
Abstract
Leukemias depend on transformed stem cells for their growth and thus these cells represent important therapeutic targets. However, leukemic stem cells resemble normal hematopoietic stem cells (HSCs) with respect to most surface markers, gene expression patterns, and ability to be transplanted. Furthermore, the microenvironment that supports healthy HSCs non-hematopoietic populations, and immune cells correspondingly, the cytokines, adhesion molecules and signal transduction pathways are also impaired during leukemogenesis. This altered environment promotes leukemic growth specifically through pro-inflammatory cytokines. Here, we characterize normal and leukemic signaling, as well as the instructive cues from the neighboring hematopoietic cells and the microenvironment that promote stem cell self-renewal and differentiation.
Similar content being viewed by others


Introduction
All mature blood cells are derived from rare hematopoietic stem cells (HSCs) that reside in the adult bone marrow [1]. Evidence shows that proper gene expression and signaling transduction in HSCs is essential for homeostasis. Several transcription factors and signaling pathways are critical for the formation and function of HSCs, as well as for the differentiation to specific blood lineages. However, malfunctioning HSCs through dysregulated transcription factor expression can result in malignancies. Furthermore, following hematologic injuries or leukemia, alterations in non-transformed HSC result in life threatening complications from infection, bleeding, and anemia. In this review, we focus on work that strives to understand the cells and signaling pathways that regulate, maintain, and interact with HSCs and leukemic stem cells.
Cytokines and immune regulation of HSCs and LSCs
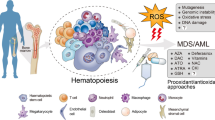
The hematopoietic stem cells (HSC) rely on mature myeloid and lymphoid lineage cells to help regulate their differentiation and self-renewal. One function is to protect stem cells and allow them to undergo adequate maturation; another is to eliminate HSCs that have become malignant [2]. The transition from normal to aberrant hematopoiesis is accompanied by a spectrum of immunological alterations (Fig. 1). During leukemogenesis vital cell functions such as proliferation, differentiation, and signal transduction become dysregulated [3, 4]. Furthermore, the cytokines that regulate normal hematopoiesis are also perturbed. The interchange of these molecules is needed to maintain the hematopoietic microenvironment and immune homeostasis [5]. While cytokine networks mediate communication across numerous cell types, one of their principal functions is to serve as immunomodulating agents; they are specifically important for coordinating the class and magnitude of the immune response [6]. Downstream effects can be antagonistic, additive, or synergistic. Currently, cytokine immunotherapy is being applied to combat leukemic stem cell (LSC) pathophysiology [7, 8]. However, the pleiotropic nature of cytokine signaling makes it difficult for treatments to be specific and have a high efficacy; off target effects are a major obstacle. To move forward, a more detailed and precise mapping of the cytokine landscape is necessary.

a Immune cells act as homeostatic plasticizers of the hematopoietic niche. A number of resident immune cells cross-talk with HSCs, stroma, and other BM populations via cytokine, chemokine, and soluble peptide release. They protect cells, allow them to undergo adequate maturation, and are also responsible for trimming of malignant clones. b MDS malignancies often transition to leukemia and disease progression involves deterioration of immune system functionality. c Leukemic stem cells release suppressive cytokines and chemokines to evade immune detection. This allows expansion of malignant clones, and hampers the ability of immune cells to respond to aberrant hematopoiesis
LSCs utilize specific mechanisms to escape immune surveillance; they secrete various co-stimulatory molecules and suppressive cytokines[9]. This hinders effector immune cell function, and deleteriously reconstitutes the hematopoietic microenvironment. At the molecular level LSCs and their differentiated progeny exhibit unique genetic signatures and expression profiles [10, 11] that allow them to express different antigens [12]. LSCs also target metabolic pathways [13], and create an anti-apoptotic environment which facilitates their own proliferation while distorting the recycling of immune cells [14]. Additionally, several surface proteins common to immune cells, including T cell immunoglobulin mucin-3 (TIM-3) have been associated with LSC self-renewal and leukemic transformation [15].
The bone marrow is an important immunological transit point, as previously reviewed [16]. In addition to serving as a primary organ for hematopoiesis, the bone marrow is also an integral site for lymphocyte circulation [17]. The cytokine networks that run through the bone marrow are important for HSC maturation, pluripotency, and contact with immune cells. Alterations of this milieu within the bone marrow have been implicated in clonal stem cell disorders, as well as expansion LSCs [7]. For both LSCs and immune cells, the bone marrow serves as a supportive apparatus for growth and development. Correspondingly, it is the principal site for crosstalk between these cells. Thus, it is necessary to tease out the cell–cell contact mechanisms and soluble factors that are exchanged between LSC and immune cells within the bone marrow microenvironment. The trafficking of various immune cells within this microenvironment has been observed. These immune cell populations include, conventional T cells, regulatory T cells (T-regs), B cells, Natural Killer cells (NKs), Neutrophils, Dendritic Cells (DCs), and myeloid derived suppressor cells (MDSCs) [18]. Not only does the bone marrow act as a reservoir for immune cells, it also serves as a differentiation regulatory organ. In thymectomized animals, the bone marrow provides necessary architectural elements to support T cell maturation [19, 20]. In addition to modulating HSC compartment, the immune cell trafficked through the bone marrow also cross-talk with other resident cells. Therefore, it can be expected that, the dysregulation of immune networks within the bone marrow has sweeping effects on the niche populations.
During hematopoiesis, immune cells play a requisite modulatory role. Resident cells maintain HSC homeostasis through various mechanisms; they eradicate aberrant clones, balance cell proliferation, and protect stem cells from apoptosis [21]. T-regs are key mediators in this process. Notably, T-reg frequency in the bone marrow is higher than the spleen and lymph nodes; they have been shown to comprise as much as 30% of CD4+ T cells in the bone marrow [21]. T-regs are a dynamic class of cells that temper the immune response throughout the body. T-regs shield the stem cell compartment from autoimmunity, excessive inflammation, and apoptosis. Thus, the bone marrow serves as a site of immune ‘privilege’ where regulatory cells establish a gradient of immune suppression and regulation [22]. T-regs suppress CD4+ and CD8+ T cells cytokine production, prominently IL-2 [23]. They also secrete various inhibitory cytokines, such as IL-35 to regulate T cell activity and apoptosis [24, 25]. Consequently, a growing body of evidence suggests that T-regs are hijacked by LSCs for protection and survival. It is likely that LSCs recruit T-regs and exploit their modulatory capacities to escape detection [26]. The mechanism by which LSC target T-regs remains to be elucidated; they may act on them directly, or employ signals from the periphery.
In particular, myelodysplastic syndrome (MDS) is a useful model for characterizing the immune regulation of LSCs. MDS is marked by various defects which encumber normal hematopoiesis; this includes autoimmunity, excess inflammation, dysregulated cell death, impaired signaling. Accumulation of these events leads to aggressive clonal proliferation within the bone marrow that goes undetected by the immune system. Given the intersectionality between MDS and leukemia, as well as the propensity to proceed from one to the other, it is worth noting that there are common pathologies associated with both diseases. Two key features are the dysregulation of innate immunity, and an increase in inflammation [27]. In leukemia, miR-29b has been shown to alter the capacities of NK cells, which disrupts the innate immune response and allows LSCs to escape surveillance [28]. Correspondingly, reduced NK function is seen in MDS patients as the disease progresses [29], and synthetic reactivation of NK cell activity has been explored as a potentially therapeutic treatment [30]. With respect to signaling, cytokine stimulation and differentiation of NK cell populations has been shown to enhance NK cell response and functionality [31]. It has also been shown that LSCs employ the CD160 signaling axis, a pathway common to NK cells and CD8 T cells, to facilitate their expansion [32].
Mast cells, which serve as activators of inflammation, have also been implicated. Mast cell growth factor, a ligand for c-kit, has been shown to promote hematopoiesis and proliferation of leukemic precursors [33, 34]. Dysregulation of this receptor tyrosine kinase, accompanies the corrosion of the stem cell niche and the hematopoietic system in MDS and other stem cell malignancies. Likewise, the regulation of c-kit receptor by Ran-binding protein microtubule-organizing center appears to function in the development of bone marrow progenitors [35]. Leukemia-initiating-cells have also been associated with a mast cell signature; the IL2/CD25 axis may serve as a key regulator in leukemic cell activation [36]. As with leukemia, T-reg functionality is distorted in MDS; T-reg receptor profiles as well as migratory and homing capacities to the bone marrow are altered to favor LSC development [37]. Both MDS and leukemia are marked by unique cytokine and chemokine expression patterns [9] and it is likely that this alters the communication landscape between immune cells, stem cells, and stromal cells of the hematopoietic system. Similarly, disequilibrium of the signal transduction pathway and malignant cell survival has also been shown to be correlated [38]. A notable feature of leukemia and MDS development is the deregulation of Toll-like Receptors (TLRs); these receptors are critical for innate immune signaling [39] and they are important for the cooperative regulation of pro-inflammatory cytokines and chemokines. In MDS, a significant upregulation of TLR1, 2 and 6 has been observed; specifically, the TLR2 signaling axis has been shown to mediate inflammation via the activation of activate IL-8 and histone demethylase JMJD3. Likewise, levels of tumor necrosis factor alpha (TNF-α), interferon gamma (IFN-γ), transforming growth factor beta (TGF-β), and interleukins IL-6 and IL-8 are also elevated [40–42]. Moreover, TLR dysfunction has been implicated in the progression to leukemia where signal transduction of important proliferative and apoptotic pathways is not properly activated [43]. The cytokine imbalances and dysregulated activation of TLRs observed in MDS stifles immune cell function, increases inflammation, and disrupts cellular communication. Collectively, these changes contribute to the formation of the leukemic stem cell niche.
Many hematological disorders, such as MDS and leukemia are pathologies concomitant with aging. This increase in age causes significant remodeling of the bone marrow microenvironment. One feature is the gradual replacement of the bone marrow compartment with adipose tissue [44]. Another component of aging is immunosenescence. The decline of immune function that results from aging has various consequences on the stem cell lineages; it hinders the bone marrow’s ability to nurture precursors, leads to dysplastic changes in resident cells, and promotes defective apoptosis, and cytopenia [45]. Loss of functionality disrupts cytokine and hormone networks necessary to maintain the various hematopoietic lineages [46, 47]. Because HSCs depend on adequate signaling from the bone marrow for proper quiescence and self-renewal, the age-related decline of immune function is often a trigger for leukemic transformation [48]. Cellular senescence increases chronic inflammation through the senescence-associated secretory phenotype (SASP). This phenotype engages cell cycle and metabolism related genes, soluble factors (chemokines, growth factors, and interleukins), secreted proteases and ECM components [49–51]. IL-1, IL-6, and IL-8 are some of the most significantly upregulated interleukins [52]. The insulin-like growth factor (IGF)/IGF receptor signaling axis also contributes to SASP profile and an overall inflammatory phenotype [53]. Senescent cells also amplify the CXCR2 signaling networks via increased secretion of CXCR-2 (IL-8RB)-binding chemokines [54]. The aggregate effects of disrupted cellular signaling in combination with the loss of immunological capacity create a climate permissive of LSC expansion. Aging and cellular senescence introduces changes to the bone marrow niche, and resident populations which alter the landscape of soluble and insoluble factors in the microenvironment. These changes synergistically impede proper hematopoietic maturation.
Under normal conditions, HSCs move through a series of steps as they evolve into specialized cells. The burden of regulating these transitions falls heavily on the immune system. Proper clonal expansion and trimming of malignant cells is carried out under the directive of immune cells. In hematopoietic pathologies, such as leukemia, the transition from HSCs to LSCs is accompanied by a loss of immune function. When immune regulation fails, HSCs lose the ability to balance self-renewal and lineage commitment; hallmarks of LSC development include abnormal expansion and dysregulated proliferation. Given the dependency between these cell types, an investigation of immune components is required to determine the regulatory checkpoints that go awry during LSC biogenesis. The balance between immune surveillance and immune evasion is a critical topic of exploration regarding LSC development and disease survival.
LSCs crosstalk with the microenvironment
The bone marrow niche is a complex arrangement of cells such as osteoblasts, endothelial cells, mesenchymal stem cells, stromal cells, and the above-mentioned immune cells. Within the bone marrow (BM) two distinct niches have been observed, the endosteum and vascular niche. The majority of HSCs reside in the endosteum, inner surface of the BM, which is lined with cells such as osteoblasts and osteoclasts. The sinusoidal (vascular) niche is comprised of thin vessels along with reticular cells, stromal cells, mesenchymal stromal cells (MSCs), and neurons, all of which can regulate and sustain HSCs and allow for communication with the rest of the body. Similar to immune cells within the microenvironment, niche cells communicate with HSCs other through cytokines, chemokines, and adhesion molecules that regulate self-renewal, quiescence, and mobilization.
To define the cells that constitute the niche, fate-mapping models have shown that stromal tissue, bone, and cartilage are derived from increasing lineage-restricted stem and progenitor cells [55]. Growth plate studies reveal a high frequency of non-hematopoietic, non-endothelial cells that contain 8 distinct subpopulations with differential expression of CD105, Thy, 6C3, and CD200. The populations resolved from this staining are: skeletal stem cells (SSC), bone, cartilage, and stromal progenitor (BCSP), pre-BCSP, B-lymphocyte stromal progenitors (BLSP) 6C3 (as defined by expression of Ly-51), hepatic leukemia factor expressing cells (HEC), Thy (based on CD90/Thy1 expression), and pro-chondrogenic progenitor (PCP). SSC, pre-BCSP, and BCSP have the potential to give rise to bone, cartilage, and marrow, while BLSP, 6C3, HEC, and Thy primarily make bone. As suggested by its name, PCP gave rise to cartilage [55]. Similar to the hematopoietic system of differentiation, the skeletal system has a hierarchy of progenitor–precursor relationships that are just now being discovered.
Several factors known to support hematopoietic stem cells have been used to resolve subsets of cells within the bone marrow microenvironment (BMM). These include CXCL-12, angiopoietin-1, stem cell factor (SCF), jagged-1, and thrombopoietin [56]. Recent work has taken this a step further using single cell analysis to define populations in close proximity to HSCs [57]. Cells within short distances of HSPCs had upregulation of cell surface proteins Vcam1, Adam9, and Amot as well as immune response genes Map3K14, CXCL12, and IL-18. Additionally, these proximal cells were found to be lineage committed but still immature. Conditional knockout-models have showed that mature osteoblastic cell derived RNase angiogenin (ANG) regulates lymphoid progenitors, while mesenchymal and osteolineage progenitor cell derived ANG aids in HSC quiescence and repopulation. Furthermore, it was shown that proximal cells secrete IL-18 and employ this cytokine to regulate quiescence of short-term progenitors. Finally, it has been demonstrated that antibody inhibition of cell adhesion molecule, such as Embigin, promote an increase in the frequency of HSCs, progenitors, and colony-forming cells. Taken together, these results establish that the BMM is a highly organized unit of cells, and that these populations crosstalk with HSCs to regulate hematopoiesis.
The study of the BMM on the pathogenesis of hematopoietic malignancies is not well understood. Current literature points to an interaction between LSC and MSCs. This interaction may impact both populations, perhaps modulating the molecular pathways that regulate normal HSCs. Understanding the niche in leukemia will provide insight on how to alter the microenvironment to target malignant cells. Disruption of the niche generates an array of problems, which help LSCs evade therapeutic agents [58]. In xenograft models, LSCs disrupt the microenvironment and normal HSCs behavior through chemokine signaling. Stromal Cell Derived Factor 1 (SDF-1), also known as CXCL12, is a chemokine secreted by the microenvironment to maintain HSCs and facilitate their homing; it is upregulated during times of hypoxia and inflammation. However, upon serial transplantation, acute lymphoblastic leukemia LSCs were found to home to SDF-1 negative areas. Through cytokine signaling, these altered niches are able to dysregulate the microenvironment and exclude HSCs [59]. Confocal microscopy studies reveal that healthy HSCs compete with acute myeloid leukemia (AML) LSCs for physical niche occupancy in competitive transplantation models [60]. Mice receiving higher doses of either whole BM or HSCs were able to survive longer with LSCs due to either competition for occupancy or resources. Notably, the two cell types were not seen to inhabit the same environment. Together these studies demonstrate that the niche is altered during leukemia and suggests that LSCs physically outcompete HSCs.
The specific ways in which leukemic cells alter the microenvironment to disrupt normal hematopoiesis is not well defined. As mentioned previously, CXCL12 is decreased in some leukemia [59]; however, the role CXCL12 plays in LSCs maintenance is still unknown. High resolution imaging of fate-mapping mouse models reveal that LSCs home to the endothelial region of the BMM where CXCL12 production is stable [61]. Additionally, the burden of LSCs decreases upon the deletion of the CXCL12 gene from endothelial cells. Moreover, deletion or inhibition of CXCR4 from LSCs decreases the leukemic burden. To demonstrate therapeutic potential, a small molecule AMD3465, has been used as an antagonist to the receptor in both murine and human leukemic xenograft models. In both models, leukemic burden decreased upon addition of this small molecule therapy [61]. Similarly, AML mouse models have shown that the LSCs induce transcriptional changes in normal MSCs. BM stromal cells from AML patients show a shift in differentiation with more osteoblastic cells and less undifferentiated cells [62]. Additionally, the AML BM was also unable to form colonies and proliferate as readily as healthy BM. They also showed that cell-signaling molecules such as Jagged-1 and CXCL12 were altered in the BM microenvironment. Jagged-1 percentage was decreased in leukemic MSCs compared to healthy MSCs when co-cultured with normal HSCs. Moreover, there was a significant suppression of Hes-1 and Hes-5, downstream targets of notch signals. A similar trend was seen with CXCL12 expressing cells. Besides, healthy HSCs are unable to expand when cultured on leukemic MSCs [62].
Notch is a plasma membrane protein that functions in cell signaling by having its extracellular domain and intracellular domain cleaved. There are four types of Notch and majority of acute lymphoid leukemia patients test positive for a gain of function mutation in the intracellular domain of Notch1. A mouse model of T-ALL was used to determine the mechanism behind the leukemic expansion in the BM [58]. As with other leukemia, these leukemic cells were able to displace normal cells for space within the BMM. Additionally, there was a reduction of osteoblasts in the marrow. This correlated with decreased expression of osteoblast transcription factors such as Runx2, osterix, osteocalcin, and ostegrin, as well as CXCL12. Meanwhile, stromal cells showed an increased expression of IL-6, SCF, HIF1a, VEGF, and Jag1. Furthermore, Notch1 was found to be able to bind to the promoter of the CXCL12 gene and affect expression [58]. Notch1 activation suppresses osteoblasts by decreasing CXCL12 that results in inhibition of normal hematopoiesis. This mechanism suggests that niche-targeted approach can be used to regain osteoblast function and restore normal hematopoiesis.
LSCs use the factors of different niche environments to resist chemo-targeting. During treatment, LSCs ‘home’ to adipose tissue to escape detection [63]. The adipose niche induces a pro-inflammatory response from the LSCs. This causes the cells to secrete cytokines which increase lipolysis and the release of fatty acids. LSCs then take up these the fatty acids to increase fatty acid oxidation (FAO). The expression of CD36 on LSCs seems to confer an advantage for the uptake of fatty acids. Additionally, the CD36+ LSCs have lower ATP levels than the CD36− cells even with comparable mitochondrial mass. Given the high FAO, low ATP levels, and their increased dependence on glycolysis, CD36+ LSCs resemble quiescent HSCs. Since chemotherapy targets cycling cells, CD36+ cells are more resistant to chemotherapeutics [63]. Taken together, these illustrate the role the leukemic microenvironment plays in LSCs maintenance and drug resistance.
The majority of leukemic research has focused on genetic alterations that increase the risk of developing leukemia or that drive the leukemogenesis. Another recent emphasis is determining mutations that lead to a stressed environment for the HSCs. In Shwachman–Diamond Syndrome (SDS) and Myelodyplastic Syndrome (MDS), mesenchymal inflammation has been shown to stress HSCs and increase the risk of leukemic transformation [64]. Furthermore, a conditional knockout specifically in mesenchymal cells for the Sbds gene results in similar bone formation as in human SDS. Additionally, these targeted mice increased mitochondrial dysfunction and increased DNA damage in the HSCs. However, in the Sbds KO mice, unlike ROS induced damage in healthy controls, the pathways for the DNA damage, such as base excision repair and ataxia telangiectasia Rad3, are transcriptionally altered. p53, a tumor suppressor gene, is overexpressed in the mutant niche cells as a potential mechanism for this genotoxic stress [64]. This upregulation of p53 causes an increase in secretion of S100A8/9 heterodimer, which is a member of the S100 family and involved in cellular processes such as differentiation. S100A8/9 in turn binds to Toll-Like Receptor 4 (TLR4) to cause the increase in ROS and DNA damage of HSCs. Similar analysis was done with Noonan Syndrome (NS) to study the genetic elements which increase the risk of developing leukemia [65]. In these patients, the gene PTPN11 encodes SHP2, tyrosine phosphatase, a positive regulator of the RAS signaling pathway, is mutated in 50% of the patients. Using niche specific expression of a mutant Ptpn11 E 76 K/+ gene showed that Erk, Akt, NF-κB intracellular signaling pathways were highly upregulated in the HSCs of the mutant mice. Along with increasing intracellular signaling, the BM plasma show elevated levels of pro-inflammatory cytokines such as IL-1β, TREM-1, CCL3, CCL12, CCL4, and TIMP-1, but a decrease in CXCL12. Moreover, the anti-inflammatory cytokine IL-1ra was also increased. Patients with elevated CCL3 have increased complications from NS compared to healthy patients. However, when looking at the HSCs, they found that IL-1β causes the HSCs to differentiate towards myeloid cells and monocytes, conversely CCL3, CCL4, and CCL12 appears to show no differentiation effect. Meanwhile, the impact of these factors on the MSCs results an increase in proliferation. Furthermore, these perturbed MSCs recruit monocytes that produce pro-inflammatory cytokines. This causes the HSCs to be hyper-activated and displaced from the niche causing myeloproliferative neoplasm (MPN). These studies demonstrate the role of the mutant niche in causing disruption of the hematopoietic system. The combined effects of high stress and inflammation cause HSCs to be displaced and acquire mutations leading to leukemic disease.
As depicted in Fig. 2 many molecules play a role in the complex network of communication between the BMM and either HSCs or LSCs. The microenvironment secretes signals that maintain and regulate HSCs. LSCs hijack the HSC niche to fulfill their demands, and promote their own survival. Changes within the leukemic microenvironment promote LSC survival and chemoresistance. LSCs remain more quiescent in the altered niche making targeted therapies problematic. Therefore, therapeutic treatment for leukemia needs to evaluate niche environment holistically. Understanding how LSCs alter the microenvironment will aid in finding new opportunities to treat the illness.

Bone Marrow Microenvironment Cross-talk Molecules: the BMM is comprised of cells such as osteoblasts and endothelial cells that secrete chemokines to maintain normal hematopoiesis. When genetic changes occur to alter HSCs to LSCs, cross-talk molecules are altered in such a way that the microenvironment is unable to easily support HSCs causing competition. The opposite is also true when the niche cells are able to stress HSCs into morphing into LSCs. A vast array of molecules such as NOTCH and CXCL12 play a role in the delicate balance of normal versus leukemic niche
Dysregulated signaling pathways contributing to LSCs
As mentioned above, recent studies have tried to dissect the genetic modifications which increase the risk of developing leukemia or promote transformation of normal HSCs. A combination of genetic and epigenetic alterations, signaling pathway disturbances, and changes to the bone marrow microenvironment permit the survival of LSCs over HSCs within the leukemic niche [66]. Furthermore, these pathways modify signaling pathways from upstream cytokines receptors to determine self-renewal and differentiation. Both LSCs and HSCs have been defined by their ability to reconstitute normal hematopoiesis or induce leukemia following transplantation. This requires differentiation and self-renewal of the stem cells. The signaling pathways controlling self-renewal have been characterized and defined using mouse models. These include hedgehog ligands that promote HSC proliferation [67]. Another pathway is Wnt signaling. Wnt ligands bind to their cell-surface receptors to stabilize β-catenin; this leads to HSC expansion [68]. Furthermore, similar to expression within the BMM, expression of the intracellular domain of Notch (ICN) within hematopoietic cells leads to HSC expansion [69]. Therefore, several signaling pathways are known to regulate the self-renewal and maintenance of HSCs and LSCs (Fig. 3).

Crosstalk between conservative signalling pathways. Interplay between the signalling proteins activate the transcription factors for transcribing the genes related to apoptosis, cell cycle, inflammation, self-renewal and migration of cells. Up-regulation or down-regulation of these proteins could lead to development of leukemia. HH hedgehog, IL interleukin, NICD Notch intracellular domain, PTCH patched, SMO smoothened, TFs transcription factors, THPO thrombopoetin, TNF tumor necrosis factor
Proper regulation of Wnt signaling is required to maintain a balance between the quiescent and active form of HSCs. This is done by balancing self-renewal and differentiation. Recently, WNT5A, a main stimulator of Wnt signaling, was found within the inflammatory niche [70]. Moreover, haploinsufficiency of WNT5A led to impaired actin polarization due to dysregulation of the zeb1 transcription factor. This affects the homing, migration and adhesion of LSCs [70], suggesting that alterations of this signaling pathway contribute to the recurrence of disease. Furthermore, Ctnnb1 and Wnt receptors frizzled 4/6 and the target gene, cyclin D2, were upregulated in LSC populations [71]. While the knockdown in expression of β-catenin results in the reduced growth of these cells; ablation or conditional deletion eliminates the oncogenic potential of the MLL-transformed cells [66]. Consistent with this idea, the expression of β-catenin can be down-regulated by p53, a commonly dysregulated pathway in various types of leukemia. Based on these findings, a protein that negatively regulates the self-renewal of stem cells is Aspp1 (apoptosis-stimulating protein of p53), a co-factor of p53 that enhances its pro-apoptotic effects was recently discovered [72]. In fact, HSCs deficient in both Aspp1 and p53 accumulate DNA damage and develop hematological malignancies. However, Aspp1 can regulate the pool of HSC via p53 dependent and p53 independent mechanisms. Also, downregulation of Aspp1 by promotor hypermethylation has been observed in leukemic cells [72]. Additional studies that support these findings show that dual targeting of p53 and c-MYC simultaneously improves the selectivity for targeting LSCs in CML as compared to tyrosine kinase inhibitors alone that are unable to eradicate the LSCs responsible for relapse [73]. Moreover, resistance to BET inhibitors (Bromodomain and extra terminal protein that bind acetylated chromatin marks) as seen in leukemic cells, is partially due to activation of Wnt/β-catenin signaling, and targeting this pathway restored the sensitivity of cells towards these inhibitors [74].
Interestingly, the Notch signaling is also regulated by β-catenin protein (Ctnnb1) [66]. Within this pathway, the complex γ-secretase and the target genes of Hes family, c-Myc, cyclins D1 and D3, Notch1 and 3 have all been proven to be important in HSCs in quiescence, while Notch1 has tumor suppressor activity in AML [66, 75]. Hes1, a target gene for Notch as well as Transforming Growth Factor (TGF-β) signaling are transcriptionally regulated by the factor JunB to control the proliferation of HSCs for production of myeloid progenitors [75]. Meanwhile, HSCs deficient in junB have reduced activity of many downstream effectors for this pathway, like Smad7 and p57. Supporting this idea, reduction in Hes1 causes the loss of quiescence and hyperproliferation of the LSCs [75]. These data confirm that Wnt/β-catenin and Notch signaling are critical regulators of HSCs and LSCs.
Not surprising, that GSK-3β has been considered a key therapeutic target for leukemia because of the variety of pathways with which it interacts, including Wnt/β-catenin, Notch, PI3K/PTEN/Akt, and Hedgehog (Hh) [76–78]. Indeed, miR-126 regulates LSCs quiescence by targeting the components of this PI3K/AKT/GSK3β signaling axis in leukemia resulting in disease relapse and treatment resistance [79]. However, others have shown that the inhibition of miR-126 leads to LSC differentiation and cell cycle activation improving the sensitivity of anti-proliferative drugs [79]. Additionally, the negative regulator of PI3K pathway, Pten is also important to maintain the quiescent state of the HSCs [80]. That is, cells deficient in Pten are sensitized to IFNα and G-CSF. This results in hyper-activation of PI3 K pathway, increase of protein synthesis and tumor suppression expression and depletion of HSCs [80].
Providing further evidence to the interplay between these signaling pathways, Pten deficient B cells activate the hedgehog (HH) pathway resulting in an expansion of B lineage cells but do not display a CLL-like phenotype [81]. In this model, activation of the Hh/Gli1 along with PI3 K/AKT signaling was unable to cause expansion of the B lineage cell. This proves that his pathway does not play a role in the onset of CLL, but is rather required for their survival as proven by both stroma-derived HH signaling and PI3 K/AKT pathway [81]. In another model of lymphoid leukemia, using a JAK3 mutant mouse model [82], an increase in the expression of stimulatory ligands, Dll4, Il7 and Vegf, that promote proliferation, and cell survival was observed. Similar to CLL-like model, this was due to the upregulation in expression of the GLI1 transcription factor and ectopic expression of Sonic Hedgehog (SHH) and Indian Hedgehog (IHH), ligands of the hedgehog pathway in the T-cell progenitors. These JAK3 mutant mice were also shown to be sensitive to treatment of the Shh pathway include smoothened (SMO) and glioma-associated oncogene homolog (GLI) inhibitors [82]. Furthermore, cases of AML having FMS-like tyrosine kinase (FLT3) internal tandem duplication (ITD) mutation had an increased expression of GLI2, an effector of the HH pathway, which regulates the transcription of CyclinD1 and N-Myc. The constitutive activation of Hh/Gli led to enhancement of STAT5 and hence proliferation of myeloid progenitors. And, the combined use of SMO and FLT3 antagonists inhibited the growth of AML cells in vitro as well as in vivo [83]. Targeting of the these pathways, including Wnt/β-catenin and Hedgehog (Hh), have proven difficult, as these self-renewal and stem cell pathways are essential in other tissues throughout the body.
Apart from these signaling pathways described above, the JAK/STAT pathway plays a mechanistic role in the development of leukemia due to the interplay of the pathway with the cytokines and the inflammatory niche. The constitutive activation of the JAK/STAT pathway drives leukemogenic potential as observed in BCR/ABL+ CML, BCR/ABL− myeloproliferative neoplasms and is even associated with poor prognosis of AML LSCs [84–86]. Janus kinases (JAKs), cytoplasmically associated with cytokine receptors are phosphorylated at their tyrosine residues and are activated when cytokines and growth factors bind to their receptors. Mutated and constitutively active JAK, cause the phosphorylation and dimerization of STATs that are transcription factors regulating genes important for cell survival and proliferation. There have been significant advances in targeting these pathways in leukemia and solid tumors [84, 87]. Interestingly, tyrosine-unphosphorylated STAT5 (uSTAT5) represses megakaryocytic transcriptional program in the absence of thrombopoietin (THPO) and restricted the access of ERG, a transcriptional factor to its target genes, proving the role of cytokine in megakaryocytic differentiation [87]. Therefore, although the focus has been on active or phosphorylated STAT protein, the inactive form also regulates gene expression and cell fate. Additionally, downregulating the expression of THPO receptor MPL reduced the THPO-stimulated STAT3/5 activation in turn decreasing the expression of STAT target genes Bcl-XL, Bcl2, Hif2a, Myc, Osm and Pim1 responsible for cell cycle progression and inhibition of apoptosis [85]. Moreover, UT2, an endogenous transmembrane protein upstream-of-mTORC2 that negatively regulates STAT3, inhibiting the phosphorylation of STAT3 at tyrosine 705 and reducing the cytokine IL-6 signaling, was decreased in multiple myeloma [88]. This suggests that phosphorylated STAT in the JAK/STAT pathway leads to activation of genes related to progression of cell cycle and inhibition of apoptosis aiding in the development and survival of LSCs, and hence can be therapeutically targeted for treatment.
Proinflammatory cytokine are typically elevated during myeloid malignancies. A few of these elevated factors includes CCL2, CCL3, CCL5, CSF2, IFN-γ, IL-1, IL-3, IL-6, IL-8, IL-10, IL-12, IL-13, THPO, TNFα and stem cell factor [86, 89–91]. Increased levels of these factors, as well as increased cell proliferation are observed in leukemic patients and people suffering from other myeloproliferative disorders [9, 86]. IL-1, a proinflammatory cytokine triggers the phosphorylation and ubiquitination of downstream transcriptional targets of NF-κB, p38, JNK, MAPK and AP-1 signaling pathways. The target genes of this activation being the IL-1α, IL-1β, IL-6, IL-8, MCP-1, MKP-1 and TNFα, stimulating cell division and myeloid cell differentiation of HSCs while inhibiting lymphopoiesis and erythropoiesis [91, 92]. Furthermore, IL-1β and CXCL2 were upregulated in AML due to a positive feedback mechanism phosphorylating GATA-2 via activation of the p38/ERK signaling promoting transcriptional activation and hence cell proliferation [93]. Tumor suppressive action of RIPK3, a target of the TNF receptor signaling pathway, promotes cell-death of LSCs in AML from inflammasome-mediated release of IL-1β that restricts myeloid leukemogenesis [94]. Additionally, chronic exposure to IL-1 reversibly impaired self-renewal ability of HSC similar to long-term exposure of IFNα while its acute production led to myeloid differentiation for blood regeneration observed during a physiological emergency state [92]. Meanwhile, an enhanced sensitivity to IL-1 induced NF-κB signaling and an upregulation IL-1 receptor Type 1 (IL-1-R1) and IL-1 receptor accessory protein (IL-1-RAP) was observed in CML LSCs [91]. A significant inhibition of the NF-κB signaling was observed after IL1-RA (IL1 receptor antagonist) on treatment with tyrosine kinase inhibitors in CML aiming to maintain treatment-free remissions by targeting the LSCs [91]. Apart from IL-1, the role of IL-6 in CML has been shown to be crucial for leukemic maintenance [89, 90]. Non-transformed progenitors and normal HSCs were restored with anti-IL-6 treatment of chimeric CML mice. Additionally, changes in cytokine levels from the leukemia in this chimeric model affect the normal stem and progenitor cell function to transformation of the neighboring cells [89]. Furthermore, BCR-ABL translocation regulated the expression of IL-6 through BCL6, a transcription regulator of IL-6 gene via the paracrine loop, reprogramming differentiation towards the myeloid lineage proving its importance in CML development. This finding was confirmed by the restoration of the normal low IL-6 levels on treatment with tyrosine kinase inhibitors. Disruption of the paracrine loop led to delays in the onset of CML with persistent BCR-ABL activity, proving the role of cytokines in the sustenance of LSCs [90].
It can be concluded that for development, survival, and self-renewal of LSCs several signaling pathways, transcription factors and cytokines play crucial roles. Numerous transcription factors known to regulate the genes required for cell cycle activation and inhibition of apoptosis for cell proliferation are activated in leukemia, either by the oncogene or by changes to the microenvironment. Recent studies have revolved around the pro-inflammatory cytokines important to the development of LSCs and maintenance of the leukemic environment.
As discussed here, hematopoietic stem cells rely on cytokines from the bone marrow niche and neighboring hematopoietic cells to maintain their dormancy, as well as differentiate to mature blood lineages to meet the needs of the individual. However, during malignancies, alteration in secreted factors and the niche contribute to maintenance of leukemic stem cells and disease progression. Here, we described the alterations in immune regulation, pro-inflammatory cytokines, self-renewal pathways and the microenvironment from leukemia. Findings in the past few years suggest that the future of treating leukemia will likely involve restoring the diseased microenvironment and inhibition of cytokines and secreted factors that promote leukemic maintenance.
References
Orkin SH, Zon LI. Hematopoiesis: an evolving paradigm for stem cell biology. Cell. 2008;132:631–44.
Dadi S, Chhangawala S, Whitlock BM, Franklin RA, Luo CT, Oh SA, Toure A, Pritykin Y, Huse M, Leslie CS, Li MO. Cancer Immunosurveillance by tissue-resident innate lymphoid cells and innate-like T cells. Cell. 2016;164:365–77.
Buggins AG, Milojkovic D, Arno MJ, Lea NC, Mufti GJ, Thomas NS, Hirst WJ. Microenvironment produced by acute myeloid leukemia cells prevents T cell activation and proliferation by inhibition of NF-kappaB, c-Myc, and pRb pathways. J Immunol. 2001;167:6021–30.
Kitagawa Y, Wing JB, Sakaguchi S. Transcriptional and epigenetic control of regulatory T cell development. Prog Mol Biol Transl Sci. 2015;136:1–33.
Li L, Neaves WB. Normal stem cells and cancer stem cells: the niche matters. Cancer Res. 2006;66:4553–7.
Croft M. Co-stimulatory members of the TNFR family: keys to effective T-cell immunity? Nat Rev Immunol. 2003;3:609–20.
Moudra A, Hubackova S, Machalova V, Vancurova M, Bartek J, Reinis M, Hodny Z, Jonasova A. Dynamic alterations of bone marrow cytokine landscape of myelodysplastic syndromes patients treated with 5-azacytidine. Oncoimmunology. 2016;5:e1183860.
Arellano M, Waller EK. GM-CSF + INF-alpha induce a graft-versus-leukemia effect in BMT patients with relapsed AML and ALL. J Clin Oncol. 2004;22:6634.
Kornblau SM, McCue D, Singh N, Chen W, Estrov Z, Coombes KR. Recurrent expression signatures of cytokines and chemokines are present and are independently prognostic in acute myelogenous leukemia and myelodysplasia. Blood. 2010;116:4251–61.
Gentles AJ, Plevritis SK, Majeti R, Alizadeh AA. Association of a leukemic stem cell gene expression signature with clinical outcomes in acute myeloid leukemia. JAMA. 2010;304:2706–15.
Jung N, Dai B, Gentles AJ, Majeti R, Feinberg AP. An LSC epigenetic signature is largely mutation independent and implicates the HOXA cluster in AML pathogenesis. Nat Commun. 2015;6:8489.
Korsmeyer SJ, Arnold A, Bakhshi A, Ravetch JV, Siebenlist U, Hieter PA, Sharrow SO, LeBien TW, Kersey JH, Poplack DG, Leder P, Waldmann TA. Immunoglobulin gene rearrangement and cell surface antigen expression in acute lymphocytic leukemias of T cell and B cell precursor origins. J Clin Investig. 1983;71:301–13.
Curti A, Pandolfi S, Valzasina B, Aluigi M, Isidori A, Ferri E, Salvestrini V, Bonanno G, Rutella S, Durelli I, Horenstein AL, Fiore F, Massaia M, Colombo MP, Baccarani M, Lemoli RM. Modulation of tryptophan catabolism by human leukemic cells results in the conversion of CD25- into CD25+ T regulatory cells. Blood. 2007;109:2871–7.
Milojkovic D, Devereux S, Westwood NB, Mufti GJ, Thomas NS, Buggins AG. Antiapoptotic microenvironment of acute myeloid leukemia. J Immunol. 2004;173:6745–52.
Kikushige Y, Miyamoto T, Yuda J, Jabbarzadeh-Tabrizi S, Shima T, Takayanagi S, Niiro H, Yurino A, Miyawaki K, Takenaka K, Iwasaki H, Akashi K. A TIM-3/Gal-9 autocrine stimulatory loop drives self-renewal of human myeloid leukemia stem cells and leukemic progression. Cell Stem Cell. 2015;17:341–52.
Zhao E, Xu H, Wang L, Kryczek I, Wu K, Hu Y, Wang G, Zou W. Bone marrow and the control of immunity. Cell Mol Immunol. 2012;9:11–9.
Massberg S, Schaerli P, Knezevic-Maramica I, Kollnberger M, Tubo N, Moseman EA, Huff IV, Junt T, Wagers AJ, Mazo IB, von Andrian UH. Immunosurveillance by hematopoietic progenitor cells trafficking through blood, lymph, and peripheral tissues. Cell. 2007;131:994–1008.
Riether C, Schurch CM, Ochsenbein AF. Regulation of hematopoietic and leukemic stem cells by the immune system. Cell Death Differ. 2015;22:187–98.
Garcia-Ojeda ME, Dejbakhsh-Jones S, Weissman IL, Strober S. An alternate pathway for T cell development supported by the bone marrow microenvironment: recapitulation of thymic maturation. J Exp Med. 1998;187:1813–23.
Dejbakhsh-Jones S, Jerabek L, Weissman IL, Strober S. Extrathymic maturation of alpha beta T cells from hemopoietic stem cells. J Immunol. 1995;155:3338–44.
Zou L, Barnett B, Safah H, Larussa VF, Evdemon-Hogan M, Mottram P, Wei S, David O, Curiel TJ, Zou W. Bone marrow is a reservoir for CD4+ CD25+ regulatory T cells that traffic through CXCL12/CXCR4 signals. Cancer Res. 2004;64:8451–5.
Fujisaki J, Wu J, Carlson AL, Silberstein L, Putheti P, Larocca R, Gao W, Saito TI, Lo Celso C, Tsuyuzaki H, Sato T, Cote D, Sykes M, Strom TB, Scadden DT, Lin CP. In vivo imaging of Treg cells providing immune privilege to the haematopoietic stem-cell niche. Nature. 2011;474:216–9.
Sakaguchi S, Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T. Regulatory T cells: how do they suppress immune responses? Int Immunol. 2009;21:1105–11.
Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, Cross R, Sehy D, Blumberg RS, Vignali DA. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature. 2007;450:566–9.
Pandiyan P, Zheng L, Ishihara S, Reed J, Lenardo MJ. CD4+ CD25+ Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nat Immunol. 2007;8:1353–62.
Ustun C, Miller JS, Munn DH, Weisdorf DJ, Blazar BR. Regulatory T cells in acute myelogenous leukemia: is it time for immunomodulation? Blood. 2011;118:5084–95.
Albitar M, Manshouri T, Shen Y, Liu D, Beran M, Kantarjian HM, Rogers A, Jilani I, Lin CW, Pierce S, Freireich EJ, Estey EH. Myelodysplastic syndrome is not merely “preleukemia”. Blood. 2002;100:791–8.
Mundy-Bosse BL, Scoville SD, Chen L, McConnell K, Mao HC, Ahmed EH, Zorko N, Harvey S, Cole J, Zhang X, Costinean S, Croce CM, Larkin K, Byrd JC, Vasu S, Blum W, Yu J, Freud AG, Caligiuri MA. MicroRNA-29b mediates altered innate immune development in acute leukemia. J Clin Investig. 2016;126(12):4404–16.
Epling-Burnette PK, Bai F, Painter JS, Rollison DE, Salih HR, Krusch M, Zou J, Ku E, Zhong B, Boulware D, Moscinski L, Wei S, Djeu JY, List AF. Reduced natural killer (NK) function associated with high-risk myelodysplastic syndrome (MDS) and reduced expression of activating NK receptors. Blood. 2007;109:4816–24.
Gleason MK, Ross JA, Warlick ED, Lund TC, Verneris MR, Wiernik A, Spellman S, Haagenson MD, Lenvik AJ, Litzow MR, Epling-Burnette PK, Blazar BR, Weiner LM, Weisdorf DJ, Vallera DA, Miller JS. CD16xCD33 bispecific killer cell engager (BiKE) activates NK cells against primary MDS and MDSC CD33+ targets. Blood. 2014;123:3016–26.
Wagner JA, Berrien-Elliott MM, Rosario M, Leong JW, Jewell BA, Schappe T, Abdel-Latif S, Fehniger TA. Cytokine-induced memory-like differentiation enhances unlicensed natural killer cell antileukemia and FcgammaRIIIa-triggered responses. Biol Blood Marrow Transplant. 2017;23(3):398–404.
Liu FT, Giustiniani J, Farren T, Jia L, Bensussan A, Gribben JG, Agrawal SG. CD160 signaling mediates PI3K-dependent survival and growth signals in chronic lymphocytic leukemia. Blood. 2010;115:3079–88.
Goselink HM, Williams DE, Fibbe WE, Wessels HW, Beverstock GC, Willemze R, Falkenburg JH. Effect of mast cell growth factor (c-kit ligand) on clonogenic leukemic precursor cells. Blood. 1992;80:750–7.
Valent P, Spanblochl E, Bankl HC, Sperr WR, Marosi C, Pirc-Danoewinata H, Virgolini I, Eichler HG, Agis H, Sillaber C, et al. Kit ligand/mast cell growth factor-independent differentiation of mast cells in myelodysplasia and chronic myeloid leukemic blast crisis. Blood. 1994;84:4322–32.
Puverel S, Kiris E, Singh S, Klarmann KD, Coppola V, Keller JR, Tessarollo L. RanBPM (RanBP9) regulates mouse c-Kit receptor level and is essential for normal development of bone marrow progenitor cells. Oncotarget. 2016;7(51):85109–23.
Kobayashi CI, Takubo K, Kobayashi H, Nakamura-Ishizu A, Honda H, Kataoka K, Kumano K, Akiyama H, Sudo T, Kurokawa M, Suda T. The IL-2/CD25 axis maintains distinct subsets of chronic myeloid leukemia-initiating cells. Blood. 2014;123:2540–9.
Kotsianidis I, Bouchliou I, Nakou E, Spanoudakis E, Margaritis D, Christophoridou AV, Anastasiades A, Tsigalou C, Bourikas G, Karadimitris A, Tsatalas C. Kinetics, function and bone marrow trafficking of CD4+ CD25+ FOXP3+ regulatory T cells in myelodysplastic syndromes (MDS). Leukemia. 2009;23:510–8.
Nguyen AH, Berim IG, Agrawal DK. Chronic inflammation and cancer: emerging roles of triggering receptors expressed on myeloid cells. Expert Rev Clin Immunol. 2015;11:849–57.
Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–20.
Kitagawa M, Saito I, Kuwata T, Yoshida S, Yamaguchi S, Takahashi M, Tanizawa T, Kamiyama R, Hirokawa K. Overexpression of tumor necrosis factor (TNF)-alpha and interferon (IFN)-gamma by bone marrow cells from patients with myelodysplastic syndromes. Leukemia. 1997;11:2049–54.
Sawanobori M, Yamaguchi S, Hasegawa M, Inoue M, Suzuki K, Kamiyama R, Hirokawa K, Kitagawa M. Expression of TNF receptors and related signaling molecules in the bone marrow from patients with myelodysplastic syndromes. Leuk Res. 2003;27:583–91.
Wei Y, Dimicoli S, Bueso-Ramos C, Chen R, Yang H, Neuberg D, Pierce S, Jia Y, Zheng H, Wang H, Wang X, Nguyen M, Wang SA, Ebert B, Bejar R, Levine R, Abdel-Wahab O, Kleppe M, Ganan-Gomez I, Kantarjian H, Garcia-Manero G. Toll-like receptor alterations in myelodysplastic syndrome. Leukemia. 2013;27:1832–40.
Kuninaka N, Kurata M, Yamamoto K, Suzuki S, Umeda S, Kirimura S, Arai A, Nakagawa Y, Suzuki K, Kitagawa M. Expression of Toll-like receptor 9 in bone marrow cells of myelodysplastic syndromes is down-regulated during transformation to overt leukemia. Exp Mol Pathol. 2010;88:293–8.
Takeshita S, Fumoto T, Naoe Y, Ikeda K. Age-related marrow adipogenesis is linked to increased expression of RANKL. J Biol Chem. 2014;289:16699–710.
Lambert C, Wu Y, Aanei C. Bone marrow immunity and myelodysplasia. Front Oncol. 2016;6:172.
Tsuboi I, Morimoto K, Hirabayashi Y, Li GX, Aizawa S, Mori KJ, Kanno J, Inoue T. Senescent B lymphopoiesis is balanced in suppressive homeostasis: decrease in interleukin-7 and transforming growth factor-beta levels in stromal cells of senescence-accelerated mice. Exp Biol Med (Maywood). 2004;229:494–502.
Wang CQ, Udupa KB, Xiao H, Lipschitz DA. Effect of age on marrow macrophage number and function. Aging (Milano). 1995;7:379–84.
Braig M, Pallmann N, Preukschas M, Steinemann D, Hofmann W, Gompf A, Streichert T, Braunschweig T, Copland M, Rudolph KL, Bokemeyer C, Koschmieder S, Schuppert A, Balabanov S, Brummendorf TH. A ‘telomere-associated secretory phenotype’ cooperates with BCR-ABL to drive malignant proliferation of leukemic cells. Leukemia. 2014;28:2028–39.
Cahu J, Bustany S, Sola B. Senescence-associated secretory phenotype favors the emergence of cancer stem-like cells. Cell Death Dis. 2012;3:e446.
Tchkonia T, Zhu Y, van Deursen J, Campisi J, Kirkland JL. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Investig. 2013;123:966–72.
Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729–40.
Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, Desmet CJ, Aarden LA, Mooi WJ, Peeper DS. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008;133:1019–31.
Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5:99–118.
Acosta JC, O’Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, Fumagalli M, Da Costa M, Brown C, Popov N, Takatsu Y, Melamed J, d’Adda di Fagagna F, Bernard D, Hernando E, Gil J. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. 2008;133:1006–18.
Chan CK, Seo EY, Chen JY, Lo D, McArdle A, Sinha R, Tevlin R, Seita J, Vincent-Tompkins J, Wearda T, Lu WJ, Senarath-Yapa K, Chung MT, Marecic O, Tran M, Yan KS, Upton R, Walmsley GG, Lee AS, Sahoo D, Kuo CJ, Weissman IL, Longaker MT. Identification and specification of the mouse skeletal stem cell. Cell. 2015;160:285–98.
Nwajei F, Konopleva M. The bone marrow microenvironment as niche retreats for hematopoietic and leukemic stem cells. Adv Hematol. 2013;2013:953982.
Silberstein L, Goncalves KA, Kharchenko PV, Turcotte R, Kfoury Y, Mercier F, Baryawno N, Severe N, Bachand J, Spencer JA, Papazian A, Lee D, Chitteti BR, Srour EF, Hoggatt J, Tate T, Lo Celso C, Ono N, Nutt S, Heino J, Sipila K, Shioda T, Osawa M, Lin CP, Hu GF, Scadden DT. Proximity-based differential single-cell analysis of the niche to identify stem/progenitor cell regulators. Cell Stem Cell. 2016;19:530–43.
Zhou HS, Carter BZ, Andreeff M. Bone marrow niche-mediated survival of leukemia stem cells in acute myeloid leukemia: yin and Yang. Cancer Biol Med. 2016;13:248–59.
Colmone A, Amorim M, Pontier AL, Wang S, Jablonski E, Sipkins DA. Leukemic cells create bone marrow niches that disrupt the behavior of normal hematopoietic progenitor cells. Science. 2008;322:1861–5.
Glait-Santar C, Desmond R, Feng X, Bat T, Chen J, Heuston E, Mizukawa B, Mulloy JC, Bodine DM, Larochelle A, Dunbar CE. Functional niche competition between normal hematopoietic stem and progenitor cells and myeloid leukemia cells. Stem Cells. 2015;33:3635–42.
Pitt LA, Tikhonova AN, Hu H, Trimarchi T, King B, Gong Y, Sanchez-Martin M, Tsirigos A, Littman DR, Ferrando AA, Morrison SJ, Fooksman DR, Aifantis I, Schwab SR. CXCL12-producing vascular endothelial niches control acute T cell leukemia maintenance. Cancer Cell. 2015;27:755–68.
Kim JA, Shim JS, Lee GY, Yim HW, Kim TM, Kim M, Leem SH, Lee JW, Min CK, Oh IH. Microenvironmental remodeling as a parameter and prognostic factor of heterogeneous leukemogenesis in acute myelogenous leukemia. Cancer Res. 2015;75:2222–31.
Ye H, Adane B, Khan N, Sullivan T, Minhajuddin M, Gasparetto M, Stevens B, Pei S, Balys M, Ashton JM, Klemm DJ, Woolthuis CM, Stranahan AW, Park CY, Jordan CT. Leukemic stem cells evade chemotherapy by metabolic adaptation to an adipose tissue niche. Cell Stem Cell. 2016;19:23–37.
Zambetti NA, Ping Z, Chen S, Kenswil KJ, Mylona MA, Sanders MA, Hoogenboezem RM, Bindels EM, Adisty MN, Van Strien PM, van der Leije CS, Westers TM, Cremers EM, Milanese C, Mastroberardino PG, van Leeuwen JP, van der Eerden BC, Touw IP, Kuijpers TW, Kanaar R, van de Loosdrecht AA, Vogl T, Raaijmakers MH. Mesenchymal inflammation drives genotoxic stress in hematopoietic stem cells and predicts disease evolution in human pre-leukemia. Cell Stem Cell. 2016;19:613–27.
Dong L, Yu WM, Zheng H, Loh ML, Bunting ST, Pauly M, Huang G, Zhou M, Broxmeyer HE, Scadden DT, Qu CK. Leukaemogenic effects of Ptpn11 activating mutations in the stem cell microenvironment. Nature. 2016;539:304–8.
Heidel FH, Arreba-Tutusaus P, Armstrong SA, Fischer T. Evolutionarily conserved signaling pathways: acting in the shadows of acute myelogenous leukemia’s genetic diversity. Clin Cancer Res. 2015;21:240–8.
Trowbridge JJ, Scott MP, Bhatia M. Hedgehog modulates cell cycle regulators in stem cells to control hematopoietic regeneration. Proc Natl Acad Sci USA. 2006;103:14134–9.
Reya T, Duncan AW, Ailles L, Domen J, Scherer DC, Willert K, Hintz L, Nusse R, Weissman IL. A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature. 2003;423:409–14.
Karanu FN, Murdoch B, Gallacher L, Wu DM, Koremoto M, Sakano S, Bhatia M. The notch ligand jagged-1 represents a novel growth factor of human hematopoietic stem cells. J Exp Med. 2000;192:1365–72.
Schreck C, Istvanffy R, Ziegenhain C, Sippenauer T, Ruf F, Henkel L, Gartner F, Vieth B, Florian MC, Mende N, Taubenberger A, Prendergast A, Wagner A, Pagel C, Grziwok S, Gotze KS, Guck J, Dean DC, Massberg S, Essers M, Waskow C, Geiger H, Schiemann M, Peschel C, Enard W, Oostendorp RA. Niche WNT5A regulates the actin cytoskeleton during regeneration of hematopoietic stem cells. J Exp Med. 2017;214:165–81.
Yeung J, Esposito MT, Gandillet A, Zeisig BB, Griessinger E, Bonnet D, So CW. beta-Catenin mediates the establishment and drug resistance of MLL leukemic stem cells. Cancer Cell. 2010;18:606–18.
Yamashita M, Nitta E, Suda T. Aspp1 preserves hematopoietic stem cell pool integrity and prevents malignant transformation. Cell Stem Cell. 2015;17:23–34.
Abraham SA, Hopcroft LE, Carrick E, Drotar ME, Dunn K, Williamson AJ, Korfi K, Baquero P, Park LE, Scott MT, Pellicano F, Pierce A, Copland M, Nourse C, Grimmond SM, Vetrie D, Whetton AD, Holyoake TL. Dual targeting of p53 and c-MYC selectively eliminates leukaemic stem cells. Nature. 2016;534:341–6.
Fong CY, Gilan O, Lam EY, Rubin AF, Ftouni S, Tyler D, Stanley K, Sinha D, Yeh P, Morison J, Giotopoulos G, Lugo D, Jeffrey P, Lee SC, Carpenter C, Gregory R, Ramsay RG, Lane SW, Abdel-Wahab O, Kouzarides T, Johnstone RW, Dawson SJ, Huntly BJ, Prinjha RK, Papenfuss AT, Dawson MA. BET inhibitor resistance emerges from leukaemia stem cells. Nature. 2015;525:538–42.
Santaguida M, Schepers K, King B, Sabnis AJ, Forsberg EC, Attema JL, Braun BS, Passegue E. JunB protects against myeloid malignancies by limiting hematopoietic stem cell proliferation and differentiation without affecting self-renewal. Cancer Cell. 2009;15:341–52.
Welham MJ, Kingham E, Sanchez-Ripoll Y, Kumpfmueller B, Storm M, Bone H. Controlling embryonic stem cell proliferation and pluripotency: the role of PI3K- and GSK-3-dependent signalling. Biochem Soc Trans. 2011;39:674–8.
Gandillet A, Park S, Lassailly F, Griessinger E, Vargaftig J, Filby A, Lister TA, Bonnet D. Heterogeneous sensitivity of human acute myeloid leukemia to beta-catenin down-modulation. Leukemia. 2011;25:770–80.
Wickremasinghe RG, Prentice AG, Steele AJ. p53 and Notch signaling in chronic lymphocytic leukemia: clues to identifying novel therapeutic strategies. Leukemia. 2011;25:1400–7.
Lechman ER, Gentner B, Ng SW, Schoof EM, van Galen P, Kennedy JA, Nucera S, Ciceri F, Kaufmann KB, Takayama N, Dobson SM, Trotman-Grant A, Krivdova G, Elzinga J, Mitchell A, Nilsson B, Hermans KG, Eppert K, Marke R, Isserlin R, Voisin V, Bader GD, Zandstra PW, Golub TR, Ebert BL, Lu J, Minden M, Wang JC, Naldini L, Dick JE. miR-126 regulates distinct self-renewal outcomes in normal and malignant hematopoietic stem cells. Cancer Cell. 2016;29:602–6.
Porter SN, Cluster AS, Signer RA, Voigtmann J, Monlish DA, Schuettpelz LG, Magee JA. Pten cell autonomously modulates the hematopoietic stem cell response to inflammatory cytokines. Stem Cell Rep. 2016;6:806–14.
Kern D, Regl G, Hofbauer SW, Altenhofer P, Achatz G, Dlugosz A, Schnidar H, Greil R, Hartmann TN, Aberger F. Hedgehog/GLI and PI3K signaling in the initiation and maintenance of chronic lymphocytic leukemia. Oncogene. 2015;34:5341–51.
Dagklis A, Demeyer S, De Bie J, Radaelli E, Pauwels D, Degryse S, Gielen O, Vicente C, Vandepoel R, Geerdens E, Uyttebroeck A, Boeckx N, de Bock CE, Cools J. Hedgehog pathway activation in T-cell acute lymphoblastic leukemia predicts response to SMO and GLI1 inhibitors. Blood. 2016;128:2642–54.
Lim Y, Gondek L, Li L, Wang Q, Ma H, Chang E, Huso DL, Foerster S, Marchionni L, McGovern K, Watkins DN, Peacock CD, Levis M, Smith BD, Merchant AA, Small D, Matsui W. Integration of Hedgehog and mutant FLT3 signaling in myeloid leukemia. Sci Transl Med. 2015;7:291ra296.
de Freitas RM, da Costa Maranduba CM. Myeloproliferative neoplasms and the JAK/STAT signaling pathway: an overview. Rev Bras Hematol Hemoter. 2015;37:348–53.
Zhang B, Li L, Ho Y, Li M, Marcucci G, Tong W, Bhatia R. Heterogeneity of leukemia-initiating capacity of chronic myelogenous leukemia stem cells. J Clin Investig. 2016;126:975–91.
Kleppe M, Kwak M, Koppikar P, Riester M, Keller M, Bastian L, Hricik T, Bhagwat N, McKenney AS, Papalexi E, Abdel-Wahab O, Rampal R, Marubayashi S, Chen JJ, Romanet V, Fridman JS, Bromberg J, Teruya-Feldstein J, Murakami M, Radimerski T, Michor F, Fan R, Levine RL. JAK-STAT pathway activation in malignant and nonmalignant cells contributes to MPN pathogenesis and therapeutic response. Cancer Discov. 2015;5:316–31.
Park HJ, Li J, Hannah R, Biddie S, Leal-Cervantes AI, Kirschner K, Flores Santa Cruz D, Sexl V, Gottgens B, Green AR. Cytokine-induced megakaryocytic differentiation is regulated by genome-wide loss of a uSTAT transcriptional program. EMBO J. 2016;35:580–94.
Lee D, Wang YH, Kalaitzidis D, Ramachandran J, Eda H, Sykes DB, Raje N, Scadden DT. Endogenous transmembrane protein UT2 inhibits pSTAT3 and suppresses hematological malignancy. J Clin Invest. 2016;126:1300–10.
Welner RS, Amabile G, Bararia D, Czibere A, Yang H, Zhang H, Pontes LL, Ye M, Levantini E, Di Ruscio A, Martinelli G, Tenen DG. Treatment of chronic myelogenous leukemia by blocking cytokine alterations found in normal stem and progenitor cells. Cancer Cell. 2015;27:671–81.
Reynaud D, Pietras E, Barry-Holson K, Mir A, Binnewies M, Jeanne M, Sala-Torra O, Radich JP, Passegue E. IL-6 controls leukemic multipotent progenitor cell fate and contributes to chronic myelogenous leukemia development. Cancer Cell. 2011;20:661–73.
Zhang B, Chu S, Agarwal P, Campbell VL, Hopcroft L, Jorgensen HG, Lin A, Gaal K, Holyoake TL, Bhatia R. Inhibition of interleukin-1 signaling enhances elimination of tyrosine kinase inhibitor treated CML stem cells. Blood. 2016;128(23):2671–82.
Pietras EM, Mirantes-Barbeito C, Fong S, Loeffler D, Kovtonyuk LV, Zhang S, Lakshminarasimhan R, Chin CP, Techner JM, Will B, Nerlov C, Steidl U, Manz MG, Schroeder T, Passegue E. Chronic interleukin-1 exposure drives haematopoietic stem cells towards precocious myeloid differentiation at the expense of self-renewal. Nat Cell Biol. 2016;18:607–18.
Katsumura KR, Ong IM, DeVilbiss AW, Sanalkumar R, Bresnick EH. GATA factor-dependent positive-feedback circuit in acute myeloid leukemia cells. Cell Rep. 2016;16:2428–41.
Hockendorf U, Yabal M, Herold T, Munkhbaatar E, Rott S, Jilg S, Kauschinger J, Magnani G, Reisinger F, Heuser M, Kreipe H, Sotlar K, Engleitner T, Rad R, Weichert W, Peschel C, Ruland J, Heikenwalder M, Spiekermann K, Slotta-Huspenina J, Gross O, Jost PJ. RIPK3 restricts myeloid leukemogenesis by promoting cell death and differentiation of leukemia initiating cells. Cancer Cell. 2016;30:75–91.
Author information
Authors and Affiliations
Corresponding author
About this article

Cite this article
Camacho, V., McClearn, V., Patel, S. et al. Regulation of normal and leukemic stem cells through cytokine signaling and the microenvironment. Int J Hematol 105, 566–577 (2017). https://doi.org/10.1007/s12185-017-2184-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12185-017-2184-6