
Abstract
Dekkera bruxellensis is a non-conventional Crabtree-positive yeast with a good ethanol production capability. Compared to Saccharomyces cerevisiae, its tolerance to acidic pH and its utilization of alternative carbon sources make it a promising organism for producing biofuel. In this study, we developed an auxotrophic transformation system and an expression vector, which enabled the manipulation of D. bruxellensis, thereby improving its fermentative performance. Its gene ADH3, coding for alcohol dehydrogenase, was cloned and overexpressed under the control of the strong and constitutive promoter TEF1. Our recombinant D. bruxellensis strain displayed 1.4 and 1.7 times faster specific glucose consumption rate during aerobic and anaerobic glucose fermentations, respectively; it yielded 1.2 times and 1.5 times more ethanol than did the parental strain under aerobic and anaerobic conditions, respectively. The overexpression of ADH3 in D. bruxellensis also reduced the inhibition of fermentation by anaerobiosis, the “Custer effect”. Thus, the fermentative capacity of D. bruxellensis could be further improved by metabolic engineering.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The wine and beer yeast Dekkera bruxellensis (anamorph Brettanomyces bruxellensis) is the main cause of wine spoilage worldwide, thereby causing immense economic losses (Boulton et al. 1996; Fugelsang 1996; Delfini and Formica 2001; Loureiro and Malfeito-Ferreira 2003; Schifferdecker et al. 2014). Infected wines develop distinctive and unpleasant aromas due to volatile phenols produced by this species, also called “Brett’ taints” (Chatonnet et al. 1995) and normally associated with the smell of barnyard, burnt plastic, wet animal and horse-sweat (Licker et al. 1998). This species is also known for its contribution to the flavour composition of Belgium’s Lambic and Gueuze beers (Dequin et al. 2003; Dufour et al. 2003), specialized red wines (Château Musar and Château de Beaucastel), feta cheese and Kombucha tea (Mayser et al. 1995; Teoh et al. 2004). It is often associated with ethanol production plants, where it can act as an ethanol producer (Passoth et al. 2007) or as a spoiler (de Souza Liberal et al. 2007).
Whole genome sequences of 10 Dekkera/Brettanomyces bruxellensis isolates originating from wine, beer or soft drinks were recently reported (Curtin et al. 2012; Piskur et al. 2012; Borneman et al. 2014; Crauwels et al. 2014; Valdes et al. 2014; Crauwels et al. 2015), which is a valuable tool to enhance our understanding of this yeast. The ploidy of the sequenced strains ranges from diploids (CBS 2499, VIB X9085, AWRI 1613, MUCL 49865 and ST05.12/48) to triploids (AWRI 1608, AWRI 1499, CBS 6055 and ST05.12/53); the ploidy of the Chilean wine isolate (LAMAP 2480) is not yet available. Comparative genomics surprisingly placed D. bruxellensis as a sister species to the methylotrophic yeast species Pichia (Komatagaella) pastoris, Ogataea angusta/polymorpha and Kuraishia capsulata (Piskur et al. 2012; Curtin et al. 2012; Curtin and Pretorius 2014); these species are aerobic, Crabtree-negative and poor ethanol producers. Despite its phylogenetic position, D. bruxellensis is a good ethanol producer, Crabtree-positive and a facultative anaerobic yeast, and it exhibits a fermentative lifestyle even in the presence of excess glucose and oxygen, traits it shares with baker’s yeast Saccharomyces cerevisiae. D. bruxellensis was shown to employ a promoter rewiring that was evolved in parallel to S. cerevisiae as one of the molecular mechanisms for the development of the ‘make-accumulate-consume’ life strategy (Rozpedowska et al. 2011). Unlike S. cerevisiae, D. bruxellensis is more resistant to acidic pH (Rozpedowska et al. 2011). It also utilizes alternative carbon sources, for example, cellobiose and pentoses such as xylose and l-arabinose (Toivola et al. 1984; Galafassi et al. 2011; Moktaduzzaman et al. 2015); these carbon sources are plentiful and inexpensive in lignocellusic feedstocks. This yeast can also utilize nitrate as a sole nitrogen source due to the presence of the genes of the nitrate assimilation pathway coding for nitrate transporter, nitrite and nitrate reductase and nitrate assimilation transcription factors (Woolfit et al. 2007; Steensels et al. 2015). This enables D. bruxellensis to outcompete S. cerevisiae in industrial fermentations (de Barros Pita et al. 2011), since S. cerevisiae is unable to utilize the abundant nitrate in the major biofuel industry substrate sugarcane juice (de Souza Liberal et al. 2007; Vaughan-Martini and Martini 2011). Like S. cerevisiae, D. bruxellensis can adapt to fermentation inhibitors in lignocellulose hydrolysates (Blomqvist et al. 2011). These traits make D. bruxellensis attractive for biofuels.
Because of their importance for food and biofuel, D. bruxellensis’s natural strains have been studied for ethanol production. However, genetically engineered strains have yet to be applied to ethanol production. In this study, we developed auxotrophic strains of D. bruxellensis (ura3 and ura5) and we analysed the activity of four D. bruxellensis promoters (ADH3, TEF1, GAL7 and PHO5). To investigate the effects of the overexpression of alcohol dehydrogenase gene on the fermentative performance of D. bruxellensis, we constructed recombinant strains carrying an expression cassette for the D. bruxellensis ADH3 gene with the selected promoter TEF1 and analysed them under both aerobic and anaerobic conditions.
Material and methods
Strains and growth conditions
Yeast strains used in this study (Y997 (ura3), Y1009 (ura5) and Y1010 (ura5) listed in Table S1, Supplementary Material) were grown at 25 °C in a complete rich medium YPD (5-g/L yeast extract, 10 g/L peptone, 20 g/L glucose, pH 6.2) or a defined minimal medium (1.7 g/L yeast nitrogen base without amino acids and ammonium sulphate, 5 g/L ammonium sulphate) supplemented with glucose (20 g/L) and uracil (50 mg/L) for uracil auxotrophs. During the characterisation of other putative auxotrophic mutants, any single amino acids were supplemented in the minimal medium, following the standard yeast medium protocols. Yeast transformants were selected on solid minimal medium supplemented with glucose (20 g/L). Yeast transformants were further grown in defined minimal medium (1.7 g/L Yeast Nitrogen Base without amino acids and ammonium sulphate, 5 g/L ammonium sulphate) with the supplementation of either glucose (20 g/L), ethanol (10 g/L) or galactose (20 g/L). For phosphate starvation conditions, cells were grown in defined minimal medium (1.5 g/L yeast nitrogen base without amino acids and without phosphate, 5 g/L ammonium sulphate) supplemented with glucose (20 g/L) and KH2PO4 (0.005 g/L). This medium was designated as YNB + 10 % phosphate.
The Escherichia coli strain TOP10 (Stratagene, Agilent Technologies, Santa Clara, USA) was used in all experiments that needed a bacterial host. The strain was grown at 37 °C in Luria-Bertani (LB) medium (5 g/L yeast extract, 10 g/L NaCl, 15 g/L peptone, pH 7.4). Transformed E. coli cells were selected on LB medium containing 100 mg/L of ampicillin.
Molecular biology techniques
Plasmid DNA isolations from E. coli transformants were carried out using a GeneJET Plasmid Miniprep Kit (Thermo Fisher Scientific, Waltham, USA). All the enzymes that were used for cloning (Phusion DNA polymerase, T4 DNA ligase and restriction enzymes) were purchased from Thermo Scientific (Waltham, USA).
Generation of auxotrophic mutants
Several D. bruxellensis strains (Table S1, Supplementary Material) were mutagenized by UV or ethane methyl sulfonate (EMS), following the standard yeast mutagenesis protocols. For each strain, several thousand colonies were screened for auxotrophy. Putative ura3 and ura5 strains were selected on defined minimal medium supplemented with both uracil (50 mg/L) and 5′- fluoroorotic acid (FOA 1 g/L). One of the identified ura3 mutants, D. bruxellensis Y997, was used in most of the transformation experiments.
Plasmid construction
All plasmid constructs generated during this study were confirmed by sequencing. All primers used in plasmid construction are listed in Table S2 (Supplementary Material). The D. bruxellensis URA3 gene was sub-cloned from the genomic DNA (originating from CBS 2499 strain) and re-sequenced; the obtained sequence was deposited at GenBank with the accession number AY964183 (http://www.ncbi.nlm.nih.gov/nuccore/AY964183). The primer pair OL7 and OL8 amplified a 2.2-kb fragment carrying the URA3 gene, which was digested with both SalI and PstI and then sub-cloned into the plasmid pUC57, which resulted in P892 (Fig. S1 and Table S3, Supplementary Material).
For the overexpression of D. bruxellensis ADH3 gene (DbADH3), the open reading frame of gene gm1.2868_g (Table S5, Supplementary Material) with 300-bp downstream of the ORF was fused by overlap PCR with the D. bruxellensis promoter TEF1 (DbTEF1) using the primers DbTEF1-XbaI-sense, DbTEF1-ADH3-antisense, DbADH3-TEF1-sense and DbADH3-SalI-antisense (Table S2, Supplementary Material). The fragment obtained by overlap PCR was then digested with both SalI and XbaI and then cloned into P892, which resulted in plasmid P1227 (Fig. S4, Supplementary Material).
Transformation system
To transform D. bruxellensis yeast, a lithium acetate electrotransformation procedure based on previous protocols by Becker and Guarente (1991) and Boretsky et al. (2007) was developed. An overnight culture was inoculated into 200 mL of YPD and grown overnight at 25 °C. The cells were harvested, washed with water and resuspended in LiAc/TE buffer (0.1 M lithium acetate, 10 mM Tris-HCl, 1 mM EDTA, pH 7.5). After incubation (1 h at 25 °C), the cells were pelleted, washed with 1 M sucrose and diluted 1:1 in 1 M sucrose. Aliquots of 100 μL of this suspension were mixed with 10–20 μg of plasmid DNA and transferred into chilled 2-mm electroporation cuvettes. Electroporation was carried out in a Bio-Rad Gene Pulser II (BioRad Laboratories, Hercules, USA) (resistance 100 Ω, capacitance 50 μF, voltage 2.3 kV) with a Bio-Rad Pulse Controller II (BioRad Laboratories, Hercules, USA) included in the circuit. Directly after the pulse, 900 μL of YPD was added. The cells were then transferred into 1.5-mL tubes and incubated at 25 °C for 1 h. After the incubation, the cells were pelleted, washed once with water and were resuspended in 350 mL of water. Then, 100 μL of suspension was plated out on selective medium and incubated at 25 °C for 5–15 days.
Stability assay of yeast transformants
In this procedure, cells from single colonies of yeast transformants were inoculated into rich nonselective medium (liquid YPD) and grown for 60 generations at 25 °C. Following growth, the cultures were diluted and plated on YPD plates to obtain single colonies. The dilutions that gave rise to 100–200 colonies were replica-plated on selective minimal medium. The transformants, of which 100 % of the colonies remained prototrophic after this type of cultivation, were assumed stable, selected for further analysis and checked by PCR for the presence of desirable constructs.
Sequencing and bioinformatic analysis
For the promoter expression studies, the sequences of D. bruxellensis genes were identified in the D. bruxellensis CBS 2499 strain database JGI (http://genome.jgi.doe.gov/Dekbr2/Dekbr2.home.html) using S. cerevisiae protein sequences (http://www.yeastgenome.org) as reference running tblastn. The gene promoter sequences were searched for putative binding sites for transcription factors using a homemade Python script. The D. bruxellensis ADH3 protein sequence was searched for mitochondrial-targeting motifs by pairwise alignment with S. cerevisiae ADH3 sequence using EMBOSS ClustalW2 (http://www.ebi.ac.uk/Tools/msa/clustalw2/).
The sequencing of all generated constructs was performed by MWG Biotech (Ebersberg, Germany).
Alcohol dehydrogenase activity assay in cell-free extracts
For alcohol dehydrogenase activity assay, yeast cells were collected from fermentation experiments from early logarithmic growth phase. Cell-free extracts were prepared using glass beads (Sigma-Aldrich, St. Louis, USA, G8772) as described earlier (Ishchuk et al. 2008). The obtained extracts were used for measuring the enzyme activity spectrophotometrically as described by Postma et al. (1989) with some modifications. The reaction mixture with ethanol as a substrate contained 50 mM potassium phosphate buffer pH 7.5, 0.25 mM NAD+ and 100 mM ethanol. The reaction mixture with acetaldehyde as a substrate contained 50 mM potassium phosphate buffer pH 7.5, 0.25 mM NADH and 10 mM acetaldehyde. Adding cell-free extract started the reaction. The protein concentration was determined by the Bradford method (Bradford 1976).
Gene expression analysis
To study the promoter activity, the transcription levels of the corresponding genes were investigated using D. bruxellensis strain Y997 transformed with plasmid P892 (prototroph of Y997). Eight clones of this prototroph transformant were inoculated in 50 mL of YNB supplemented with ethanol (10 g/L), KH2PO4 (0.005 g/L), galactose (20 g/L) or glucose (20 g/L) and were grown at 25 °C until OD600nm 1–1.5. Cells were harvested (3000×g, 10 min, 4 °C), washed once with diethylpyrocarbonate (DEPC)-treated water and stored at −80 °C for further analysis. RNA was isolated using the Ambion PureLink RNA Mini Kit manual (Life Technologies, Carlsbad, USA). The concentration of the RNA was measured by NanoDrop spectrophotometer (Thermo Fisher Scientific, Waltham, USA). One microgramme of isolated RNA was further used for cDNA synthesis using a Superscript III Reverse Transcriptase kit (Invitrogen, Thermo Fisher Scientific, Waltham, USA) with RNaseOUT Ribonuclease Inhibitor and random primers (Invitrogen, Thermo Fisher Scientific, Waltham, USA). SYBR GreenER qPCR SuperMix (Invitrogen, Thermo Fisher Scientific, Waltham, USA) was used with the complementary (cDNA) as template and the specific primers for the genes ADH3, GAL7, PHO5 and TEF1 (Table S2, Supplementary Material). PCRs were performed in duplicates using a RotorGene 2000 cycler real-time PCR machine (Corbett Research, Cambridgeshire, UK) with the conditions specified by Invitrogen. The expression of the analysed promoters in each condition was calculated using REST 2009 software v2.0.13 with RG mode (Pfaffl et al. 2002). The α-tubulin gene (YML085C) served as an endogenous control (untreated), and its Ct and amplification data were used to normalize each sample. The influence of the media on the promoter’s expression was additionally analysed using Minitab 17.2.1.0 software (Minitab Inc., State College, USA).
DNA copy number estimation of D. bruxellensis ADH3 overexpression strains
To detect the DNA copy number of the ADH3 gene in strain Y997 carrying plasmid P1227 integrated into the genome, the total DNA was isolated using a standard zymolyase and phenol-chloroform extraction from cultures grown in selective medium (YNB). Thirty nanogramme of purified genomic DNA was used as template in a 20-μL PCR reaction using SYBR GreenER qPCR SuperMix (Invitrogen, Thermo Fisher Scientific, Waltham, USA) in triplicates using a RotorGene 2000 cycler real-time PCR machine (Corbett Research, Cambridgeshire, UK). The relative copy number of ADH3 gene was calculated using ratios of DNA copy number of the ADH3 to α-tubulin (YML085C) genes in transformants versus the parental strain Y997 (Table S8, Supplementary Material).
Aerobic and anaerobic glucose fermentation in bioreactors
Aerobic batch cultivations were performed in Multifors (Infors HT, Bottmingen, Switzerland) bioreactors, with a working volume of 1 L using minimal defined media (Verduyn et al. 1992) supplemented with 20 g/L glucose as carbon source and 5 g/L ammonium sulphate as nitrogen source. Dissolved oxygen (monitored using an InPro 6800S sensor from Mettler Toledo, Greifensee, Switzerland) was maintained above 30 % using stirrers in a cascade mode, varying the stirring speed between 200 and 1200 rpm, at 25 °C with airflow set at 1 L/min. The pH was maintained at 5 (±0.5) through automatic addition of 2 M KOH and 1 M H2SO4 and monitored with a 405-DPAS-SC-K8S/225pH sensor (Mettler Toledo, Greifensee, Switzerland). Samples were taken during exponential growth phase at appropriate intervals and the metabolites analysed by HPLC. The cells were centrifuged (2 min, 16.000×g) and filtered through a 0.2-μm membrane filter and analysed using an HPLC 1200 Series System (Agilent Technologies, Santa Clara, USA) to determine the relative concentrations of the residual glucose and metabolites (ethanol, acetic acid and glycerol). The HPLC components and column specifications are reported by Dashko et al. (2015). Product yields, specific glucose consumption rates and metabolites production rates were calculated as reported before (van Hoeck et al. 2000).
Anaerobic experiments were performed with the same bioreactors, instead fitted with Norprene tubings (Cole-Parmer, Vernon Hills, USA) to reduce diffusion of O2. Fermentors were flashed with N2 (<3 ppm O2) at a flow rate of 0.1 L N2/min at a constant steering speed of 300 rpm. Synthetic minimal media with 2 % glucose was supplemented with 420-mg/L Tween-80 and 10-mg/L ergosterol. Neither amino acids nor pyrimidines were supplemented.
Results
Selection of auxotrophic strains and transformation system
Several D. bruxellensis strains were mutagenized by UV or ethane methyl sulfonate (EMS), and thousand colonies of each strain screened for auxotrophic mutants. The frequency of obtained auxotrophs was lower than 0.1 %, only in one third of the tested strains we could obtain a limited number of mutants having one or another growth requirement (Table S1, Supplementary Material). Even in the ‘auxotroph-positive’ strains, like Y879 and Y897, which provided several mutants, the frequency of the obtained auxotrophs was lower than 0.1 %. Among all strains used, only strain Y879 (D. bruxellensis CBS 2499) was recently sequenced and is diploid (Piskur et al. 2012; Borneman et al. 2014), the ploidy status of the rest of the strains is unknown. On the other hand, diploid and triploid genomes are common among D. bruxellensis isolates (Curtin and Pretorius 2014). Thus, causes for low number of the auxotrophic mutants in our collection could be an increased ploidy of some of the strains or particular gene copy number. Indeed, some of the strains studied (Y865, Y901, Y883 and Y891) carry few copies of URA3 gene (Fig. S2, Supplementary Material). Only in five strains (Y867, Y869, Y871, Y881, Y897) we could obtain FOA-resistant colonies that also exhibited uracil requirement. One of these mutants, Y997 (Table S4, Supplementary Material), originating from parental strain Y881, was sequenced for the URA3 locus and shown to harbour two point mutations (one single base pair deletion located at +599, one single basepair substitution located at +600) within the URA3 open reading frame (Fig. S3, Supplementary Material). The Y997 mutant and other FOA-resistant uracil auxotrophs were used in transformation experiments.
The transformation system for D. bruxellensis yeast, which is available, is based on the non-homologous integration using a dominant selective marker (Miklenic et al. 2013). In our study, for transformation procedure of this yeast, we have developed an alternative protocol, lithium acetate-electrotransformation (Materials and Methods), which is based on previous methods described by Becker and Guarente (1991) and Boretsky et al. (2007). This protocol was used to transform auxotrophic mutants obtained in our study. The Y997 ura3 mutant was transformed with plasmid P892 carrying D. bruxellensis URA3 gene (Fig. S1, Supplementary Material) linearized with restriction enzyme HindIII, which is not present within the URA3 gene. While the linearized P892 plasmid yielded 68 transformants on average between transformation experiments, circular P892 did not give rise to any transformants (Fig. 1). Among 20 randomly picked P892 transformants, 10 transformants proved to be stable after propagation for 60 generations in non-selective medium (YPD) in a transformants stability assay (see Materials and Methods section).

Transformation efficiency of D. bruxellensis ura3 and ura5 mutants by linear and circular plasmids. Circular and by HindIII linearized plasmids were used for transformation. P892 plasmid is carrying D. bruxellensis URA3 gene, P1228—S. cerevisiae URA5 gene. a Transformation efficiency. b Colonies of transformants on selective medium YNB with 2 % glucose
Other selected FOA-resistant mutants required uracil for growth, but did not give rise to any transformants when being transformed with a plasmid carrying D. bruxellensis URA3 gene. In S. cerevisiae, there are two genes (URA5 and URA10) coding for major and minor isozymes of orotate phosphorybosyltransferase, and URA10 partially complements the mutations in URA5 gene (de Montigny et al. 1990); thus, 5-FOA-resistant mutants in this yeast are mostly ura3. The genome of D. bruxellensis CBS 2499 (Y879, http://genome.jgi.doe.gov/Dekbr2/Dekbr2.home.html) was found to carry URA5, but lacked URA10. Two of the FOA-resistant D. bruxellensis mutants obtained in our study from Y871 (CBS 1942) strain (Table S4, Supplementary Material) were proved to be ura5 by complementation with the S. cerevisiae URA5 gene with a transformation efficiency of 14 transformants per microgram of DNA (Fig. 1).
Promoter analysis
To develop an expression plasmid for D. bruxellensis, we have experimented with four promoters, which are known to be strong or regulative in other yeast species: ADH3 (alcohol dehydrogenase isoform III), TEF1 (translational elongation factor 1A), PHO5 (acid phosphatase) and GAL7 (α-d-galactose-1-phosphate uridyltransferase). To evaluate the strength of these promoters, we studied the expression of the corresponding genes using the D. bruxellensis strain Y997. One of the three S. cerevisiae ADH3 orthologs was selected (gm1.2868_g (JGI v2.0), and we further refer to it as ADH3 (Table S5, Supplementary Material).
To study the effects of carbon sources on the promoter activity, their sequences (1000 bp upstream of ATG) were searched for motifs known to be involved in the response to specific carbon source (Mig1, Cph1, Gal4, Adr1, Cat8) or to phosphate limitation (Pho4) (Thukral et al. 1991; Cheng et al. 1994; Lundin et al. 1994; Treitel and Carlson 1995; Gancedo 1998; Shao et al. 1998; Traven et al., 2006; Weinhandl et al. 2014). The Python script highlighted the occurrence of the Mig1 binding site inside the ADH3 promoter sequence at position 71 bp. The GAL7 promoter carries two binding sites for Mig1 at positions 144 and 437 bp as described before (Moktaduzzaman et al. 2015). A putative binding site for the Pho4 transcription factor was found at position 835 bp in the PHO5 promoter sequence. A binding motif for Gcr1 transcription factor was found at positions 638 and 929 bp in the TEF1 promoter sequence (Table S9, Supplementary Material).
The selected promoters were studied by analysing the expression of the corresponding genes under conditions of different media by RT-qPCR (Fig. 2, Table S7, Supplementary Material) using primer pairs DbADH3-RT-sense and DbADH3-RT-antisense for ADH3 gene, DbGAL7-RT-sense and DbGAL7-RT-antisense for GAL7 gene, DbPHO5-RT-sense and DbPHO5-RT-antisense for PHO5 gene, DbTEF1-RT-sense and DbTEF1-RT-antisense for TEF1 gene, α-tubulin-sense and α-tubulin-antisense for YML085C gene (Table S2, Supplementary Material). The RT-qPCR reactions of eight prototroph transformants of the Y997 strain were run in duplicates. The study’s dataset was analysed by two approaches (multivariate analysis (Johnson and Wichern 2002) and relative gene expression analysis (Pfaffl et al. 2002)) to estimate the effects of different media on gene expression.

The expression levels of D. bruxellensis promoters DbADH3, DbGAL7, DbPHO5 and DbTEF1 in Y997 strain grown in YNB media supplemented with either 1 % ethanol, 2 % galactose, 2 % glucose or phosphate depletion medium (YNB + 10 % phosphate). The values shown represent means of the four expression ratios obtained with REST 2009 v2.0.13 software with RG mode from eight clones. The α-tubulin gene Ct and amplification data was used to normalize samples. The error bars represent standard deviation. The Y-axis has a logarithmic scale
For each of the eight clones, the four media were (deterministically) applied to clone samples, and so the dataset was analysed with a two-way linear model without interaction (Anderson 2003). Clones were modelled as blocks, and media were modelled as treatments. Using this model, the dataset was analysed with MINITAB 17.2.1.0 (Minitab Inc., State College PA, USA). The media had a significant effect on the combined (Ct and Amplification factor) response, according to the trace test of the analysis of variance (ANOVA) (Lawley 1938; Hotelling 1951; Anderson 2003). The media had a significant effect on the gene expression measured by Ct (p ≤ 0.0005) but not amplification factor (p ≈ 0.282), according to the lambda test (Wilks 1932; Anderson 2003). Then, for each gene’s Ct, a univariate ANOVA (without interaction) is reported in Table S6, Supplementary Material. The media factor was insignificant for the genes YML085C (p ≈ 0.627), PHO5 (p ≈ 0.210) and TEF1 (p ≈ 0.147) and significant for ADH3 (p ≤ 0.0005) and GAL7 (p ≤ 0.0005) genes. For the genes with significant effects (ADH3 and GAL7), estimates and t tests of the effects of the media are shown in Table 1. For the ADH3 gene, the ethanol and galactose media were highly significant (each p ≤ 0.0005) in comparison to glucose, which has zero effect by parametrization. For the GAL7 gene, the galactose medium was highly significant (p ≤ 0.0005). The largest effects occurred for the ADH3 gene Ct, and for this gene, the galactose and ethanol media had large and significant effects (each p ≤ 0.0005). For the GAL7 Ct, the galactose medium had a large and significant effect (p ≤ 0.0005) (Table 1). The statistical results provide strong evidence that ADH3 and GAL7 are differentially expressed when the medium varies. In contrast, the expression of YML085C, PHO5 and TEF1 is practically constant regardless of the media (Table 1, Table S6, Supplementary material).
In the second approach, the REST 2009 software v2.0.13 was used to calculate the gene expression ratios, where YML085C was used to normalize the samples. For this program, the blocking (as discussed for ANOVA) was not taken into account, and the ratios of all eight clones obtained were averaged (Fig. 2). It indicates that ADH3 and GAL7 are differentially expressed but PHO5 and TEF1 appear unchanged between different media (Fig. 2). The ADH3 gene’s highest expression levels were obtained when cells were grown in the medium containing the non-fermentable carbon sources galactose (ratio of 0.141) and ethanol (ratio of 0.102) (Fig. 2). When glucose was used as a carbon source, the ADH3 of D. bruxellensis was expressed but had the lowest value, which decreased 2.3 times and 3.0 times (in YNB + 2 % glucose and YNB + 10 % phosphate, respectively) in comparison to the medium with ethanol. As in S. cerevisiae, this gene in D. bruxellensis was found to carry recognition sites for the transcription factor Mig1 (Table S9, Supplementary Material) and is subjected to glucose repression. The expression of ADH3 of D. bruxellensis was not fully repressed on glucose, similar to S. cerevisiae ADH3 expression results (Young and Pilgrim 1985). Thus, ADH3 of D. bruxellensis was suspected of influencing the ethanol production from glucose. The DbGAL7 gene was also differentially expressed and showed its highest expression level when cells were grown in medium containing galactose (ratio of 0.159), which is similar to the data reported before for D. bruxellensis CBS 2499 strain (Moktaduzzaman et al. 2015). The effect of glucose repression in the growth medium on the expression of the GAL7 gene was weaker than that on ADH3, as the GAL7 expression on glucose was 1.4 times higher compared to ADH3 (Fig. 2). The DbPHO5 promoter did not manifest differences between the tested media, which indicates that this gene in the Y997 strain is not activated by phosphate depletion (Fig. 2).
The D. bruxellensis promoter TEF1, which showed the highest expression of all analysed promoters under all tested conditions, was further selected to build the expression vector.
Overexpression of D. bruxellensis ADH3 gene
We chose the D. bruxellensis ADH-encoding gene, gm1.2868_g (ADH3, Table S5, Supplementary Material) for the studies on its impact on ethanol production during aerobic and anaerobic glucose fermentation in D. bruxellensis as it was expressed albeit at low levels on glucose (Fig. 2). We were prompted to further investigate the role of ADH3 in the fermentation of glucose by overexpressing this gene in D. bruxellensis. For this purpose, we cloned this gene, D. bruxellensis ADH3, under the strong and constitutive promoter DbTEF1 (Fig. S4, Supplementary Material). The constructed plasmid P1227 (Fig. S4, Supplementary Material) was linearized by PstI and used to transform the ura3 mutant Y997. Among the obtained stable transformants (see Material and Methods and Stability assay of yeast transformants sections), the plasmid was shown to be present in one or two copies (Table S8, Supplementary Material). When we assume that the ADH3 gene is present in the control strain in one copy, then the relative DNA copy number is between 1.25 and 2.15 (Table S8, Supplementary Material). These results suggest that the recipient strain is diploid and originally has two copies of ADH3 in the genome. Then, the integration of one additional ADH3 copy in the diploid genome will result in three copies and the relative ratio of 1.5 (ADH3 transformants no.5, no.9 and no.11). The integration of two copies will result in four gene copies in the genome and in the relative copy ratio of 2 (ADH3 transformants no.3 and no.10) (relative copy number, Table S8, Supplementary Material).
To verify the plasmid insertion site in the genome, the total DNA of one of the transformants (ADH3 transformant Y997_no. 3) was digested with BglII (absent on the plasmid), self-ligated and transformed into bacteria to isolate the plasmid carrying genomic loci around the integration site (Fig. S6, Supplementary Material). As expected from the plasmid copy estimation (it is assumed that it carries two plasmid copies in the genome, Table S8, Supplementary Material), two kinds of plasmids were isolated carrying different genomic loci of D. bruxellensis. The sequencing of both plasmids confirmed that two copies of the integrated plasmid did not disrupt any genes. One of the plasmids carried the choline phosphate cytydyltransferase and the second plasmid carried a hypothetical protein with both genes found in scaffold 2 in D. bruxellensis CBS 2499 (http://genome.jgi.doe.gov/Dekbr2/Dekbr2.home.html) (Fig. S7, Supplementary Material).
The transformants carrying the ADH3 expression plasmids displayed an elevated specific activity of alcoholdehydrogenase (Fig. 3), however, this increase was not statistically significant probably due to the activity of multiple isoforms of alcohol dehydrogenase.

Specific activity of alcohol dehydrogenase in cell-free extracts measured in D. bruxellensis transformants. Ethanol (a) and acetaldehyde (b) were used as substrate. ADH3_no.3—ADH3 transformant no.3, Y997_control—prototroph transformant carrying P892 (empty vector) integrated into the genome
These transformants were grown under aerobic conditions in flasks or under controlled aerobic and anaerobic conditions in bioreactors and compared with Y997 strain carrying P892 integrated (empty vector, Y997_control). The results of the fermentation experiments in flasks showed that all stable transformants tested, carrying the ADH3 expression plasmid, produced more ethanol than the control strain (data not shown). Since shake flasks conditions are not constant, the fluctuation in the concentration of dissolved oxygen is unpredictable over time (McDaniel et al. 1965; Sommerville and Proctor 2013), which may effect the alcoholic fermentation that depends on oxygen tension. We therefore studied one of the transformants (ADH3 transformant no. 3) under fully controlled aerobic and anaerobic batch culture conditions in bioreactors. Under aerobic conditions, we observed that the ADH3 transformant no. 3 had a 1.3 times higher specific growth rate than the control strain (Fig. 4. Table S10, Supplementary Material). In addition, the rate of depletion of glucose was 1.4 times faster in the same respective order, culminating in glucose being depleted in the medium much earlier (approximately after 33 h for ADH3 transformant no. 3 (Fig. 4a) as compared to control (after 57 h, Fig. 4b)). The transformant carrying the ADH3 expression plasmid produced 1.5 times more ethanol in g/L than the control strain. The ethanol yield of ADH3 transformant no. 3 was 1.2 times higher than the control strain. There was no clear difference in yield of biomass between ADH3 transformant no.3 and the control strain (Fig. 4a and Fig. 4b; Table S10, Supplementary Material). The ADH3 overexpressing transformant also had a slightly lower yield of acetate, suggesting the redirection of the carbon flux towards ethanol, perhaps a more efficient conversion of acetaldehyde than in the control strain (Fig. 4a, b).

Glucose fermentation profiles of D. bruxellensis transformants in bioreactors in glucose-based minimal medium. Growth profiles of ADH3 transformant no. 3 and Y997_control under aerobic (a and b) and anerobic (c and d) conditions (colour indications: black (glucose); red (ethanol); green (acetate), blue (OD600nm), glycerol (grey)). Yields of metabolites and maximum specific growth rate (μmax) under aerobic (e) and anaerobic (f) conditions (ethanol, acetate, glycerol and biomass) are shown in grammes per gramme of consumed glucose (g/g)
Under anaerobic conditions, we observed the inhibition of fermentation, the “Custer effect”, which had been reported to occur for other D. bruxellensis strains under the same conditions (Pereira et al. 2012), as the tested strains displayed lower specific glucose consumption rates in comparison with the aerobic conditions. It is noteworthy that no supplements such as amino acids were added to the medium. While there was no clear difference in biomass yield between the transformant overexpressing the ADH3 gene and the control, the ethanol yield was 1.5 times higher in the transformant (Fig. 4f). The difference between the glucose consumption rates between strains suggests that in the transformant the overexpression of the ADH3 gene helps to ease the “Custer effect”, as the transformant consumed glucose 1.7 times faster than the control (Table S10, Supplementary Material), and this difference is bigger when we compare the aerobic bioreactor data (Table S10, Supplementary Material). Under anaerobic conditions, the ADH3 transformant no. 3 depleted the glucose after 87 h of fermentation (Fig. 4c), while the control strain did not finish the glucose earlier than 183 h (Fig. 4d).
Discussion
In our study, we developed new molecular biology tools (an auxotrophic transformation system and an expression vector) that we used for the metabolic engineering of D. bruxellensis.
Although D. bruxellensis is a yeast species of interest for both industry and basic research, the molecular biology tools for the manipulation of this yeast are not well developed. The transformation system for D. bruxellensis developed by Miklenic et al. (2013) is based on a dominant selection marker and non-homologous integration of DNA, resulting in stable transformants. This system’s transformation efficiency is 20 transformants per microgram of DNA (Miklenic et al. 2013). In our study, we developed an alternative protocol for the development of a transformation system using auxotrophic markers. Although 30 D. bruxellensis strains were mutagenized in our study, only 10 gave rise to auxotrophic mutants, which had a frequency below 0.1 %. D. bruxellensis lacks a simple haploid organization of the genome (Hellborg and Piskur 2009; Piskur et al. 2012), and diploid or triploid genomes are common states among different isolates of D. bruxellensis (Curtin et al. 2012; Borneman et al. 2014). This polyploidy suggests why it was difficult to isolate auxotrophic mutants. Among a few stable D. bruxellensis 5-FOA-resistant colonies, we identified one ura3 and two ura5 mutants by sequencing of the gene and/or by mutation complementation. Using a plasmid carrying the D. bruxellensis URA3 gene, we obtained the transformation efficiency, which is 3.4 times higher on average comparing to the transformation system using a dominant selective marker by Miklenic et al. (2013). The transformation of D. bruxellensis ura5 mutants by the heterologous S. cerevisiae URA5 gene resulted on average in 14 transformants per microgram of DNA, suggesting that the heterologous URA5 gene is not efficiently expressed in D. bruxellensis. These results indicate that to achieve higher transformation efficiency by integrative plasmids, native D. bruxellensis genes are useful as selective markers.
To develop expression vectors that can be used for expression of the gene of interest (e.g., D. bruxellensis ADH3 gene), we analysed the activity of D. bruxellensis promoters. Promoters, both constitutive and inducible, have been intensively studied and analysed in the yeast S. cerevisiae (Ciriacy 1975; Blumberg et al. 1988; Lohr et al. 1995; Walther and Schüller 2001; Peng and Hopper 2002; de Smidt et al. 2008); however in D. bruxellensis, this was not explored before, due to the unavailability of the genome sequence until recently. Ten D. bruxellensis complete genome sequences (Curtin et al. 2012; Piskur et al. 2012; Borneman et al. 2014; Crauwels et al. 2014; Valdes et al. 2014) have become available during the last few years, contributing to the development of molecular biology tools for D. bruxellensis. Recent research by Rozpedowska et al. (2011) generated data on the expression of rapid growth- and respiration-associated genes; Moktaduzzaman et al. (2015) reported on the expression of genes involved in hexose transport, galactose metabolism, respiration, TCA and glyoxylate cycles and gluconeogenesis. In our study, among the four D. bruxellensis promoters studied, we selected the promoter TEF1 to build an integrative expression vector. The TEF1 gene proved to have the highest expression level, which was not significantly changed between the different conditions tested. The expression of another studied gene, ADH3, which was not fully repressed by glucose, prompted us to investigate its impact on the glucose metabolism and the fermentation capacity of D. bruxellensis.
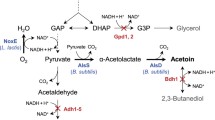
Alcohol dehydrogenase genes are known to be the key enzymes of alcoholic fermentation. D. bruxellensis is a yeast species that did not undergo the whole genome duplication event and has a very small number of gene families duplicated (Piskur et al. 2012). The ADH1, 2, 3 and 5 group is among these duplicated gene families, which in S. cerevisiae is important for the reversible conversion of aldehydes to ethanol. It was also found to have a lineage-specific duplication in D. bruxellensis (Piskur et al. 2012). In D. bruxellensis, there are three genes of ADH1-5 group, which were recently duplicated and show high sequence similarity between each other (gm1.961_g, gm.1.3583 and gm1.2868_g) and are more phylogenetically related to S. cerevisiae ADH3 (Piskur et al. 2012). In S. cerevisiae, the ADH1, 3, 4 and 5 genes are responsible for the reduction of acetaldehyde to ethanol during glucose fermentation; whereas, ADH2 catalyses the oxidation of ethanol to acetaldehyde (Piskur et al. 2012). The alignment of the D. bruxellensis ADH3 amino acid sequence with the protein sequences of S. cerevisiae ADH1, ADH2 and ADH3 using EMBOSS ClustalW2 highlights the presence of mitochondrial-targeting motifs characteristic for group I long-chain alcoholdehydrogenases (Box II and Box III) (Pilgrim and Young 1987; Reid and Fewson 1994) and a GroES-like domain (Murzin 1996; Taneja and Mande 1999) for both S. cerevisiae and D. bruxellensis ADH3 genes. In addition, D. bruxellensis Adh3 was found to carry a sequence similar to S. cerevisiae N-terminal mitochondrial-targeting signal Box I with some amino acid substitutions, which could indicate a mitochondrial localization. In order to overexpress one of the three S. cerevisiae ADH3 orthologs of D. bruxellensis (gm1.2868_g), a plasmid carrying URA3 as a selective marker and the corresponding ADH3 gene with promoter TEF1 driving its expression were integrated into the genome of ura3 host strain Y997. The created stable transformants had the ADH3 gene constitutively expressed at high level and had glucose fermentation capacity substantially improved. Our results showed an increase of 1.2 to 1.5 times in the ethanol yield from glucose under both aerobic and anaerobic conditions when cells carry two additional copies of ADH3 gene integrated into the genome. The overexpression of the ADH3 gene enhanced the glucose consumption and ethanol production rates and resulted in a higher ethanol yield. On the other hand, the acetate yield was slightly lowered comparing to the control strain under aerobic conditions.
Ethanol and acetaldehyde freely diffuse across membranes. In yeast mitochondria, Adh3 was shown to be involved in (i) the conversion of acetaldehyde to ethanol with reoxidation of mitochondrial NADH (ethanol-acetaldehyde shuttle) which is important under anaerobic growth (Bakker et al. 2000; Lertwattanasakul et al. 2009), (ii) protection from the toxic effects of ethanol, and (iii) cofactors recycling by the conversion of ethanol to acetaldehyde (Saliola et al. 2006; Suwannarangsee et al. 2012). Our data show that the overexpression of D. bruxellensis ADH3 enhances the conversion of acetaldehyde to ethanol (higher ethanol and lower acetate yields) under aerobic glucose fermentation. Reoxidation of NADH by this enzyme could also improve the glucose consumption rate by providing more NAD+ for glycolysis. It is interesting to note that the overexpression of the ADH3 in O. polymorpha also strongly activated glucose alcoholic fermentation (Suwannarangsee et al. 2012). The stronger effect on ethanol yield and glucose consumption rates of ADH3 overexpression was observed under anaerobic fermentation. The overexpression of ADH3 gene under anaerobiosis improved the glucose consumption by 1.7 times and made the “Custer effect” (inhibition of alcoholic fermentation under anaerobiosis (Scheffers 1966)) less pronounced. Under oxygen-limited conditions, the level of enzymes involved in the central carbon metabolism, which generate NADH, is higher than that oxidizing NADH in D. bruxellensis (Tiukova et al. 2013). These findings suggest that the overexpression of ADH3 of D. bruxellensis contributes to resolving the NADH imbalance during the conditions of the “Custer effect” (van Dijken and Scheffers 1986; Tiukova et al. 2013), as it improves the NADH oxidation through ethanol synthesis from acetaldehyde.
Our study presents a set of new molecular biology tools for D. bruxellensis that can be used to manipulate this yeast. The creation of strains carrying the alcohol dehydrogenase expression plasmid is the first success in the improvement of ethanol fermentation by this industrially important yeast.
References
Anderson TW (2003) An introduction to multivariate statistical analysis (3rd ed). John Wiley and Sons, New York, USA
Bakker BM, Bro C, Kötter P, Luttik MA, van Dijken JP, Pronk JT (2000) The mitochondrial alcohol dehydrogenase Adh3p is involved in a redox shuttle in Saccharomyces cerevisiae. J Bacteriol 182(17):4730–4737
Becker DM, Guarente L (1991) High-efficiency transformation of yeast by electroporation. Methods Enzymol 194:182–187
Blomqvist J, South E, Tiukova I, Momeni MH, Hansson H, Ståhlberg J, Horn SJ, Schnürer J, Passoth V (2011) Fermentation of lignocellulosic hydrolysate by the alternative industrial ethanol yeast Dekkera bruxellensis. Lett Appl Microbiol 53(1):73–78
Blumberg H, Hartshorne TA, Young ET (1988) Regulation of expression and activity of the yeast transcription factor ADRI. Mol Cell Biol 8(5):1868–1876
Boretsky YR, Pynyaha YV, Boretsky VY, Kutsyaba VI, Protchenko OV, Philpott CC, Sibirny AA (2007) Development of a transformation system for gene knock-out in the flavinogenic yeast Pichia guilliermondii. J Microbiol Methods 70(1):13–19
Borneman AR, Zeppel R, Chambers PJ, Curtin CD (2014) Insights into the Dekkera bruxellensis genomic landscape: comparative genomics reveals variations in ploidy and nutrient utilisation potential amongst wine isolates. PLoS Genet 10(2):e1004161
Boulton R, Singleton V, Bisson L, Kunkee R (1996) Principles and practices of winemaking. Chapman & Hall, New York, USA
Bradford MM (1976) A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Chatonnet P, Dubourdieu D, Boidron JN (1995) The influence of Brettanomyces/Dekkera spp. yeasts and lactic acid bacteria on ethylphenol content of red wines. Am J Enol Vitic 46(4):463–468
Cheng C, Kacherovsky N, Dombek KM, Chamier S, Thukral SK, Rhim E, Young ET (1994) Identification of potential target genes for Adr1p through characterization of essential nucleotides in UAS1. Mol Cell Biol 14(6):3842–3852
Ciriacy M (1975) Genetics of alcohol dehydrogenase in Saccharomyces cerevisiae. II. Two loci controlling synthesis of the glucose-repressible ADHII. Mol Gen Genet 138(2):157–164
Crauwels S, Van Assche A, de Jonge R, Borneman AR, Verreth C, Troels P, De Samblanx G, Marchal K, Van de Peer Y, Willems KA, Verstrepen KJ, Curtin CD, Lievens B (2015) Comparative phenomics and targeted use of genomics reveals variation in carbon and nitrogen assimilation among different Brettanomyces bruxellensis strains. Appl Microbiol Biotechnol 99(12):9123–9134
Crauwels S, Zhu B, Steensels J, Busschaert P, De Samblanx G, Marchal K, Willems KA, Verstrepen KJ, Lievens B (2014) Assessing genetic diversity among Brettanomyces yeasts by DNA fingerprinting and whole-genome sequencing. Appl Environ Microbiol 80(14):4398–4413
Curtin CD, Borneman AR, Chambers PJ, Pretorius IS (2012) De-novo assembly and analysis of the heterozygous triploid genome of the wine spoilage yeast Dekkera bruxellensis AWRI1499. PLoS ONE 7(3):e33840
Curtin CD, Pretorius IS (2014) Genomic insights into the evolution of industrial yeast species Brettanomyces bruxellensis. FEMS Yeast Res 14(7):997–1005
Dashko S, Zhou N, Tinta T, Sivilotti P, Sternad Lemut M, Trost K, Gamero A, Boekhout T, Butinar L, Vrhovsek U, Piskur J (2015) Use of non-conventional yeast improves the wine aroma profile of Ribolla Gialla. J Ind Microbiol Biotechnol 42(7):997–1010
de Barros Pita W, Leite FC, de Souza Liberal AT, Simoes DA, de Morais MA Jr (2011) The ability to use nitrate confers advantage to Dekkera bruxellensis over S. cerevisiae and can explain its adaptation to industrial fermentation processes. Antonie Van Leeuwenhoek 100(1):99–107
Delfini C, Formica J (2001) Wine microbiology: science and technology. Marcel Dekker, Inc., New York, USA
de Montigny J, Kern L, Hubert JC, Lacroute F (1990) Cloning and sequencing of URA10, a second gene encoding orotate phosphoribosyl transferase in Saccharomyces cerevisiae. Curr Genet 17(2):105–111
Dequin S, Salmon JM, Nguyen HV, Blondin B (2003) Wine yeasts. In: Boekhout T, Robert V (eds) Yeasts in food: beneficial and detrimental aspects. Behr’s Verlag, Hamburg, Germany, pp 389–412
de Smidt O, du Preez JC, Albertyn J (2008) The alcohol dehydrogenases of Saccharomyces cerevisiae: a comprehensive review. FEMS Yeast Res 8(7):967–978
de Souza Liberal AT, Basílio AC, do Monte Resende A, Brasileiro BT, da Silva-Filho EA, de Morais JO, Simoes DA, de Morais MA Jr (2007) Identification of Dekkera bruxellensis as a major contaminant yeast in continuous fuel ethanol fermentation. J Appl Microbiol 102(2):538–547
Dufour JP, Verstrepen K, Derdelinckx G (2003) Brewing yeasts. In: Boekhout T, Robert V (eds) Yeasts in food: beneficial and detrimental aspects. Behr’s Verlag, Hamburg, Germany, pp 347–388
Fugelsang KC (1996) Wine microbiology. Chapman & Hall, New York, USA
Galafassi S, Merico A, Pizza F, Hellborg L, Molinari F, Piskur J, Compagno C (2011) Dekkera/Brettanomyces yeasts for ethanol production from renewable sources under oxygen-limited and low-pH conditions. J Ind Microbiol Biotechnol 38(8):1079–1088
Gancedo JM (1998) Yeast carbon catabolite repression. Microbiol Mol Biol Rev:334–361
Hellborg L, Piskur J (2009) Complex nature of the genome in a wine spoilage yeast, Dekkera bruxellensis. Eukaryot Cell 8(11):1739–1749
Hotelling H (1951) A generalized T test and measure of multivariate dispersion. In: Neyman J (ed) Proceedings of the second Berkeley symposium on mathematical statistics and probability. University of California Press, Berkeley, Calif., pp 23–41. http://projecteuclid.org/euclid.bsmsp/1200500217
Ishchuk OP, Voronovsky AY, Stasyk OV, Gayda GZ, Gonchar MV, Abbas CA, Sibirny AA (2008) Overexpression of pyruvate decarboxylase in the yeast Hansenula polymorpha results in increased ethanol yield in high-temperature fermentation of xylose. FEMS Yeast Res 8(7):1164–1174
Johnson RA, Wichern DW (2002) Applied multivariate statistical analysis (5th ed). Pearson Education International, Upper Saddle River, USA
Lawley DN (1938) A generalization of Fisher’s z test. Biometrika 30(1–2):180–187. doi:10.1093/biomet/30.1-2.180
Lertwattanasakul N, Shigemoto E, Rodrussamee N, Limtong S, Thanonkeo P, Yamada M (2009) The crucial role of alcohol dehydrogenase Adh3 in Kluyveromyces marxianus mitochondrial metabolism. Biosci Biotechnol Biochem 73(12):2720–2726
Licker JL, Acree TE, Henick-Kling T (1998) What is ‘Brett’ (Brettanomyces) flavor? A preliminary investigation. In: Waterhouse AL, Ebeler SE (eds) Chemistry of wine flavor. American Chemical Society, Washington, DC, USA, pp 96–115
Lohr D, Venkov P, Zlatanova J (1995) Transcriptional regulation in the yeast GAL gene family: a complex genetic network. FASEB J 9(9):777–787
Loureiro V, Malfeito-Ferreira M (2003) Spoilage yeasts in the wine industry. Int J Food Microbiol 86:23–50
Lundin M, Nehlin JO, Ronne H (1994) Importance of a flanking AT-rich region in target site recognition by the GC box-binding zinc finger protein MIG1. Mol Cell Biol 14:1979–1985
Mayser P, Fromme S, Leitzmann C, Gründer K (1995) The yeast spectrum of the ‘tea fungus Kombucha’. Mycoses 38:289–295
McDaniel LE, Bailey EG, Zimmerli (1965) Effect of oxygen-supply rates on growth of Escherichia coli. II. Comparison of results in shake flasks and 50-liter fermentor. Appl Microbiol 13:115–119
Miklenic M, Strafa A, Bajic A, Zunar B, Lisnic B, Svetec IK (2013) Genetic transformation of the yeast Dekkera/Brettanomyces bruxellensis with non-homologous DNA. J Microbiol Biotechnol 23(5):674–680
Moktaduzzaman M, Galafassi S, Capusoni C, Vigentini I, Ling Z, Piskur J, Compagno C (2015) Galactose utilization sheds new light on sugar metabolism in the sequenced strain Dekkera bruxellensis CBS 2499. FEMS Yeast Res 15(2):fou009–9. doi:10.1093/femsyr/fou009
Murzin AG (1996) Structural classification of proteins: new superfamilies. Curr Opin Struct Biol 6(3):386–394
Passoth V, Blomqvist J, Schnürer J (2007) Dekkera bruxellensis and Lactobacillus vini form a stable ethanol-producing consortium in a commercial alcohol production process. Appl Environ Microbiol 73:4354–4356
Peng G, Hopper E (2002) Gene activation by interaction of an inhibitor with a cytoplasmic signaling protein. Proc Natl Acad Sci U S A 99(13):8548–8553
Pereira LF, Bassi AP, Avansini SH, Neto AG, Brasileiro BT, Ceccato-Antonini SR, de Morais MA Jr (2012) The physiological characteristics of the yeast Dekkera bruxellensis in fully fermentative conditions with cell recycling and in mixed cultures with Saccharomyces cerevisiae. Antonie Van Leeuwenhoek 101(3):529–539
Pfaffl MW, Horgan GW, Dempfle L (2002) Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res 30(9):e36
Pilgrim D, Young ET (1987) Primary structure requirements for correct sorting of the yeast mitochondrial protein ADH III to the yeast mitochondrial matrix space. Mol Cell Biol 7(1):294–304
Piskur J, Ling Z, Marcet-Houben M, Ishchuk OP, Aerts A, LaButti K, Copeland A, Lindquist E, Barry K, Compagno C, Bisson L, Grigoriev IV, Gabaldón T, Phister T (2012) The genome of wine yeast Dekkera bruxellensis provides a tool to explore its food-related properties. Int J Food Microbiol 157(2):202–209
Postma E, Verduyn C, Scheffers WA, Van Dijken JP (1989) Enzymic analysis of the crabtree effect in glucose-limited chemostat cultures of Saccharomyces cerevisiae. Appl Environ Microbiol 55(2):468–477
Reid MF, Fewson CA (1994) Molecular characterization of microbial alcohol dehydrogenases. Crit Rev Microbiol 20(1):13–56
Rozpedowska E, Hellborg L, Ishchuk OP, Orhan F, Galafassi S, Merico A, Woolfit M, Compagno C, Piskur J (2011) Parallel evolution of the make-accumulate-consume strategy in Saccharomyces and Dekkera yeasts. Nat Commun 2:302
Saliola M, De Maria I, Lodi T, Fiori A, Falcone C (2006) KlADH3, a gene encoding a mitochondrial alcohol dehydrogenase, affects respiratory metabolism and cytochrome content in Kluyveromyces lactis. FEMS Yeast Res 6(8):1184–92
Scheffers WA (1966) Stimulation of fermentation in yeasts by acetoin and oxygen. Nature 210:533–534
Schifferdecker AJ, Dashko S, Ishchuk OP, Piskur J (2014) The wine and beer yeast Dekkera bruxellensis. Yeast 31(9):323–332
Sommerville GA, Proctor RA (2013) Cultivation conditions and the diffusion of oxygen into culture media: the rationale for the flask-to-medium ratio in microbiology. BMC Microbiol 13:9. doi:10.1186/1471-2180-13-9
Shao D, Creasy CL, Bergman LW (1998) A cysteine residue in helixII of the bHLH domain is essential for homodimerization of the yeast transcription factor Pho4p. Nucleic Acids Res 26(3):710–714
Steensels J, Daenen L, Malcorps P, Derdelinckx G, Verachtert H, Verstrepen KJ (2015) Brettanomyces yeasts - From spoilage organisms to valuable contributors to industrial fermentations. Int J Food Microbiol 206:24–38
Suwannarangsee S, Kim S, Kim OC, Oh DB, Seo JW, Kim CH, Rhee SK, Kang HA, Chulalaksananukul W, Kwon O (2012) Characterization of alcohol dehydrogenase 3 of the thermotolerant methylotrophic yeast Hansenula polymorpha. Appl Microbiol Biotechnol 96(3):697–709
Taneja B, Mande SC (1999) Conserved structural features and sequence patterns in the GroES fold family. Protein Eng 12(10):815–818
Teoh AL, Heard G, Cox J (2004) Yeast ecology of Kombucha fermentation. Int J Food Microbiol 95:119–126
Thukral SK, Eisen A, Young ET (1991) Two monomers of yeast transcription factor ADR1 bind a palindromic sequence symmetrically to activate ADH2 expression. Mol Cell Biol 11(3):1566–1577
Tiukova IA, Petterson ME, Tellgren-Roth C, Bunikis I, Eberhard T, Pettersson OV, Passoth V (2013) Transcriptome of the alternative ethanol production strain Dekkera bruxellensis CBS 11270 in sugar limited, low oxygen cultivation. PLoS ONE 8(3):e58455. doi:10.1371/journal.pone.0058455
Toivola A, Yarrow D, van den Bosch E, van Dijken JP, Scheffers WA (1984) Alcoholic fermentation of D-xylose by yeasts. Appl Environ Microbiol 47(6):1221–1223
Traven A, Jelicic B, Sopta M (2006) Yeast Gal4: a transcriptional paradigm revisited. EMBO Rep 7(5):496–499
Treitel MA, Carlson M (1995) Repression by SSN6-TUP1 is directed by MIG1, a repressor/ activator protein. Genetics 92:3132–3136
Valdes J, Tapia P, Cepeda V, Varela J, Godoy L, Cubillos FA, Silva E, Martinez C, Ganga MA (2014) Draft genome sequence and transcriptome analysis of the wine spoilage yeast Dekkera bruxellensis LAMAP2480 provides insights into genetic diversity, metabolism and survival. FEMS Microbiol Lett 364:104–106
Van Dijken J, Scheffers A (1986) Redox balances in the metabolism of sugars by yeasts. FEMS Microbiol Rev 32:199–224
van Hoek P, van Dijken JP, Pronk JT (2000) Regulation of fermentative capacity and levels of glycolytic enzymes in chemostat cultures of Saccharomyces cerevisiae. Enzyme Microb Technol 26(9–10):724–736
Vaughan-Martini A, Martini A (2011) Saccharomyces. In: Kurtzman CP, Fell JW, Boekhout T (eds) The yeasts, a taxonomic study, 5th edn. Elsevier, London, UK, pp 733–746
Verduyn C, Postma E, Scheffers WA, Van Dijken JP (1992) Effect of benzoic acid on metabolic fluxes in yeasts: a continous-culture study on the regulation of respiration and alcoholic fermentation. Yeast Chichester Engl 8:501–517
Walther K, Schüller HJ (2001) Adr1 and Cat8 synergistically activate the glucose-regulated alcohol dehydrogenase gene ADH2 of the yeast Saccharomyces cerevisiae. Microbiology 147(Pt8):2037–2044
Weinhandl K, Winkler M, Glieder A, Camattari A (2014) Carbon source dependent promoters in yeasts. Microb Cell Fact 13:5. doi:10.1186/1475-2859-13-5
Wilks SS (1932) Certain generalizations in the analysis of variance. Biometrika 24(3/4):471–494
Woolfit M, Rozpedowska E, Piskur J, Wolfe KH (2007) Genome survey sequencing of the wine spoilage yeast Dekkera (Brettanomyces) bruxellensis. Eukaryot Cell 6:721–733
Young ET, Pilgrim D (1985) Isolation and DNA sequence of ADH3, a nuclear gene encoding the mitochondrial isozyme of alcohol dehydrogenase in Saccharomyces cerevisiae. Mol Cell Biol 5(11):3024–3034
Acknowledgments
We would like to thank Georgia Angelidou for her help with the Python script analysis. This study was funded by the Marie Curie Initial Training Network Cornucopia (FP7-PEOPLE-2010-ITN, grant agreement nr. 264717), Fysiografen and Lindström Foundations and FEMS Research Fellowship 2005–2. Support of Polish grant of National Scientific Center (NCN) DEC-2012/05/B/NZ1/01657 awarded to AAS is also acknowledged. This work is dedicated to the blessed memory of Professor Jure Piškur (1960–2014).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Human and animal rights and informed consent
Research involving Human Participants and/ or Animals/ Informed consent: not applicable for this study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 2638 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Schifferdecker, A.J., Siurkus, J., Andersen, M.R. et al. Alcohol dehydrogenase gene ADH3 activates glucose alcoholic fermentation in genetically engineered Dekkera bruxellensis yeast. Appl Microbiol Biotechnol 100, 3219–3231 (2016). https://doi.org/10.1007/s00253-015-7266-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-015-7266-x