, et al. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood. 2002;99:754–8.
Rothe A, Sasse S, Topp MS, Eichenauer DA, Hummel H, et al. A phase 1 study of the bispecific anti-CD30/CD16A antibody construct AFM13 in patients with relapsed or refractory Hodgkin lymphoma. Blood. 2015;125:4024–31.
Kohrt HE, Thielens A, Marabelle A, Sagiv-Barfi I, Sola C, et al. Anti-KIR antibody enhancement of anti-lymphoma activity of natural killer cells as monotherapy and in combination with anti-CD20 antibodies. Blood. 2014;123:678–86.
Hu Y, Tian ZG, Zhang C (2017) Chimeric antigen receptor (CAR)-transduced natural killer cells in tumor immunotherapy. Acta Pharmacol Sin.
Imai C, Iwamoto S, Campana D. Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood. 2005;106:376–83.
Liu H, Yang B, Sun T, Lin L, Hu Y, et al. Specific growth inhibition of ErbB2expressing human breast cancer cells by genetically modified NK92 cells. Oncol Rep. 2015;33:95–102.
Zhang C, Burger MC, Jennewein L, Genssler S, Schonfeld K, et al. ErbB2/HER2-specific NK cells for targeted therapy of glioblastoma. J Natl Cancer Inst. 2016;. https://doi.org/10.1093/jnci/djv375.
Oelsner S, Wagner J, Friede ME, Pfirrmann V, Genssler S, et al. Chimeric antigen receptor-engineered cytokine-induced killer cells overcome treatment resistance of pre-B-cell acute lymphoblastic leukemia and enhance survival. Int J Cancer. 2016;139:1799–809.
Giuliani M, Janji B, Berchem G. Activation of NK cells and disruption of PD-L1/PD-1 axis: two different ways for lenalidomide to block myeloma progression. Oncotarget. 2017;8:24031–44.
Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med. 2015;372:311–9.
Benson DM Jr, Bakan CE, Mishra A, Hofmeister CC, Efebera Y, et al. The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: a therapeutic target for CT-011, a novel monoclonal anti-PD-1 antibody. Blood. 2010;116:2286–94.
Acknowledgements
This work was supported in part by the MD Anderson Cancer Center AML Moonshot Program, High Impact Clinical Research Support Program and McKee Family Foundation.
Author information
Authors and Affiliations
Corresponding author
About this article

Cite this article
Van Elssen, C.H.M.J., Ciurea, S.O. NK cell therapy after hematopoietic stem cell transplantation: can we improve anti-tumor effect?. Int J Hematol 107, 151–156 (2018). https://doi.org/10.1007/s12185-017-2379-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-017-2379-x