
Abstract
Considerable efforts have been devoted to the development of rapid and sensitive methods allowing the detection of viral nucleic acid. We herein describe an assay for identification of a specific influenza sequence. The suggested method was based on isolation using paramagnetic particles coupled with electrochemical detection of isolated product. Peptide nucleic acid (PNA) was used as a probe for hybridization and identification of the influenza-derived specific sequence. The use of PNA can show numerous benefits: PNA probe is not degradable by enzymes and the duplex of PNA with RNA/DNA is more thermostable and more resistant to pH changes than DNA/DNA or RNA/RNA duplexes. This PNA probe assay can be applied as a magnetically guidable tool for detection of DNA/RNA samples under different conditions.
Similar content being viewed by others

Introduction
Influenza viruses, members of the family Orthomyxoviridae, are respiratory pathogens that can cause sporadic illnesses, seasonal epidemics or less frequently pandemics [1]. The impact of seasonal and pandemic influenza includes significant morbidity and mortality, as well as a socio-economic burden in medical care costs and loss of productivity [2]. Spanish flu (1918), which is considered as the greatest pandemic in the twentieth century, was responsible for 20–50 million victims [3–5]. However, the economic impact is unknown, because the Spanish influenza left so few traces in public memory as it was ‘overshadowed’ by the First World War, and hence its historiographical characterisation became the ‘forgotten’ pandemic [4]. It is difficult to estimate how the great consequences should have new pandemic. Influenza viruses pose a constant potential pandemic threat for public health [6, 7], because of the high speed of mutational changes of influenza genome which provides a constant risk of creation of the new pandemic subtype, and could have a greater mortality and huge economic impact than all of known pandemics in history. In the battle against the spread of epidemics, it is the most important to have accurate and well-timed diagnosis. The conventional methods of influenza detection are virus isolation [8, 9], real-time polymerase chain reaction (RT-PCR) [10–12] and immunoassay [13–15], but they have some disadvantages (virus isolation is sensitive, but time consuming and laborious, and RT-PCR requires appropriate facilities and trained personnel). Therefore, it is necessary to seek new improved methods for the detection of influenza.
Electrochemical biosensors for nucleic acid detection are a promising tool in comparison with the conventional methods, because they are sensitive, cost effective, with simple and fast sample analysis [16–18]. Peptide nucleic acid (PNA) is widely used as a tool for biosensing of target DNA or RNA [19–24], because the binding properties of PNA have high affinity for its complementary DNA or RNA molecules, mainly because of the lack of electrostatic repulsion between the uncharged PNA backbone and that of the natural nucleic acid [25]. PNA is a synthetized oligo amide reported for the first time by Nielsen et al. [26]. Its backbone is composed of N-(2-aminoethyl)-glycine units bounded by peptide bonds, and each nucleobase is attached to the PNA backbone by a methylenecarbonyl linker. PNA is capable of forming double-stranded complexes [27], and triple-stranded complexes [28] with complementary poly- or oligonucleotide. In spite of the fact that PNA is currently utilized in different applications as molecularly biological procedures, diagnostic assays and antisense therapies, one may suggest that PNA could find a leading role in rapid detection of pathogens due to their unique properties such as stability toward enzyme degradation (proteases and nucleasis), ability to distinguish single mismatch and high stability over wide range of pH [25]. Moreover, these properties make them a perfect tool for diagnostic assays and/or anti-sense therapies.
In this study, we suggested a biosensor for the detection of influenza-derived DNA oligonucleotide, where a complementary PNA sequence was used as the probe. The assay contains two parts: (1) isolation using paramagnetic particles and (2) electrochemical detection of isolated products.
Experimental Section
Chemicals
The PNA oligonucleotide (ODN) was synthesized by Biosynthesis (Biosynthesis, Texas, USA). The sequence of PNA probe (from N-terminus to C-terminus) was cysteine-CCTCAAGGAG. The Influenza-derived DNA sequence (5′ AAAAACTCCTTGAGG 3′) was purchased from Sigma-Aldrich (Sigma-Aldrich, USA). A model for the target sequence was taken from Malecka et al. [29]. PNA/DNA duplex was prepared by incubation (92 °C, 5 min) and slow cooling (at room temperature) of aliquot mixture of PNA and DNA. Other chemicals were purchased from Sigma-Aldrich (Sigma-Aldrich, USA) in ACS purity unless noted otherwise. Stock solutions were prepared with ACS water. Deionised water underwent demineralization by reverse osmosis using Aqua Osmotic 02 device (Aqua Osmotic, Tisnov, Czech Republic) and after that purified using Millipore RG (MiliQ water, 18 MΏ, Millipore Corp., USA). Deionised water was used for rinsing cell, washing and buffer preparation.
Characterization of PNA, DNA and PNA/DNA Duplex
Prior to isolation process, PNA, DNA, and PNA/DNA duplex were characterized by matrix-assisted laser desorption/ionization time of flight mass spectrometry (MALDI-TOF MS). The mass spectra of PNA, DNA and PNA/DNA duplex (50 µg/mL) were measured using a MALDI-TOF/TOF mass spectrometer Bruker ultrafleXtreme (Bruker Daltonik GmbH, Germany) equipped with a laser at a wavelength of 355 nm with an accelerating voltage of 25 kV, cooled with nitrogen and with a maximum energy of 43.2 µJ. The matrix used in the MALDI method was 3-hydroxypicolinic acid (3-HPA; Sigma-Aldrich). The saturated solution of 3-HPA was prepared in 50 % acetonitrile with 10 mg/mL diammonium hydrogen citrate. Matrix mixtures were vortexed and ultrasonicated using Bandelin 152 Sonorex Digital 10P ultrasonic bath (Bandelin electronic GmbH, Germany) for 2 min at 50 % intensity at room temperature. A dried-droplet method was used for sample preparation. Briefly, the sample solution was mixed with matrix solution in volume ratio of 1:1. After obtaining a homogeneous solution, 1 µL of mixture was applied on the target plate and dried under atmospheric pressure at room temperature. A mixture of protein and peptide calibration standards was used to externally calibrate the instrument. All measurements were performed in the linear positive mode in the m/z range 1–20 kDa. The mass spectra were acquired by averaging 2,500 subspectra from a total of 2,500 shots of the laser. Laser power was set 5–10 % above the threshold.
Electrochemical Analysis
Cyclic voltammetry (CV), square wave voltammetry (SWV) and square wave voltammetry coupled with adsorptive transfer technique (AdT SWV) were used for electrochemical characterisation of PNA, DNA and duplex.
The measurements were carried out at 663 VA Stand with original accessories: 800 Dosino, 846 Dosing Interface (Metrohm, Switzerland) in standard electrochemical cell with three electrode set up. Hanging mercury drop electrode (HMDE) was used as a working electrode, Ag/AgCl/3M KCl electrode was used as a reference electrode and glassy carbon electrode as an auxiliary electrode. The analysed samples were deoxygenated prior to analysis by purging with argon (99.999 %). All measurements were performed at room temperature in the presence of acetate buffer (0.2 M CH3COOH + 0.2 M CH3COONa, pH 5.0). To evaluate the result GPES 4.9 software was employed.
Cyclic Voltammetry (CV)
Parameters of CV were as follows: purge time 30 s, duration 240 s; equilibration time 2 s; number of scans 1; initial potential 0 V; end potential −1.8 V; step potential 0.00495 V; scan rate 0.5 V/s.
Square Wave Voltammetry (SWV)
Parameters of SWV were as follows: purge time 60 s; frequency 280 Hz; initial potential 0 V; end potential −1.8 V; step potential 49.5 V; amplitude 25 mV.
Adsorptive Transfer Technique Coupled with Square Wave Voltammetry (AdT SWV)
Parameters of AdT SWV were as follows: time of accumulation 120 s, frequency 280 Hz, initial potential 0 V, end potential −1.8 V, step potential 49.5 mV, amplitude 25 mV.
Isolation of Influenza Target Sequence using Paramagnetic Particles (MPs)
Automatic pipetting station EP Motion 5,075 (Eppendorf, Germany) with original devices [microplate holder, tips holder, tips (150, 300 and 1,000 μL), rack tubes, reservoir holder, container for used tips, thermo adapter, PCR plate 96, magnetic adapter] was used for the fully automated influenza-derived DNA sequence isolation process. Buffers used for the isolation were as follows: phosphate buffer I (pH 6.5, 0.1 M NaCl, 0.05 M Na2HPO4, and 0.05 M NaH2PO4), phosphate buffer II (0.2 M NaCl, 0.1 M Na2HPO4, and 0.1 M NaH2PO4), hybridisation buffer (100 mM Na2HPO4, 100 mM NaH2PO4, 0.5 M NaCl, 0.6 M Guanidium thiocyanate, and 0.15 M Trizma base adjusted by HCl on pH of 7.5). 10 µL of paramagnetic particles Dynabeads oligo dT25 (Invitrogen, Oslo) was dispensed in selected wells in the plate (PCR 96, Eppendorf, Germany). Subsequently the plate was transferred to the magnet and the stored solution from MPs was aspirated to waste. Afterward, the MPs were washed three times with 100 μL of phosphate buffer I. This step was followed by the first hybridisation. 10 μL of DNA specific sequence and 10 μL of hybridisation buffer were added in selected wells, and subsequently the plate was incubated (30 min, 25 °C, 450 RPM). This procedure was followed by a three times washing (100 μL of phosphate buffer I). The next step was the second hybridisation. 10 µL of PNA probe and 10 µL of hybridisation buffer were added into selected well and the plate was incubated (30 min, 25 °C, 450 RPM), followed by three times washing (100 μL of phosphate buffer I). Then, 30 µL of elution solution (phosphate buffer II) was added into selected wells and the plate was incubated (5 min, 85 °C, 450 RPM). Immediately, the plate was transferred to the magnet, and the isolated product from the selected wells was transferred to a new well and electrochemically analysed.
Descriptive Statistics
All the data were processed using Microsoft Excel® (USA) and Statistica.Cz Version 8.0 (Czech Republic). The results are expressed as mean ± standard deviation (SD) unless noted otherwise (Excel®). Differences with p < 0.05 were considered significant and were determined using one-way ANOVA test (particularly Scheffe test), which was applied for means comparison.
Results and Discussion
Accurate and rapid diagnosis in the early-phase of disease promises better therapeutic strategy in progression of the health care of the human population. To achieve a precise and efficient diagnosis, an important goal in biosensor evolution is to produce nanoscale assemblies capable of continuously monitoring concentrations of a specific analyte in a simple, reliable, highly-sensitive, and highly-specific manner [30]. The research in the field of biosensing technologies has gained enormous attention in recent decades [31–34]. The development of sensitive and selective biosensors is important in biological studies, clinical diagnostics, and biodefense applications [32]. Biosensors are formed from two basic components—a bio recognition element and a signal transducer [35–37]. For nucleic acids targets, the recognition is accomplished by hybridisation to complementary strands through specific base pairing [38–40]. In this study, we suggested and developed a hybridisation biosensor for the influenza nucleic acid, where complementary PNA sequence was used as an electrochemical probe instead of conventionally used nucleic acids.
Characterization by MALDI-TOF
Mass spectra of DNA and PNA are shown in Fig. 1a, b, whereas the scheme of differences between the backbone structure of DNA and PNA was inserted there. The highest peak detected with a mass of 4,586.306 Da in DNA spectrum was assigned to molecular ion [DNA + H]+. The peaks with masses 2,840.600, 5,682.918 and 5,934.495 Da in PNA spectrum were assigned to molecular ions [PNA+H]+, [2PNA+H]+ and [2PNA+dA+H]+, where dA is deoxyadenosine.

MALDI-TOF mass spectra of a DNA, b PNA and c PNA/DNA duplex. Captions of peaks were assigned manually, only the peak of m/z 2,778.036 has no caption because the assignment was not clear; dA is deoxyadenosine. Concentrations of samples were 50 µg/mL. As a matrix a 3-hydroxypicolinic acid was used. Measurements were performed in the linear positive mode. Each spectrum was averaged from 2,500 sub-spectra. Scheme of difference in backbone structure of PNA and DNA is presented as inset
The formation of PNA/DNA duplex was confirmed in Fig. 1c. The molecular mass of PNA/DNA duplex is approximately 7,425 Da. The peaks with masses 2,778.036, 4,586.306, 5,682.918 and 7,425.902 Da in PNA/DNA spectrum were assigned to molecular ions [PNA-62.546 Da+H]+, [DNA+H]+, [2PNA+H]+ and [PNA+DNA+H]+. Smaller peaks in mass spectra belong to adducts or fragments of detected peaks of PNA, DNA and PNA/DNA duplex. We were not able to assign the mass difference of 62.546 Da, which could be the loss of four oxygen atoms. It is clear from the green spectrum that the PNA/DNA duplex was formed, however in the PNA/DNA sample separate peaks of PNA and DNA were detected which probably indicates that the formation of duplex was not 100 % effective.
Electrochemical Analysis: Cyclic Voltammetry
Cyclic voltammetry monitors both reduction and oxidation process in one system and through this, it is considered as the most commonly used electrochemical method. Generally, nucleic acid bases could be oxidized or reduced; however, we determined reduction only. In Fig. 2a, there are voltammograms of all samples mutually compared. CA peak (reduction signal of adenosine and cytosine) was observed at the potential around −1.5 V. The difference in the potentials of CA peaks demonstrates various backbone structures, which bind to the nucleobases. The potential of CA peak related to DNA single strand (−1.504 ± 0.005 V) was close to the potential of CA peak related to duplex DNA/PNA (−1.509 ± 0.005 V). The small difference between potentials of CA peaks related to DNA and DNA/PNA duplex is probably caused by the minor influence of non-charged backbone of PNA to the electron transfer on the electrode surface. The CA peak related to PNA alone was detected at the most negative potential (−1.538 ± 0.005 V). The difference of potential between CA peaks related to PNA and DNA was 0.034 V.

Characterization of PNA, DNA and PNA/DNA duplex by cyclic voltammetry. a Typical voltammograms of PNA, DNA and PNA/DNA measured by CV, relative CA peak heights are presented measured at the time of accumulation 300 s as inset. b Dependence of current of hydrogen evolution on time of accumulation of tested samples (PNA/DNA, PNA, and DNA; filled marks). In minor axis there is shown the dependence of potential of hydrogen evolution on time of accumulation (unfilled marks). c Dependence of relative CA peak height on time of accumulation (filled marks) and dependence of CA peak potential on time of accumulation (unfilled marks) on minor axis. Parameters of CV measurements were as follows: purge time 30 s, first conditioning potential 0 V, duration 0–600 s; equilibration time 2 s; initial potential 0 V; end potential −1.8 V; step potential 0.00495 V; scan rate 0.5 V/s
Differences were also observed in the height of the obtained CA peaks (Fig. 2a). These differences are caused mainly by two factors. Primarily, it is the influence of single or double strand structure of detected nucleotide and secondarily it is the influence of the various charging of individual strands. DNA has negative charge (due to negative charge of the phosphate backbone), while PNA has no charge. The highest CA peak was detected on voltammogram of DNA and other peaks were related to them. The lowest peak was observed in voltammogram of PNA/DNA (18.25 %). The CA peak height of PNA was slightly higher than duplex (28 %). Figure 2b (filled markers) presents the dependence of current of hydrogen evolution (difference between currents of first vertex potential and point in oxidation scan, where current did not change significantly) on time of accumulation (0, 60, 120, 240, 360, 480 and 600 s were tested times of accumulation). We demonstrated that the current of hydrogen evolution was increased with the increasing time of accumulation in all samples. In the minor axis of Fig. 2b (unfilled markers), the dependence of potential of hydrogen evolution (potential, where no current change significantly on oxidation scan) on time of accumulation was shown. Figure 2c (filled markers) described the dependence of relative CA peak height on time of accumulation. At DNA determination, the relative CA peak height was increased with the increasing time of accumulation; a significant increase was observed up to 240 s of time of accumulation. Over 240 s time of accumulation, the relative CA peak height increasing was comparatively smaller. In the minor axis of Fig. 2c (unfilled markers), the dependence of CA peak potential on time of accumulation was described. The potential of CA peak decreased with the increasing time of accumulation.
Electrochemical Analysis: Square Wave Voltammetry
Square wave voltammetry is one of the most sensitive methods for determination of nucleic acids. Characterisation of DNA and PNA single strands and DNA/PNA duplex was done (Fig. 3a–c). The differences in potentials of CA peaks were clearly observed in Fig. 3a. The potential of CA peak related to DNA was −1.354 ± 0.005 V, potential of duplex peak was −1.365 ± 0.005 V, and peak related to PNA was the most negative (−1.390 ± 0.005 V). The differences among CA peak heights related to various nucleic acid strands are shown in the inset of Fig. 3a. PNA had the smallest value of peak height, approx. 60 %, which was related to maximum value. The maximum value was detected in the case of CA peak from DNA. The CA peak of duplex was in between them (82 %). The dependence of relative CA peak height (related to the highest CA peak for set of measurements) on time of accumulation is shown in Fig. 3b. 360 s was estimated as optimum, because further increasing of accumulation time could not bring another significant increase of obtained CA peak height. SWV method with optimized time of accumulation was used to observe the influence of concentration of nucleic acid on its CA peak height. Calibration curve of all the three nucleic acids had linear character (Fig. 3c). Regression equations are as follows: y = 4.08x − 1.87 (PNA), y = 20.89x − 8.45 (DNA) and y = 14.17x − 2.28 (DNA/PNA). Slope of PNA calibration curve is distinctly lower than two other nucleic acid samples and therefore, the sensitivity for measurement of DNA and duplex by this method is higher.

Characterization of PNA, DNA and PNA/DNA duplex by two various methods: a–c square wave voltammetry; and d–f adsorptive transfer square wave voltammetry. a Typical SWV voltammograms of PNA, DNA and PNA/DNA. Relative CA peak heights measured at time of accumulation 120 s as inset. b Dependence of relative CA peak height on accumulation time. c Dependence of CA peak height on concentration of various nucleic acid strands. d Typical voltammograms of PNA, DNA and PNA/DNA, relative CA peak heights measured at time of accumulation 120 s as inset. e Dependence of relative CA peak height on the time of accumulation. f Dependence of CA peak height on concentration of various nucleic acid strands. Parameters of SWV were as follows: purge time 60 s; frequency 280 Hz; initial potential 0 V; end potential −1.8 V; step potential 0.00495 V; amplitude 0.02505 V. Parameters of AdT SWV were as follows: time of accumulation 120 s; purge time 60 s; frequency 280 Hz; initial potential 0 V; end potential −1.8 V; step potential 0.00495 V; amplitude 0.02505 V
Adsorptive Transfer Technique Coupled with Square Wave Voltammetry
Improvement of square wave voltammetry method by adsorptive transfer technique (AdT SWV) was the next step of nucleic acid determination. This technique allows accumulation of biomolecules on the working electrode surface and increases the sensitivity of the assay. Due to the accumulation step, AdT SWV is more sensitive than SWV and is considered as the most sensitive electrochemical method for nucleic acid detection [18, 39, 41].
Characterisation of all three nucleic acids by AdT SWV method is shown in Fig. 3d–f. In Fig. 3d, typical voltammograms of all three nucleic acid strands are shown. The differences in the potentials of CA peaks among all three samples are described in insert, whereas the peak potential of DNA is most positive −1.351 ± 0.005 V. The peak potential of duplex was close to CA peak of DNA (−1.365 ± 0.005 V) and between CA peak potential of DNA and PNA. CA peak potential of PNA is the most negative (−1.436 ± 0.005 V). In inset of Fig. 3d the differences between CA peak heights are shown. The highest CA peak was detected in DNA voltammogram and other CA peaks were related to them (100 %), and the CA peak of duplex of DNA/PNA is somewhat smaller (85 %). CA peak of PNA is the smallest (52 %). Figure 3e describes the optimization of accumulation time and the dependence of relative CA peak heights on the time of accumulation (related to the highest CA peak for set of measurements). 300 s was selected as optimum, because further increase of accumulation time had no significant influence on obtained CA peak heights. AdT SWV method with optimized time of accumulation was used for monitoring the influence of concentration of nucleic acids on its CA peak heights. All three calibration curves (their parts respectively) have linear character (Fig. 3f). Regression equations of linear parts were as follows: y = 151.77x (PNA), y = 1,238.30x (PNA/DNA) and y = 2,487.60x (DNA). Analytical parameters are summarized in Table 1.
Isolation and Detection of Influenza Target Sequence Using MPs
The influenza-derived DNA oligonucleotide (5′ AAAAACTCCTTGAGG 3′) was used as a target molecule. This sequence was labelled by adenine tail for the purposes of hybridisation (based on thymine–adenine complementary) on surface of MPs modified by thymine tail. This sequence was inspired by Malecka et al. [29] and corresponds to H5 segment of A/swan/Poland/305-135V08/2006 influenza genome. The sequence of PNA probe was cysteine-CCTCAAGGAG, which was complementary to the influenza-derived oligonucleotide.
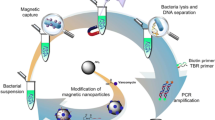
The scheme of the isolation process is shown in Fig. 4a. The cornerstone of the isolation part was in the application of paramagnetic particles modified by oligo thymine strand. The isolation procedure consists of two hybridisation steps. Firstly, target DNA sequence was hybridised on the surface of thymine modified MPs (thymine–adenine complementarity, Fig. 4a part a). Thereafter PNA probe was hybridised with DNA bounded on MPs (base pairs complementarity, Fig. 4a part b). The isolation part was terminated by the elution of the isolated molecules out of MPs. The isolated molecule was analysed by AdT SWV (Fig. 4a part c).

a Scheme of isolation process based on magnetic particles. a Binding of DNA to MPs (thymine–adenine complementarity). b Binding of PNA probe to target DNA sequence (base pairs complementarity). c Elution of isolated product out of MPs and electrochemical detection. b Dependence of relative CA peak height (related to the maximal signal intensity) on concentration of applied target DNA influenza-derived sequence. c Dependence of relative CA peak height on concentration of applied nucleic acid (for first hybridisation there was used 50 µg/mL DNA, for second hybridisation there was used 5–50 µg/mL PNA). CA peak heights were related to the highest peak. Differences in relative peak heights were with the increasing concentration of nucleic acid as follows: 0.22, 22, 64 and 88 %. All data represent mean ± SD, NS not significant, *p < 0.05. All measurements were carried out by AdT SWV at time of accumulation 300 s
Second, there was hybridised DNA sequence onto the thymine modified MPs and the effect of applied concentration of DNA on relative CA peak height (related to highest signal of set of measurements) was investigated (Fig. 4b). The CA peak of isolated DNA was increased with the increasing concentration of applied DNA. For this reason, we selected the concentration 50 µg/mL for the next study of PNA hybridisation efficiency. Various concentrations of PNA (5, 10, 25 and 50 µg/mL) were chosen and there hybridisation effect were tested. We demonstrated (Fig. 4c) that the relative CA peak (related to highest signal of isolation product after second hybridisation) increased with the increasing concentration of PNA. We also found that the CA peak of product after first isolation (DNA) was detected at potential −1.36 V and the product after second isolation (PNA probe) was detected at potential −1.38 V (potential of CA peak of duplex). Comparison between intensities of the obtained electrochemical signals related to the products of the first and second isolation is shown in Fig. 4c. The statistically significant difference between the signals of products of the first and second isolation steps is from the concentration of 10 µg/mL of PNA. At lower concentrations, the signals of PNA and DNA have similar values.
Conclusion
In this article, we described new method for the isolation and detection of a specific DNA sequence derived from the influenza genomic RNA. The procedure was based on an isolation by modified MPs and use of PNA as the electrochemical probe and marker of target DNA. An effect of isolation process was tested by four different concentrations of both applied nucleic acids. The combination between paramagnetic particles, PNA probe, automated isolation process and electrochemical detection provided a proposition of a unique tool for the detection of target nucleic acid. The suggested assay can also be used for the detection of other infectious agents.
References
Bhatnagar J, Jones T, Blau DM, Shieh WJ, Paddock CD, Drew C, Denison AM, Rollin DC, Patel M, Zaki SR (2013) J Clin Virol 56:232–237
Munro SB, Kuypers J, Jerome KR (2013) J Clin Microbiol 51:1124–1129
Gagnon A, Miller MS, Hallman SA, Bourbeau R, Herring DA, Earn DJD, Madrenas J (2013) PLoS One 8:1–9
Honigsbaum M (2013) Med Hist 57:165–185
Medina RA, Manicassamy B, Stertz S, Seibert CW, Hai R, Belshe RB, Frey SE, Basler CF, Palese P, Garcia-Sastre A (2010) Nat Commun 1:1–4
Bousse T, Shore DA, Goldsmith CS, Hossain MJ, Jang Y, Davis CT, Donis RO, Stevens J (2013) J Virol Methods 193:589–596
Wang RH, Li YB (2013) Biosens Bioelectron 42:148–155
Bui VN, Ogawa H, Ngo LH, Baatartsogt T, Abao LNB, Tamaki S, Saito K, Watanabe Y, Runstadler J, Imai K (2013) Arch Virol 158:451–455
El-Zoghby EF, Arafa AS, Kilany WH, Aly MM, Abdelwhab EM, Hafez HM (2012) Virol J 9:1–8
Tong SX, Li Y, Rivailler P, Conrardy C, Castillo DAA, Chen LM, Recuenco S, Ellison JA, Davis CT, York IA, Turmelle AS, Moran D, Rogers S, Shi M, Tao Y, Weil MR, Tang K, Rowe LA, Sammons S, Xu XY, Frace M, Lindblade KA, Cox NJ, Anderson LJ, Rupprecht CE, Donis RO (2012) Proc Natl Acad Sci USA 109:4269–4274
Charrel RN, Salez N (2013) J Infect 66:290–291
Piralla A, Daleno C, Pariani E, Conaldi P, Esposito S, Zanetti A, Baldanti F (2013) J Clin Virol 56:65–68
Li XP, Lu DL, Sheng ZH, Chen K, Guo XB, Jin ML, Han HY (2012) Talanta 100:1–6
Lupiani B, Mozisek B, Mason PW, Lamichhane C, Reddy SM (2010) Avian Dis 54:668–672
Cao C, Dhumpa R, Bang DD, Ghavifekr Z, Hogberg J, Wolff A (2010) Analyst 135:337–342
Pividori MI, Merkoci A, Alegret S (2000) Biosens Bioelectron 15:291–303
Wen YL, Liu G, Pei H, Li LY, Xu Q, Liang W, Li Y, Xu L, Ren SZ, Fan CH (2013) Methods 64:276–282
Krejcova L, Hynek D, Kopel P, Rodrigo MAM, Adam V, Hubalek J, Babula P, Trnkova L, Kizek R (2013) Viruses-Basel 5:1719–1739
Hamidi-Asl E, Raoof JB, Ojani R, Golabi SM, Hejazi MS (2013) J Iran Chem Soc 10:1075–1083
Candiani A, Bertucci A, Giannetti S, Konstantaki M, Manicardi A, Pissadakis S, Cucinotta A, Corradini R, Selleri S (2013) J Biomed Opt 18:1–5
Raoof JB, Ojani R, Golabi SM, Hamidi-Asl E, Hejazi MS (2011) Sens Actuator B-Chem 157:195–201
Hejazi MS, Pournaghi-Azar MH, Alipour E, Abdolahinia ED, Arami S, Navvah H (2011) Electroanalysis 23:503–511
Husken N, Gebala M, Schuhmann W, Metzler-Nolte N (2010) ChemBioChem 11:1754–1761
Pournaghi-Azar MH, Ahour F, Hejazi MS (2010) Anal Bioanal Chem 397:3581–3587
Briones C, Moreno M (2012) Anal Bioanal Chem 402:3071–3089
Nielsen PE, Egholm M, Berg RH, Buchardt O (1991) Science 254:1497–1500
Egholm M, Buchardt O, Nielsen PE, Berg RH (1992) J Am Chem Soc 114:1895–1897
Egholm M, Nielsen PE, Buchardt O, Berg RH (1992) J Am Chem Soc 114:9677–9678
Malecka K, Grabowska I, Radecki J, Stachyra A, Gora-Sochacka A, Sirko A, Radecka H (2012) Electroanalysis 24:439–446
Jean RD, Cheng WD, Hsiao MH, Chou FH, Bow JS, Liu DM (2014) Biosens Bioelectron 52:111–117
Rubin RL, Wall D, Konstantinov KN (2014) Biosens Bioelectron 51:177–183
Kirsch J, Siltanen C, Zhou Q, Revzin A, Simonian A (2013) Chem Soc Rev 42:8733–8768
Saftics A, Agocs E, Fodor B, Patko D, Petrik P, Kolari K, Aalto T, Furjes P, Horvath R, Kurunczi S (2013) Appl Surf Sci 281:66–72
Pires NMM, Dong T, Hanke U, Hoivik N (2013) J Biomed Opt 18:1–7
Bottari F, Oliveri P, Ugo P (2014) Biosens Bioelectron 52:403–410
Sang SB, Zhao Y, Zhang WD, Li PW, Hu J, Li G (2014) Biosens Bioelectron 51:124–135
Sekhon SS, Kim SG, Lee SH, Jang A, Min J, Ahn JY, Kim YH (2013) Mol Cell Toxicol 9:311–317
Bi S, Cui YY, Dong Y, Zhang NB (2014) Biosens Bioelectron 53:207–213
Krejcova L, Huska D, Hynek D, Kopel P, Adam V, Hubalek J, Trnkova L, Kizek R (2013) Int J Electrochem Sci 8:689–702
Ulianas A, Heng LY, Ahmad M, Lau HY, Ishak Z, Ling TL (2014) Sens Actuator B-Chem 190:694–701
Krejcova L, Hynek D, Adam V, Hubalek J, Kizek R (2012) Int J Electrochem Sci 7:10779–10801
Acknowledgments
The financial support from NanoBioTECell P102/11/1068 and CEITEC CZ.1.05/1.1.00/02.0068 is highly acknowledged.
Conflict of interest
The authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in the topical collection Advances in Chromatography and Electrophoresis and Chiranal 2014 with guest editor Jan Petr.
Rights and permissions
About this article

Cite this article
Krejcova, L., Nguyen, H.V., Hynek, D. et al. Paramagnetic Particles and PNA Probe for Automated Separation and Electrochemical Detection of Influenza. Chromatographia 77, 1425–1432 (2014). https://doi.org/10.1007/s10337-014-2737-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10337-014-2737-2