
Abstract
Increasing studies have revealed that the interaction between malignant cells and the microenvironment (so called niche) in the bone marrow can influence the development and progression of the hematopoietic malignancies. Here, we reviewed the current findings in the field, focusing the niche alterations in promoting the emergency of malignancies, in interfering with the blood reconstitution of normal hematopoietic stem and progenitor cells, and in protecting leukemic stem cells from therapy which causes disease relapse. We made efforts to discuss these aspects in view of a kind of disturbance of the microecosystem within BM and thus proposed some new concepts in therapeutics of blood malignancies.
Similar content being viewed by others


Introduction
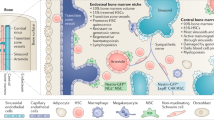
Hematopoiesis in the bone marrow (BM) produces mature blood cells on demand during an organism’s lifetime. Hematopoiesis occurs in a microenvironment with specific cellular composition and molecular signaling called niche. Found in BM, serial functional niches support hematopoietic stem cells (HSCs) and their progenies [1–11], which systemically form a functional unit of hematopoiesis in an anatomical locus (Fig. 1). Blood cells (including stem, progenitor, and mature cells) in this unit form a colony. Numerous units in a territory, such as bones, constitute a microecosystem; similar to those in a natural ecosystem, clonal size, intensity and distribution of each colony, inter-colony interaction, and interaction with niches are regulated by homeostasis [12, 13]. Deregulation in BM microecosystem balance caused by the emergence of mutated subclones, invaders (metastasis), stress factors, or autoimmune attacks may result in bone marrow failure, dysplasia, or malignancy.

Niches identified for HSPCs in the bone marrow. Quiescent HSCs are in the arteriolar niche near to the endosteum, which are mainly supported by nestin+ MSCs, sympathetic neurons, and may be osteoblasts and megakaryocytes. Cycling HSCs are in the sinusoid niches. Early lymphoid progenitor cells are in the endosteal niche being supported by osteoblasts and CXCL12-abundant reticular (CAR) cells. B-cell progenitors are in the sinusoid niche. Erythroblasts are in a niche being supported by CD169+ macrophages
In the last decade, advanced experimental systems have enabled us to better investigate BM microenvironment. Imaging systems based on two-photon (or multi-photon) confocal microscopy have been used to observe blood cell behaviors in their niche ex vivo or in vivo. Furthermore, advanced transgenic targeting techniques, including lineage-tracing [4, 7, 14], specific lineage-depletion [4], and conditional depletion of niche molecules from distinct niche cells [6, 7, 10], have been applied to identify and characterize specific niche cells and niche molecules. Computational modeling can link imaging information with three-dimensional (3D) associations between niche cells and niche-resident cells [5]. With these advanced techniques, several functional niches in BM for hematopoietic stem and progenitor cells (HSPCs) have been identified. Contribution of each technology has been reviewed elsewhere [15–23]. In a brief summary here, the cells that have been identified a composition of the niches include endothelial cells that compose arteriolar and sinusoid vessels, mesenchymal stem or progenitor cells and their mature progenies, nerve cells, and some mature hematopoietic cells (Fig. 1). Corresponding signaling circuits between these niches and HSPCs have also been revealed [6, 24–27].
The same techniques have been used for investigation of the BM niches in malignancies. In spite of some exiting findings recently, much more remain unknown in malignant BM than that in the normal due to constrains of availabilities of disease models and patient samples. Recently findings can be reviewed and attributed as follows: Oncogenesis in the abnormal BM niches; relevant alterations in the BM niches caused by malignant cell infiltration; effects of the alterations on normal hematopoiesis; protection conferred for cancer propagation cells in response to therapy; and potential therapeutic targets to circumvent the niche protection.
Oncogenesis in abnormal niches
In view of the fact that BM niches play essential roles in regulating hematopoiesis, it was presumed and tested that alterations of BM niches might lead to or promote the formation of blood malignances. In an early elegant study, mice with deficiency of retinoic acid receptor gamma (RARγ) develop Myeloproliferative neoplasms (MPNs) characterized by significantly increased granulocyte/macrophage progenitors and granulocytes in BM, peripheral blood, and spleen. Interestingly, BM from wild-type mice transplanted into the RARγ-deficient microenvironment rapidly developed the MPNs, indicating that the disorder was not intrinsic to hematopoietic cells and thus demonstrating the possibility that alterations of the microenvironment as a sole cause of hematopoietic neoplasms [28]. In a parallel study, mice with combined loss of Retinoblastoma (RB) in both hematopoietic cells and microenvironment could develop MPN, indicating that the disease resulted from an interaction between blood cells and the altered microenvironment [12].
Mesenchymal stem/progenitor cells (MSPCs) and its progenies, including osteoprogenitors, have been identified important niche cells that are functionally essential to HSPCs. Several studies found that deletion or mutation of functional genes in these niche cells led to MPNs, myelodysplasia, or leukemia [12, 28–32]. These findings and accordingly raised concepts were proved by the clinical transplantation investigations, in which donor-derived hematologic malignancies after hematopoietic stem cell transplantation were diagnosed [33].
In the other hand, intrinsically mutant HSPCs might modify BM niches to gain survival advantages over normal HSPCs in oncogenesis. These were observed in different models of MPNs.
In the BM of MPN patients and mice expressing human Janus kinase 2 (JAK2)-V617F mutation in HSPCs which causes MPN, the researchers observed that sympathetic nerve fibres, supporting Schwann cells and nestin(+) MSPCs, those are important niche cells of HSPCs, are consistently reduced. They further found that V617F mutant HSPCs highly expressed and produced interleukin-1β which induced neural damage and cell death of Schwann cells and MSPCs in the BM. The observations demonstrated that mutant HSPC-induced niche alterations essentially contribute to the emergence and development of hematopoietic malignancies [34].
In a BCR/ABL transgenic MPN model, the process in which oncogenesis modifies BM environment was observed. MPN development causes endosteal osteoblastic lineage cell (OBC) expansion and myelofibrosis development. MPN myeloid cells stimulate MSPCs to overproduce OBCs by secreting TPO and CCL3, thereby providing direct cell-to-cell interaction. Expanded OBCs are leukemia-trained, thereby effectively supporting leukemic stem cells (LSCs) but compromising the ability to maintain normal HSCs. Indeed, MPN development remodels an endosteal BM niche to form a self-reinforcing leukemic niche [35].
In summary, these findings together with the results in the other studies that are not included in this review indicate that alterations of BM niches (either primary or secondary) play essential roles in the initiation and development of leukemia. However, in most (maybe all) of these studies, only proliferative neoplasms (a pre-leukemic phase), but not overt leukemias, were developed in their animal models. It remains to be determined whether the altered BM niches provide permissive microenvironments for the pre-leukemic cells to gained leukemic mutations or selective microenvironments for the pre-leukemic cells which have had leukemic lesions to compete over other clones, in either way to achieve complete leukemia transformation. Further studies should also be conducted to determine the differential contributions of the microenvironment to the oncogenesis of distinct hematopoietic malignancies.
Dissemination of malignant cells alters normal BM niches
BM is a common place where cancer cells are distributed. BM can be considered as a ubiquitous site of malignant blood cell metastasis. For example, leukemic cells possibly migrate from origin to adjacent as well as distant locations along with the blood flow. Similar to an invader, cancer cells should find a portal to enter the BM microenvironment. In an elegant study conducted in a xenograft model of human acute lymphoblastic leukemia (ALL) cells, cell line Nalm-6, a portal was identified in a unique and spatially restricted vascular microdomain in calvarial flat bone [36]. In this microdomain, circulating cells are arrested by rolling along and binding to the endothelium, exude at these sites, and localize in perivascular sites [36]. The endothelium in this microdomain expresses E-selectin and SDF-1 to regulate this restricted leukemic cell homing. The SDF-1-CXCR4 system is essential for leukemic cells to recognize and bind permissive vasculature at the earliest homing stage. E-selectin may also enhance the homing process [36].
It remains unknown whether in all other bones, for example, long bone femurs and tibias, this identical microdomain also exists, whether in the same process or mechanism, the leukemic cells enter and localize in the perivascular sites, and whether in the same ways, all types of tumor cells metastasize into BM. Well addressing these issues will be crucial to the targeted blockage of the metastatic process in the BM.
In our own studies, we observed that leukemic cells localize and graft in the preferential microenvironment where metastatic tumor cells spread in BM, alter, and disrupt normal BM microenvironments [37].
Although the mechanism of the niche alteration or damage remains much unknown, this process may be attributed to the infiltration of distinct types of cancer cells, thereby altering BM microenvironment in different ways. In xenograft models of human acute myeloid leukemia (AML), “normal” environmental structures are restored after these models are released from leukemic burden by chemotherapy [38, 39]. Changes in BM environment may have resulted from anatomical “crowding out” of normal blood cells and mesenchymal cells. Similar to a sponge, BM can be squeezed and its shape can be restored when pressure is released (Fig. 2a).

Distinct alteration models of BM niches by leukemic cell infiltration in AML, B-ALL, or T-ALL. a In AML, the marrow structure is squeezed by heavy leukemic burden, and restored after therapy. The residual leukemic cells after therapy are in the endosteal niche. b In B-ALL, osteoblasts and vascular tissues are synchronously disrupted and damaged, and the normal structures are not restored after therapy. The survival leukemic cells are in a therapy-induced niche initiated by Nestin+ MSCs. c In T-ALL, leukemic cell infiltration damages osteoblasts and spares vascular structures. Leukemic cells evade therapy by keeping rapid migration
By contrast, different results were observed in human B-cell ALL (B-ALL) models. Leukemic dissemination synchronously disrupts osteoblasts and vascular structures, which are the main components of normal endosteal and vascular niches [37]. The extent of alteration depends on leukemic cell engraftment level. After these models are released from leukemic burden by chemotherapy, “normal” environmental structure could not be restored [37]. In B-ALL, the dissemination of leukemic cells within BM may mechanically squeeze and chemically disrupt or modify marrow tissues. Leukemic cells may produce and secrete chemokines (or enzymes) spontaneously or in response to therapy. Stromal or mesenchymal tissues are eroded, and the 3D architecture is corrupted (Fig. 2b).
Most recently, another picture was shown in T-cell ALL (T-ALL) models. Full infiltration of T-ALL in recipient BM caused dramatic shrinking, blebbing, and apoptosis of osteoblastic cells, and spared blood vessels and nestin+ perivascular cells (Fig. 2c) [40].
Together, these studies, albeit done in different laboratories, revealed that the scenarios of BM niche alteration by leukemia infiltration seem much different and dependent on leukemia types, and that the distinct scenarios may much differentially affect the behavior of leukemic cells in the disease development, progress, and relapse after therapy. We will discuss these in the later paragraphs. Further studies should be conducted regarding the mechanism by which functional BM niches in each leukemia type are altered or damaged in cellular and molecular components to address two clinically relevant issues: suppression of normal hematopoiesis and formation of minimal residual disease (MRD) in BM which causes relapse.
Dissemination of malignant cells affects normal hematopoiesis
Defects in hematopoiesis are frequently observed in patients with malignant BM. How do cancer-induced alterations in the microenvironment affect benign cellular ecosystems? The suppression of normal hematopoiesis may not necessarily result from the anatomical “crowding out” of benign cells, because suppression can occur even when relatively low cancer cell burden is present.
Changes in normal hematopoiesis in leukemic BM environment have been investigated using xenograft mouse models and congenic transplantation mouse models. Comore A et al. used a xenograft mouse model and observed that leukemic cell growth disrupts normal BM niches and creates abnormal niches that hijack transplanted human CD34+ HSPCs. A malignant niche out-competed native niches for CD34+ cell localization but failed to maintain the number of these cells. Sequestered HSPCs fail to mobilize into the peripheral blood in response to cytokine stimulation. Furthermore, leukemic cell-produced SCF, rather than SDF-1, recruits HSPCs to the leukemic niche [41].
Hu X et al. used a congenic transplantation mouse model of T-cell leukemia and observed the kinetics and functions of normal HSPCs. The overall hematopoiesis is progressively suppressed during leukemia development. In addition, leukemic environment elicits distinct effects on normal stem and progenitor cells. Normal HSCs in leukemic mice are maintained in a more quiescent state and retain their stem cell functions. Hematopoietic progenitor cells (HPCs) demonstrate accelerated proliferation and exhaustion [42]. Further studies are needed to provide insights into BM to reveal the mechanism by which leukemic cells interfere with the niches specific to HSCs and HPCs.
A similar suppression of normal hematopoiesis in leukemic BM has been observed in transgenic BCR-ABL+ leukemia/MPN mice [43]. In the same model, a suppression mechanism has been observed. Leukemic niche cells or MPN-expanded OBCs undergo compromised HSC-supportive activities that greatly downregulate numerous HSC retention factors [35].
These pilot observations enter the “gateway” of BM niche in leukemia. Further investigations should be conducted to elucidate the niches disturbed by leukemic dissemination, determining the changes in terms of its locations, cellular compositions, functional capacities, and signaling regulations.
LSCs hide in distinct preferential niches in response to therapy
LSCs evade therapy, thereby causing relapse. Studies on this issue have mainly focused on cell-intrinsic aspects encompassing the importance of differentiation stage or epigenetic programs in resistant sub-populations or the importance of genetic variation among different subclones in an individual cancer type [44–49]. However, the subclonal residence of LSCs in preferential microenvironmental niches may mainly participate in therapy evasion. These findings should be further investigated in terms of the nature of related niches and mechanisms that protect surviving subclones [50, 51]. One of the most relevant issues is to determine where LSCs find refuge to survive disruption caused by chemotherapy, usurpation of normal niches, or creation of LSC-specific niches.
LSCs may occupy normal niches. The mechanism by which BM niches support normal HSCs is possibly applied to protect LSCs; this mechanism has been investigated in xenograft leukemia models. LSCs localize and engraft in normal endosteal niches which used to keep HSCs in quiescence and chemoresistance (Fig. 2a) [38, 39]. Leukemia engrafted in HSC niches induces alteration in this niche. In addition, such alteration favors leukemic cell growth. For example, ALL blasts highly express osteopontin (OPN) when these blasts are grafted in an endosteal niche, which is a pro-dormancy niche. These blasts are tightly anchored by OPN in this niche and induced into a quiescent phase by other unknown factors in a local microenvironment [52]. Therefore, the induction of cell cycle entry facilitates cytotoxic agents to eliminate LSCs in mouse models [38, 52] and may be translated to clinical treatments.
This scenario seems more complex, because the growth of leukemic cells damages and modifies BM environments differently as discussed in the previous paragraph [35, 41, 43]. LSCs may create their own niches. We reported that ALL-LSCs hide in native niches (both endosteal and vascular types) when these niches are not substantially damaged. Once the native niches are damaged, LSCs develop a novel niche to evade therapy. A novel concept is thus conceptualized, that is, a therapy-induced niche. The interplay between a niche and niche-resident cells mediates niche protection (Fig. 2b) [37].
Most recently, a much provocative report showed that T-ALL cells do not depend on any specific BM sub-compartments in seeding and colonization of BM, and survival of chemotherapy. Instead, T-ALL cells exhibit promiscuous distribution in BM and dynamic interactions with BM microenvironments throughout disease development from the early BM seeding to response and resistance to chemotherapy [40]. And, thus, a stochastic mechanism underlying these processes is proposed (Fig. 2c) in contrast to the findings and hypothesis in other leukemias in which specifically niches support and protect resident leukemic cells [37, 39].
Together, these pilot observations of the microenvironment in the different type of leukemias indicate that distinct leukemic cells behavior differently in the BM microenvironment. Comparative investigations are required to figure out these differences and thus guide the precise identification of potential therapeutic targets to interfere with or relieve the niche protection in different leukemias.
Potential targets to circumvent niche protection
Considering that LSCs hide in “normal” BM niche in the quiescent stage, researchers induce (or mobilize) quiescent LSCs to enter cell cycle and administer to eliminate residual LSCs in BM. Ishikiwa revealed that G-CSF treatment significantly facilitates cell cycle-dependent chemotherapy to eliminate human primary AML stem cells in xenograft models [38]. Sipkins further demonstrated that the combination of a single OPN neutralization treatment before Ara-C significantly enhanced the elimination of residual leukemic cells [52].
LSC-generated niche may use specific signals to mediate niche protection for its resident cells. Signal interference, such as niche-LSC interaction protein, may provide another approach for the circumvention of niche protection and enhance the possibility of eliminating residual LSCs. We reported that GDF15 mediates the interaction between a therapy-induced niche and its resident LSCs, thereby conferring chemoresistance to residual LSCs in B-ALL. Furthermore, interfering with GDF15 expression or function significantly enhances chemotherapy efficacy [37].
Moving targets are certainly more difficult to be shot, although therapeutic interventions for T-ALL have been proposed to target the migration and promiscuous interactions of leukemic cells with the BM environment [40].
The residence of cancer stem cells (CSCs) in preferential microenvironmental niches affects therapy efficacy. Although this phenomenon should be further investigated in terms of the nature of related niches and mechanisms that protect surviving CSCs, established experimental models can be used to explore new approaches to circumvent niche protection. Ishikawa reported the research advancements in high-throughput drug screening (known and unknown compounds) using AML xenograft models [53].
Perspectives
Ecology deals with interactions between organisms and their environment. Natural ecosystem-related principles, such as evolutionary fitness and natural selection, have successfully been applied to investigate the pathogenesis and therapeutics of cancers [54–57]. Stem cells are considered as the unit of initiation and evolution of pre-cancerous and cancerous clones. The identification and characterization of stem cells in these tissues have been providing substantial insights into arresting oncogenesis and curing cancers by specifically targeting these stem cells and their resident microenvironments or niches [13, 46, 50, 51]. Leukemogenesis in BM is one of the classic examples of events occurring in microecosystems.
CSCs are closely associated with the heterogeneities of cancer cells. The history of CSC studies on aspects of cancer cell heterogeneities has been archived [46]. These aspects include morphology, immunophenotype, function, and genetics. For instance, intraclonal genetic diversities have been analyzed and results revealed that cancer exhibits a subclonal architecture. The resistance of several subclones to therapy may be intrinsic and resembles the genetic signature of cells. This resistance is also associated with niches, because each subclone may prefer specific niche. CSCs and niches dynamically evolve, and these two aspects rely on and modify each other [58–60]. As CSCs and niches mature and form a rigid and resistant system, any therapy could fail until a particular host dies. Therefore, further studies should focus on the heterogeneities of CSC niches; specific niches that protect resistant subclones of CSCs should also be identified. Further insights into these niches help reveal the mechanism of niche-CSC interaction that contributes to drug resistance and identify potential targets to circumvent niche protection.
Clinical and experimental oncologists realized that appropriate primary therapy is necessary to achieve complete remission and prevent MRD formation and relapse. Studies on CSC behavior in niches have revealed that primary therapy substantially affects and modifies CSCs and their niches. CSCs hijack and modify existing niches or create new niches to evade therapy. Hence, the characteristics of therapy-modified or therapy-induced niches and relevant mechanisms that protect surviving CSCs should be understood to prevent MRD formation and relapse.
A complete innovative concept has recently been proposed to control CSCs and thus the disease. That is to control the fate of CSCs rather than to kill them or to relieve its potential of cancer propagation [61]. A better approach to achieve this would be via manipulation of the microecosystem of BM environments. To maintain a condition in the system, normal blood cells and malignant cells mildly compete and reach a relative homeostasis. So that the host may benefit best from the treatments and has a long-lasting life with coexistence of leukemic cells in their niches.
References
Kiel MJ, Yilmaz OH, Iwashita T, Terhorst C, Morrison SJ. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005;121(7):1109–21.
Calvi LM, Adams GB, Weibrecht KW, Weber JM, Olson DP, Knight MC, Martin RP, Schipani E, Divieti P, Bringhurst FR, et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature. 2003;425(6960):841–6.
Omatsu Y, Sugiyama T, Kohara H, Kondoh G, Fujii N, Kohno K, Nagasawa T. The essential functions of adipo-osteogenic progenitors as the hematopoietic stem and progenitor cell niche. Immunity. 2010;33(3):387–99.
Mendez-Ferrer S, Michurina TV, Ferraro F, Mazloom AR, Macarthur BD, Lira SA, Scadden DT, Ma’ayan A, Enikolopov GN, Frenette PS. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature. 2010;466(7308):829–34.
Kunisaki Y, Bruns I, Scheiermann C, Ahmed J, Pinho S, Zhang D, Mizoguchi T, Wei Q, Lucas D, Ito K, et al. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature. 2013;502(7473):637–43.
Greenbaum A, Hsu YM, Day RB, Schuettpelz LG, Christopher MJ, Borgerding JN, Nagasawa T, Link DC. CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature. 2013;495(7440):227–30.
Ding L, Saunders TL, Enikolopov G, Morrison SJ. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature. 2012;481(7382):457–62.
Bruns I, Lucas D, Pinho S, Ahmed J, Lambert MP, Kunisaki Y, Scheiermann C, Schiff L, Poncz M, Bergman A, et al. Megakaryocytes regulate hematopoietic stem cell quiescence through CXCL4 secretion. Nat Med. 2014;20(11):1315–20.
Zhao M, Perry JM, Marshall H, Venkatraman A, Qian PX, He XC, Ahamed J, Li LH. Megakaryocytes maintain homeostatic quiescence and promote post-injury regeneration of hematopoietic stem cells. Nat Med. 2014;20(11):1321–6.
Ding L, Morrison SJ. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature. 2013;495(7440):231–5.
Chow A, Huggins M, Ahmed J, Hashimoto D, Lucas D, Kunisaki Y, Pinho S, Leboeuf M, Noizat C, van Rooijen N, et al. CD169(+) macrophages provide a niche promoting erythropoiesis under homeostasis and stress. Nat Med. 2013;19(4):429–36.
Walkley CR, Shea JM, Sims NA, Purton LE, Orkin SH. Rb regulates interactions between hematopoietic stem cells and their bone marrow microenvironment. Cell. 2007;129(6):1081–95.
Weissman IL. Stem cells: Units of development, units of regeneration, and units in evolution. Cell. 2000;100(1):157–68.
Lo Celso C, Fleming HE, Wu JW, Zhao CX, Miake-Lye S, Fujisaki J, Cote D, Rowe DW, Lin CP, Scadden DT. Live-animal tracking of individual haematopoietic stem/progenitor cells in their niche. Nature. 2009;457(7225):92–6.
Calvi LM, Link DC. The hematopoietic stem cell niche in homeostasis and disease. Blood. 2015;126(22):2443–51.
Park D, Sykes DB, Scadden DT. The hematopoietic stem cell niche. Front Biosci Landmark. 2012;17:30–9.
Mendelson A, Frenette PS. Hematopoietic stem cell niche maintenance during homeostasis and regeneration. Nat Med. 2014;20(8):833–46.
Boulais PE, Frenette PS. Making sense of hematopoietic stem cell niches. Blood. 2015;125(17):2621–9.
Kfoury Y, Scadden DT. Mesenchymal cell contributions to the stem cell niche. Cell Stem Cell. 2015;16(3):239–53.
Scadden DT. Nice neighborhood: emerging concepts of the stem cell niche. Cell. 2014;157(1):41–50.
Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature. 2014;505(7483):327–34.
Li LH, Clevers H. Coexistence of quiescent and active adult stem cells in mammals. Science. 2010;327(5965):542–5.
Wilson A, Trumpp A. Bone-marrow haematopoietic-stem-cell niches. Nat Rev Immunol. 2006;6(2):93–106.
Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity. 2006;25(6):977–88.
Ludin A, Gur-Cohen S, Golan K, Kaufmann KB, Itkin T, Medaglia C, Lu XJ, Ledergor G, Kollet O, Lapidot T. Reactive oxygen species regulate hematopoietic stem cell self-renewal, migration and development, as well as their bone marrow microenvironment. Antioxid Redox Signal. 2014;21(11):1605–19.
Gur-Cohen S, Kollet O, Graf C, Esmon CT, Ruf W, Lapidot T. Regulation of long-term repopulating hematopoietic stem cells by EPCR/PAR1 signaling. Ann N Y Acad Sci. 2016;1370(1):65–81.
Matsumoto Y, Suda T. Regulation of hematopoietic stem cells in niche signaling. Nihon Rinsho. 2012;70(Suppl 2):133–8.
Walkley CR, Olsen GH, Dworkin S, Fabb SA, Swann J, McArthur GA, Westmoreland SV, Chambon P, Scadden DT, Purton LE. A microenvironment-induced myeloproliferative syndrome caused by retinoic acid receptor gamma deficiency. Cell. 2007;129(6):1097–110.
Kim YW, Koo BK, Jeong HW, Yoon MJ, Song R, Shin J, Jeong DC, Kim SH, Kong YY. Defective Notch activation in microenvironment leads to myeloproliferative disease. Blood. 2008;112(12):4628–38.
Fulzele K, Krause DS, Panaroni C, Saini V, Barry KJ, Liu XL, Lotinun S, Baron R, Bonewald L, Feng JQ, et al. Myelopoiesis is regulated by osteocytes through Gs alpha-dependent signaling. Blood. 2013;121(6):930–9.
Kode A, Manavalan JS, Mosialou I, Bhagat G, Rathinam CV, Luo N, Khiabanian H, Lee A, Murty VV, Friedman R, et al. Leukaemogenesis induced by an activating beta-catenin mutation in osteoblasts. Nature. 2014;506(7487):240–4.
Dong L, Yu WM, Zheng H, Loh ML, Bunting ST, Pauly M, Huang G, Zhou MX, Broxmeyer HE, Scadden DT, et al. Leukaemogenic effects of Ptpn11 activating mutations in the stem cell microenvironment. Nature. 2016;539(7628):304–8.
Sala-Torra O, Hanna C, Loken MR, Flowers MED, Maris M, Ladne PA, Mason JR, Senitzer D, Rodriguez R, Forman SJ, et al. Evidence of donor-derived hematologic malignancies after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2006;12(5):511–7.
Arranz L, Sanchez-Aguilera A, Martin-Perez D, Isern J, Langa X, Tzankov A, Lundberg P, Muntion S, Tzeng YS, Lai DM, et al. Neuropathy of haematopoietic stem cell niche is essential for myeloproliferative neoplasms. Nature. 2014;512(7512):78–81.
Schepers K, Pietras EM, Reynaud D, Flach J, Binnewies M, Garg T, Wagers AJ, Hsiao EC, Passegue E. Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche. Cell stem cell. 2013;13(3):285–99.
Sipkins DA, Wei X, Wu JW, Runnels JM, Cote D, Means TK, Luster AD, Scadden DT, Lin CP. In vivo imaging of specialized bone marrow endothelial microdomains for tumour engraftment. Nature. 2005;435(7044):969–73.
Duan CW, Shi J, Chen J, Wang B, Yu YH, Qin X, Zhou XC, Cai YJ, Li ZQ, Zhang F, et al. Leukemia propagating cells rebuild an evolving niche in response to therapy. Cancer Cell. 2014;25(6):778–93.
Saito Y, Uchida N, Tanaka S, Suzuki N, Tomizawa-Murasawa M, Sone A, Najima Y, Takagi S, Aoki Y, Wake A, et al. Induction of cell cycle entry eliminates human leukemia stem cells in a mouse model of AML. Nat Biotechnol. 2010;28(3):275–80.
Ishikawa F, Yoshida S, Saito Y, Hijikata A, Kitamura H, Tanaka S, Nakamura R, Tanaka T, Tomiyama H, Saito N, et al. Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat Biotechnol. 2007;25(11):1315–21.
Hawkins ED, Duarte D, Akinduro O, Khorshed RA, Passaro D, Nowicka M, Straszkowski L, Scott MK, Rothery S, Ruivo N, et al. T-cell acute leukaemia exhibits dynamic interactions with bone marrow microenvironments. Nature. 2016;538(7626):518–22.
Colmone A, Amorim M, Pontier AL, Wang S, Jablonski E, Sipkins DA. Leukemic cells create bone marrow niches that disrupt the behavior of normal hematopoietic progenitor cells. Science. 2008;322(5909):1861–5.
Hu X, Shen H, Tian C, Yu H, Zheng G, XuFeng R, Ju Z, Xu J, Wang J, Cheng T. Kinetics of normal hematopoietic stem and progenitor cells in a Notch1-induced leukemia model. Blood. 2009;114(18):3783–92.
Zhang B, Ho YW, Huang Q, Maeda T, Lin A, Lee SU, Hair A, Holyoake TL, Huettner C, Bhatia R. Altered microenvironmental regulation of leukemic and normal stem cells in chronic myelogenous leukemia. Cancer Cell. 2012;21(4):577–92.
Anderson K, Lutz C, van Delft FW, Bateman CM, Guo Y, Colman SM, Kempski H, Moorman AV, Titley I, Swansbury J, et al. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature. 2011;469(7330):356–61.
Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5(4):275–84.
Dick JE. Stem cell concepts renew cancer research. Blood. 2008;112(13):4793–807.
Hong D, Gupta R, Ancliff P, Atzberger A, Brown J, Soneji S, Green J, Colman S, Piacibello W, Buckle V, et al. Initiating and cancer-propagating cells in TEL-AML1-associated childhood leukemia. Science. 2008;319(5861):336–9.
Notta F, Mullighan CG, Wang JC, Poeppl A, Doulatov S, Phillips LA, Ma J, Minden MD, Downing JR, Dick JE. Evolution of human BCR-ABL1 lymphoblastic leukaemia-initiating cells. Nature. 2011;469(7330):362–7.
Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC, Wartman LD, Lamprecht TL, Liu F, Xia J, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150(2):264–78.
Lane SW, Scadden DT, Gilliland DG. The leukemic stem cell niche: current concepts and therapeutic opportunities. Blood. 2009;114(6):1150–7.
Meads MB, Gatenby RA, Dalton WS. Environment-mediated drug resistance: a major contributor to minimal residual disease. Nat Rev Cancer. 2009;9(9):665–74.
Boyerinas B, Zafrir M, Yesilkanal AE, Price TT, Hyjek EM, Sipkins DA. Adhesion to osteopontin in the bone marrow niche regulates lymphoblastic leukemia cell dormancy. Blood. 2013;121(24):4821–31.
Saito Y, Yuki H, Kuratani M, Hashizume Y, Takagi S, Honma T, Tanaka A, Shirouzu M, Mikuni J, Handa N, et al. A pyrrolo-pyrimidine derivative targets human primary AML stem cells in vivo. Sci Transl Med. 2013;5(181):18152.
Greaves M. Darwinian medicine: a case for cancer. Nat Rev Cancer. 2007;7(3):213–21.
Greaves M, Maley CC. Clonal evolution in cancer. Nature. 2012;481(7381):306–13.
Merlo LM, Pepper JW, Reid BJ, Maley CC. Cancer as an evolutionary and ecological process. Nat Rev Cancer. 2006;6(12):924–35.
Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194(4260):23–8.
Bissell MJ, LaBarge MA. Context, tissue plasticity, and cancer: are tumor stem cells also regulated by the microenvironment? Cancer Cell. 2005;7(1):17–23.
Borovski T, De Sousa EMF, Vermeulen L, Medema JP. Cancer stem cell niche: the place to be. Cancer Res. 2011;71(3):634–9.
Rosen JM, Jordan CT. The increasing complexity of the cancer stem cell paradigm. Science. 2009;324(5935):1670–3.
Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature. 2013;501(7467):328–37.
Acknowledgements
This study was supported by Grants from the National Basic Research Program of China (2012CB967000) and the National Natural Science Foundation of China (NSFC) (91442106 and 81171888).
Author information
Authors and Affiliations
Corresponding author
Additional information
L.-L. Mu, F. Ke, X.-L. Guo, and J.-J. Cai contributed equally to this work.
About this article

Cite this article
Mu, LL., Ke, F., Guo, XL. et al. Neoplasms in the bone marrow niches: disturbance of the microecosystem. Int J Hematol 105, 558–565 (2017). https://doi.org/10.1007/s12185-017-2193-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-017-2193-5