
Abstract
The lung is a vital organ that incessantly faces external environmental challenges. Its homeostasis and unimpeded vital function are ensured by the respiratory epithelium working hand in hand with an intricate fine-tuned tissue-resident immune cell network. Lung tissue-resident immune cells span across the innate and adaptive immunity and protect from infectious agents but can also prove to be pathogenic if dysregulated. Here, we review the innate and adaptive immune cell subtypes comprising lung-resident immunity and discuss their ontogeny and role in distinct respiratory diseases. An improved understanding of the role of lung-resident immunity and how its function is dysregulated under pathological conditions can shed light on the pathogenesis of respiratory diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Located at the environmental interface, the lung constantly encounters external insults threatening host homeostasis. The multifaceted protective function of the lung relies on a delicate balance among repelling invading pathogens while in parallel tolerating harmless particulate matter and sustaining its vital function. A pivotal safeguard of this balance is the local lung tissue microenvironment consisting of the respiratory epithelium and a sophisticated network of non-circulating lung-resident immune cells [1].
In the past decade, several studies shed light on tissue-resident immunity and went beyond the “strict limits” between innate and adaptive immunity. It is now well known that, apart from circulating immune cells, which are activated upon antigen encounter by the first line of defense and then migrate to the site of inflammation, there are also innate and adaptive immune cells with protective properties, which reside in the tissue, respond fast, and effectively cope with every invading pathogen [2]. Tissue-resident cells extend across adaptive and innate immunity and, as integral part of an immune sensing network, provide first-line tissue-specific immune protection. Despite its protective function, mounting evidence also highlights the role of derailed tissue-resident immunity in disease pathogenesis. Although the role of the adaptive tissue-resident immunity in sustaining respiratory health has been significantly appraised [3, 4], innate and innate-like immune cell subtypes are also important in this context [5]. In this review, we focus on the main innate, innate-like and adaptive lung-resident immune subsets and discuss their ontogeny and role in respiratory health and disease.
Tissue-resident innate immune cells in pulmonary homeostasis
Macrophages, key orchestrators of respiratory immunity
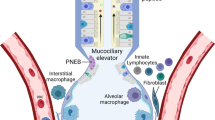
Lung-resident macrophages comprise the alveolar (AM) and interstitial (IM) macrophages [5,6,7]. AM are the majority of lung-resident macrophages and are located in the alveolar space thereby constantly facing external stimuli, while IM are fewer and can be found within the lung parenchyma [7] (Fig. 1). AM interact closely with the respiratory epithelium and remove invading pathogens and other particles [5] through phagocytosis.

Location of immune tissue-resident cells in the lungs (alveoli and bronchus). Created with BioRender.com
Lung-resident macrophages arise independently either from embryonic progenitor cells or circulating monocytes (Fig. 2) [6, 8, 9]. Studies in mice highlight a fetal origin of AM in steady state, which emerge as the end-products of the postnatal maturation of fetal liver- and yolk sac-derived monocytes residing in the lung, in response to transforming growth factor β (TGF‐β), cytokine granulocyte–macrophage colony‐stimulating factor (GM‐CSF) and peroxisome proliferator‐activated receptor γ (PPARγ) [9,10,11,12]. The life-long maintenance of AM relies on their local self-expansion and is largely independent of circulating monocytes in steady state (Fig. 2a, b) [10, 11]. However, augmented contribution of circulating monocytes to AM replenishment is observed with aging and upon respiratory infections, lung fibrosis, or regeneration (Fig. 2b) [9, 13,14,15,16,17]. On the contrary, mouse IM have a heterogeneous developmental origin, arising mainly from circulating and lung monocytes, with only minimal contribution of yolk sac precursors early in fetal life [18].

Fetal emergence and life-long maintenance of alveolar macrophages. Origin and mechanisms for a lungresident macrophage replenishment b in a healthy or diseased lung
In contrast to mouse lung macrophages, little is known about the development and homeostasis of human AM and IM. This gap in knowledge is due to the limited availability of appropriate experimental methods and can only be addressed in the context of bone marrow or lung transplantation or with the use of humanized mouse models. Studies in allogeneic bone marrow recipients demonstrated that myeloablation and thus, host AM depletion prior to transplantation induces fast AM replacement with the final AM pool deriving mostly from donor-derived circulating monocytes [19, 20]. Upon lung transplantation, AM are transferred along with the donor lung in the recipients with their longevity impacting donor-specific immune responses and potential long-term graft rejection [21, 22]. Similarly, the origin of human IM is poorly understood, although mounting evidence highlights the contribution of circulating monocytes to human lung macrophage ontogeny in general [23]. Indeed, using a humanized mouse model, Evren et al. demonstrated that CD14+CD16– circulating monocytes can extravasate into the lung and give rise to human IM and AM in adults, especially upon lung injury and inflammation [24]. Moreover, the authors showed that the monocytic maturation to IM and AM is characterized by a sequential upregulation of CD206 and CD169, a finding in agreement with human studies reporting that CD206+CD169− IM develop earlier than CD206+CD169+ AM which appear upon lung inflation shortly after birth [24, 25]. More evidence supporting the temporal and tissue state-dependent origin of human AM was recently provided by Evren et al. who characterized the embryonic origin of AM and pinpointed CD116+ fetal liver cells as human AM precursors in early life and steady-state conditions [26].
Dendritic cells professional first responders and immune response initiators
As key antigen-presenting cells (APC), pulmonary dendritic cells (DCs) act promptly upon antigen encounter and trigger adaptive immunity by transporting antigens to lung-draining lymph nodes [7]. In contrast to lung-resident macrophages, insights into prenatal and neonatal development, differentiation and maturation of pulmonary DCs are largely missing. Although DCs are not self-expanding and their tissue maintenance relies on bone marrow-derived replacement, they are considered part of lung-resident immunity given their long tissue persistence and slow replenishment [7].
Three main lung-resident DC subtypes have been identified, namely plasmacytoid DCs (pDCs), monocyte-derived DCs (moDCs), and conventional DCs (cDCs), which can be further subdivided in CD103+ cDCs (cDC1s) and CD11b+ cDCs (cDC2s) [5, 7, 27]. CD103+ cDCs are responsible for sampling and presentation of antigens from the alveolar space to CD8+ (and CD4+) T cells thereby inducing enhanced effector CD8+ T cell generation [28]. Interestingly, in the human lung, the CD103+ cDC equivalent subset seems to be the myeloid type 2 DCs (CD11c+BDCA-3+(CD141) [29, 30]. CD11b+ DCs and moDCs express many common markers and thus, can be difficultly distinguished. Nevertheless, the Fc receptors CD64 and/or FcεRIα are commonly used to further distinguish these subtypes [7]. moDCs are recruited to the lung upon inflammation, while their pulmonary existence in steady-state is unclear. Myeloid type 1 DCs (CD11c+CD1c+) are the human equivalent population to CD11b+ mouse DCs [29, 30]. The last main DC subgroup in both mouse and human lungs are pDCs. In mice, pDCs can be identified based on the expression of distinct markers like the plasmacytoid dendritic cell antigen-1 (PDCA-1) [7, 27]. In human lungs, pDCs are characterized as CD11c−BDCA-2+ [29, 30], while both in mice and humans, pDCs express the antiviral factor bone marrow stromal antigen-2 (BST-2) [31].
Innate lymphoid cells at the interface between innate and adaptive immunity
Innate lymphoid cells (ILCs) are a group of diverse innate immune cells with a common lymphoid origin that lack antigen-specific receptors and are thus, not implicated into antigen-specific immune responses [32, 33]. Though a small part of lung-resident immunity, ILCs play a pivotal role in mounting and sustaining protective immune responses against invading pathogens while safeguarding tissue homeostasis [34, 35]. If dysregulated, ILCs may also contribute to respiratory disease pathogenesis [36, 37].
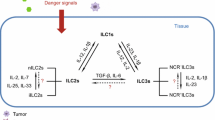
The ILC family consists of five main cell subsets with a common lymphoid origin and distinct phenotype and function, namely ILC1s, ILC2s, ILC3, natural killer (NK) cells, and lymphoid tissue inducer (LTi) cells. Based on developmental, phenotypic, and functional similarities, these subsets can be further classified into three groups, with group 1 comprising ILC1s and NK cells, group 2 referring to ILC2s and group 3 consisting of ILC3s and LTi cells [38]. ILCs are considered as innate counterparts of T lymphocytes, with ILC1s, ILC2s, and ILC3s being functionally analogous to CD4+ T helper (Th)1, Th2, and Th17 cells, respectively, while NK cells are cytotoxic cells functionally resembling to CD8+ T cells [33, 39, 40]. Finally, LTi cells induce secondary lymphoid organogenesis, with their function starting in fetal life [41].
ILCs originate from a common lymphoid progenitor, with their development mainly occurring in the fetal liver and the bone marrow postnatally [42]. Long-lasting maintenance of tissue-resident ILCs is mostly achieved by local self-renewal, but ILC replenishment by bone marrow- or lymphoid organ-derived precursors can also occur [43]. While the lung emergence of ILC1s is poorly understood, ILC2s and ILC3s populate the lung early in postnatal life, with interleukin (IL)-33 production by type II alveolar epithelial cells being critical for ILC2 emergence in the lung [44], and insulin-like growth factor 1 deriving from alveolar fibroblasts promoting ILC3 development, respectively [45]. ILCs reside in all parts of the respiratory tract (Fig. 1). Physiologically, ILC2s are the main ILC population in the mouse respiratory tract, while ILC3s are the predominant ILC subset in humans [46].
Group 1 ILCs comprise NK cells, found in blood circulation and tissues, and ILC1s, which are tissue-resident cells located in several organs, including the lung [43, 47]. Group 1 ILCs are typically implicated in antiviral and antitumor immunity [48,49,50]. They confer protection mainly through interferon (IFN)-γ secretion in response to IL-12, IL-15, or IL-18, which in turn boosts intracellular pathogen elimination and antigen presentation by other immune cells [49, 50] (Fig. 3). NK cells can additionally secrete perforin and granzyme B, two key mediators of their cytotoxic functions [51]. While expression of tissue residency markers, such as CD103 and CD69, mainly characterizes ILC1s, markers associated with blood recirculation, namely CD62L, sphingosine-1-phosphate receptor (S1PR) and CC-chemokine receptor 7 (CCR7), are mostly expressed by NK cells [33, 52].

Different subtypes of tissue-resident immune cells and their cytokine production. Created with BioRender.com
As the innate counterpart of Th2 cells, ILC2s are involved in type 2 immunity, due to their ability to produce type 2 cytokines, such as IL-5, IL-4, and IL-13, upon stimulation by epithelial alarmins, including IL-33, IL-25, and thymic stromal lymphoprotein (TSLP) (Fig. 3) [38, 53, 54]. Hence, ILC2s play key roles in asthma and helminth infection [55, 56]. In mice and humans, ILC2 development, maintenance and function depend on the transcription factor GATA3 [57, 58]. Interestingly, ILC2s exhibit tissue-specific phenotypic and functional traits, which are mostly determined perinatally [59, 60]. Schneider et al. reported the existence of fetally, perinatally, and adult-derived ILC2s in adult tissues and identified the perinatal period as a key determinant of tissue-resident ILC2s and their distinct tissue-specific gene expression signature [60]. Of note, only 5–10% of lung-resident ILC2s were of embryonic origin 2 months after birth, indicating that the pool of fetal ILC2s is constantly diluted by postnatally de novo generated ILC2s [60]. Importantly, a tissue-specific de novo generation or local expansion of adult ILC2s in physiological or pathogenic conditions, respectively, was demonstrated, thereby suggesting that the local tissue microenvironment can define the tissue-specific profile of ILC2s in adult life [60]. Apart from their temporal origin, ILC2s exhibit a tissue-dependent responsiveness to IL-33 and IL-25, with IL-33-responding ILC2s residing mainly in the lung and adipose tissue in steady-state conditions and IL-25-responding ILC2s mainly found in the intestine upon helminth infection [44, 59]. ILC2s are the main ILC population in the mouse lung. In steady-state conditions, mouse lung ILC2s express several surface markers, including IL7Ra, CD25, ST2, CD69, CD90, and CD44, while being lineage negative [34, 61]. Despite their similarities, mouse and human ILC2s differ in CD44 and CD161 expression, with mouse cells expressing the first and human ILC2s the second surface marker [33].
Group 3 ILCs, namely ILC3s and LTi cells, depend on the retinoic acid-related orphan receptor-γt (RORγt) for their activation and induce Th17-like immune responses with secretion of IL-17, IL-22, GM-CSF and/or tumor necrosis factor-α (TNFα) (Fig. 3) [38, 62]. ILC3s are characterized by functional and phenotypic heterogeneity. Among others, IL-18 and GATA3 can induce ILC3 maintenance, proliferation, and cytokine production [63, 64]. As key IL-22 producers, ILC3s play an important role in lung homeostasis by sustaining epithelial barrier integrity and function [38, 65].
Lung tissue-resident adaptive immunity
Tissue-resident memory T cells specialized sentinels of lung-specific immune memory
The lungs are enriched in both CD4+ and CD8+ tissue-resident memory T (TRM) cells [66, 67], with distinct transcriptional profiles and functional properties [68]. TRM cells are pivotal sentinels of tissue homeostasis, due to their ability to respond rapidly to secondary infections [69] and their role in antitumor local immunosurveillance [70]. However, if dysregulated, TRM cells may also lead to pathogenic immune responses, as seen in the case of allergic asthma [71,72,73,74].
Typical TRM cell characteristics include their ability to adhere to peripheral tissues such as the lung and the gut, as well as their lack of homing signals. The phenotypic identification of TRM cells relies on the differential expression of typical surface markers, with CD69 being the most commonly used one for mouse and human TRM cells. CD69 is also a key determinant of TRM cell fate, since it competitively interacts with S1PR thereby inhibiting its expression and impeding sphingosine-1-phosphate (S1P1)-mediated tissue escape [75, 76]. TRM cell tissue retention and inhibition of recirculation are also facilitated by CD44 and CD103 upregulation as well as CD62L and CCR7 downregulation, respectively [77, 78].
Lung TRM cells mainly arise from effector T cells following their DC-mediated activation and subsequent migration from lymphoid tissues into the lung [79, 80]. In the inflamed lung, recruited CD8+ effector T cells interact with the local tissue microenvironment, with subsequent differentiation into lung CD8+ TRM cells and accumulation at tissue regenerative sites [81, 82]. Lung monocytes and regulatory T cells as well as cytokines secreted by the local tissue microenvironment, such as TGF-β, IL-33, IL-15, TNF, and IFN-γ, play a pivotal role in TRM differentiation and tissue retention [75, 76, 83, 84]. Specifically, TGF-β has been shown to promote important steps for the acquisition of a tissue-resident phenotype, namely CD103 and CD69 expression along with the downregulation of Kruppel-like factor 2 (KLF2) and sphingosine-1-phosphate (S1P1) [76]. During CD8+ TRM cell differentiation, T-box transcriptional factors, comprising eomesodermin (Eomes) and T-bet, are also downregulated with minimally sustained T-bet expression required for long-term TRM survival [75]. In contrast to the generation of CD8+ TRM cells in other barrier tissues [85], important for the CD8+ effector T cell differentiation into lung TRM cells is their prior interaction with cognate antigen, which in turn induces upregulation of CD69, VLA-1, and CD103 [78, 86]. As shown also in other tissues including the liver, skin, kidney, and small intestine [87], a key player in the formation of lung CD8+ TRM cells is B-lymphocyte-induced maturation protein 1 (Blimp-1), which shifts the lineage choice of CD8+ effector T cell towards TRM and not central memory cells [88].
Despite more extensive investigation of CD8+ TRM cell biology, CD4+ TRM cells are a more abundant TRM population in the lung and were the first identified and characterized resident memory CD4+ T-cell subset [89, 90]. CD4+ and CD8+ TRM cells share many phenotypic similarities [77] but differ in their generation, surface marker expression and response to cytokines [91]. Although co-expression of CD69 and CD103 is a typical TRM signature and can be used for the identification of lung CD8+ TRM cells, lung CD4+ TRM cells exhibit high CD69 but only low or no CD103 expression [89, 90]. In contrast to their CD8+ counterparts, CD4+ TRM generation is affected mainly by IL-2 and IL-15 rather than TGF- β [90, 92].
Lung CD4+ and CD8+ TRM cells reside in specific tissue sites that support their longevity (Fig. 1). CD8+ TRM cells occupy newly constructed niches, also known as repair-associated memory depots (RAMDs), which are associated with tissue regeneration upon injury and are critical for CD8+ TRM cell survival [81, 82]. On the other hand, CD4+ TRM cells contribute to the formation of inducible bronchus-associated lymphoid tissue (iBALT), which in turn favors their maintenance while providing an immune network that can rapidly respond upon infection [93] (Fig. 1). Nevertheless, lung TRM cells are not as long-lived as in other organs and slowly decline after their generation with constant replacement by circulating effector T cells locally converting into TRM cells [94].
BRM cells, key features of tissue-specific humoral immunity
Tissue-resident B memory (BRM) cells are a subgroup of experienced B memory antigen-specific cells, which become unable to recirculate and reside in the lung by altering the expression of their receptors and chemokines [95]. Due to lack of typical BRM markers, identification of lung BRM cells can be challenging and can be mainly achieved through immunohistochemical tissue staining or with the use of intravascular staining and parabiosis animal models (Fig. 4) [3]. In mice, BRM cells share surface markers with B memory cells such as CD38 and CD73 [96], while in humans, they express CD27 [97]. Lung BRM cells express CXCR3, CXCL9, CXCL10, CXCL11, and CCR6 [98], which drive and retain them in the lung parenchyma, where they are located in the iBALT or in the basolateral surface of the respiratory epithelium [99] (Fig. 1). Of note, primary BRM cell generation depends on first antigen encounter [3], which occurs in iBALT and drives BRM precursors to stay and survive in the lung as tissue-resident cells [100]. These cells differ both phenotypically and functionally from B cells of the circulation as well as from those located in the lung-draining lymph nodes [3, 98]. Besides the expression of the above mentioned markers, BRM cells differ from the rest B memory cells by downregulating the homing receptor CD62L and upregulating tissue-resident markers, such as CD69 and CD103 [101]. BRM cells mainly produce antibodies following a secondary infection with their response being faster and much more effective compared to that of other antibody-producing B cells [3].

Models to study immune tissue-resident cells in the lung. a In vivo intravenous immune cell labelling. b In situ labelling of cells through photoconversion. c Parabiosis model. d Orthotopic lung transplantation. Created with BioRender.com
γδ-T cells, adaptive and innate-like hybrid facilitators of immune surveillance
Lung-resident γδ-T cells respond rapidly to challenges and orchestrate elicited immune responses thereby contributing to antimicrobial protection, tumor surveillance, and tissue repair [102, 103]. Their distinct nature derives from the combination of conventional adaptive properties with their ability to mount robust innate-like responses [102].
While constituting only 1–5% of blood circulating lymphocytes, γδ-T cells are highly enriched in mucosal and epithelial tissues, such as the skin and the lung, where they account for 8–20% of resident pulmonary lymphocytes [104]. During the perinatal period, Vγ6/Vδ1-expressing γδ-T cells migrate from the thymus to other tissues, including the lung [104, 105], with subsequent tissue-specific differentiation and age-dependent pattern of Vγ gene usage [106]. Specifically, Vγ6+ γδ-T cells are the main γδ-T cell population in the lung during the first 8–10 weeks of life, while Vγ4+ γδ-T cells become the most abundant γδ-T cell subset after that time point [106]. In the adult mouse lung, γδ-T cells are a heterogeneous population consisting mostly of Vγ4+ cells, two smaller Vγ6+ and Vγ1+ cell subsets, and only sparse Vγ7+ cells [107]. This programmed rearrangement of the Vγ gene pattern seems reasonable, considering that γδ-T cells interact with a vast variety of antigens, which depend both on the tissue of residence as well as on the age of the host, thereby conferring targeted conditionally adjusted immunity. Apart from the airway mucosa, lung γδ-T cell can be found in all non-alveolar regions (Fig. 1). Of note, Vγ4+ and Vγ1+ γδ-T cells are preferentially distributed in parenchymal lung areas [107].
Lung-resident γδ-T cells produce mainly IL-17 [103]. Thus, IL-17-mediated signalling seems to be a key mechanism underlying γδ-T cell contribution to pathogen elimination and pulmonary homeostasis [108], while IL-22 production by γδ-T cells underlies their protective function against lung fibrosis in mice [109]. However, lung γδ-T cells can be also involved in aberrant immune responses and thus disease pathogenesis, as seen in the case of allergic asthma [110].
Tissue-resident immunity in respiratory diseases
Tissue-resident immunity serves multiple roles in respiratory health and disease. In this section, we discuss the involvement of all above mentioned lung-resident immune cell subtypes in respiratory viral and bacterial infections, asthma, as well as cancer and metastasis (Table 1).
Respiratory viral infections
Viruses, including the influenza virus, the respiratory syncytial virus (RSV) and the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), are common causes of respiratory infections. Upon encountering the first line of local defense, namely the respiratory epithelium, a further immune response with implication of several circulating and lung-resident immune cell populations is initiated.
Influenza virus infection
Invading influenza viruses can target mouse and human lung-resident AM, which are, nevertheless, less susceptible to infection and exhibit lower virus replication and TNF production compared to monocyte-derived macrophages [10, 111]. Of note, AM depletion upon influenza infection leads to increased pulmonary viral load and worsened disease outcome, suggesting that AM enhance pathogen clearance thereby protecting from severe infection [112]. This AM function is mainly due to type I IFN production, which induces pulmonary monocyte recruitment and controls viral replication [113]. Of note, type I IFN production may also underlie the anti-inflammatory behavior of AM, since it alleviates inflammasome activation, reduces IL-1 and enhances IL-10 secretion by monocyte-derived cells and moDCs [114, 115]. However, influenza-driven AM reduction and dysfunction seem to be mediated by IFN-γ signalling thereby increasing susceptibility to bacterial superinfections [116]. Nevertheless, after influenza infection resolution in mice, protection from a secondary Streptococcus pneumoniae (S. pneumoniae) infection is conferred by newly recruited monocyte-derived AM that enrich the already lung-resident AM pool [13]. In contrast to AM, little is known about the interaction of IM and the influenza virus. A recent study in cynomolgus macaques demonstrated significantly increased IM accumulation in influenza-infected lungs, thereby indicating a potential role of these cells in antiviral defense and immune response orchestration [117].
As critical orchestrators of subsequent immune responses, DCs are also implicated in anti-influenza immunity. Distinct DC subsets exhibit differential susceptibility to influenza infection, mainly depending on the level of MHC II expression. Therefore, highly MHCII-expressing CD103+ and CD11b+ cDCs can be easily targeted by the virus, while low expressing pDCs are not affected [118]. MHCII molecules are known to serve as viral receptors or co-receptors [119], and this may likely explain the observed differences in DC susceptibility since the entire molecule or at least a region may be recognized by the influenza virus and enhance its binding to DCs. Of note, the infected migratory CD103+ cDCs (cDC1s) are the main APCs to lymph node-residing CD4+ and CD8+ T cells [79, 120]. Interestingly, cDC1 depletion in influenza-infected mice impaired viral clearance and exacerbated disease outcome [120]. Of note, cDC1s also promote CD8+ T cell survival in the lungs and development of robust memory response upon influenza infection [121]. However, at later stages of influenza infection, CD11b+ cDCs (cDC2) tend to gather in the lung-draining lymph nodes and become the main CD8+ T cell-stimulating DC subset [122]. Interestingly, Bosteels et al. recently demonstrated that, upon a respiratory viral infection, pulmonary cDC2s acquire a hybrid phenotype in a type I IFN-dependent manner, sharing expression of the Fc receptor CD64 and of interferon regulatory factor 8 with monocytes and cDC1s, respectively, and exhibit then an enhanced ability to prime CD4 and CD8 immunity [123].
Group 1 ILCs, namely ILC1s and NK cells, contribute to antiviral immunity, mainly through IFN-γ secretion [49, 50]. Of note, NK cells seem to play an ambiguous role upon influenza infection. Although several studies report higher pulmonary virus titers in influenza-infected NK cell-deficient mice [124, 125], a pathogenic impact of NK cells, exacerbating influenza-related pathology, has also been described [126]. In contrast, ILC1s serve a key role in immunosurveillance at sites of early viral infection and protect by secreting IFN-γ in response to cDC1-mediated signaling [50]. Additionally, in H1N1-influenza-infected mice, ILC2s enhance airway epithelial restoration by boosting epithelial cell proliferation and airway remodeling via amphiregulin and IL-13 production [34]. Although little is known about the role of ILC3s in viral respiratory infections, the implication of IL-22, a key ILC3 cytokine, in influenza infection has been thoroughly investigated. IL-22 contributes to lung epithelial repair following infection [127]. Additionally, IL-22 and thus IL-22-producing ILC3s confer protection against secondary bacterial lung infections [128].
Mouse and human studies identify virus-specific TRM cells in influenza-infected lungs [89, 90, 129], which fight acute infection through IFN-γ and TNF-a production [130] and support long-term protective immunity in situ [131]. Pulmonary influenza-specific CD8+ TRM cells confer also heterosubtypic protection against infection due to the presence of common or similar epitopes [69, 129, 132]. Indeed, CD8+ TRM cells with the ability to confer cross-protection against influenza A, B, and C viruses have been found in human lungs following influenza infection [129]. Interestingly, the development of lung-resident CD8+ TRM and BRM cells upon influenza virus infection is supported by a newly identified population of follicular tissue-resident CD4+ T helper (TRH) cells in an IL-21-dependent manner [133]. Apart from TRM cells, long-lived BRM cells are also found in influenza-infected mouse and human lungs [3, 98, 134]. Importantly, influenza-specific BRM cells reside in iBALT and exhibit distinct transcriptional and phenotypic traits, including increased CXCR3, CCR6 and CD69 expression, which distinguish them from populations in lung-draining lymph nodes, spleen, or blood [98]. Upon viral re-exposure, pulmonary BRM cells preferentially migrate into infected sites, where they differentiate into plasma cells with high antibody production [134].
Lung-resident γδ-T cells are also involved in antiviral immunity. Upon neonatal influenza infection in mice, epithelial cell-derived IL-33 triggered the accumulation of IL-17A-producing γδ-T cells in the infected lungs. In turn, these cells induced type 2 immunity, ILC2 and regulatory T (Treg) cell recruitment and, finally, enhanced amphiregulin release and tissue repair [135]. In humans, the main γδ-T response against influenza virus infection is exerted by IFN-γ-producing Vγ9Vδ2-T cells [136]. Indeed, activated human Vγ9Vδ2-T cells could kill influenza-infected human alveolar epithelial cells and impede viral replication in vitro [137], while they could also attenuate disease severity in immunodeficient, infected with human influenza virus strains, humanized mice [138].
RSV infection
Similar to the influenza virus infection, tissue-resident immunity is critically involved in immune responses elicited upon a RSV infection. As sentinels of respiratory barrier immunity, both AM and pDCs enhance viral clearance and thus protect from severe RSV infection [139, 140]. Indeed, AM or pDC depletion upon RSV infection led to increased lung viral load and aggravated disease pathology [139, 140]. The contribution of AM to viral clearance is mainly due to type I IFN production [113, 141]. In fact, AM seem to produce more type I IFN than pDCs, and thus play a superior role in antiviral immunity [113]. Interestingly, AM not only initiate immune response upon RSV encounter, but are also able to limit viral replication regardless of type I IFN [142].
Similar protective roles have been described for group 1 ILCs upon RSV infection [143, 144]. On the contrary, increased ILC2s have been associated with severe RSV bronchiolitis in infants [145], and correlated with high TSLP and IL-33 levels [145]. Upon mouse RSV infection, early TSLP-mediated induction of IL-13-secreting ILC2s led to airway hyperresponsiveness (AHR) and increased mucus production [144]. Similarly, Wu et al. recently demonstrated that RSV-triggered IL-33-activated ILC2s boost AHR and airway eosinophilia through IL-13 production [146]. Interestingly, increased ILC2 numbers persist in the mouse lung for several weeks post neonatal RSV infection [146, 147], and thus may account for the link between early-life RSV infection and long-term respiratory pathologies [148].
On the other hand, a protective role of virus-specific CD8+ TRM cells has been described upon RSV infection in both mice and humans [149, 150]. In RSV-immune mice, TRM cells amplified virus clearance thereby protecting against re-infection [150]. RSV infection in humans led to accumulation of virus-specific CD8+ TRM cells in the lungs during convalescence, while increased RSV-specific TRM cells in the airways before infection were associated with reduced virus titers and disease severity [149]. Virus-specific CD4+ TRM cell accumulation in the airways has also been reported upon experimental human RSV infection [151].
Upon RSV-immunization and subsequent infection, increased Vγ4+ γδ-T cells were found in mouse lungs and could produce several pro-inflammatory cytokines, including IFN-γ, TNF, IL-4, and IL-5, upon ex vivo stimulation [152]. Of note, depletion of these cells attenuated lung immunopathology and disease severity and minimally enhanced viral proliferation without affecting viral clearance [152]. Additionally, RSV-infected mouse neonates failed to mount robust IL-17A-mediated responses, in contrast to adult mice, in which γδ-T cells were the main IL-17A sources. Importantly, IL-17 suppression in adult mice exacerbated RSV-induced lung inflammation, whereas IL-17 supplementation in neonates had the opposite effect. Thus, increased RSV disease severity in infancy may be associated with an early-life deficiency or dysfunction of lung IL-17A-producing γδ-T cells [153].
SARS-CoV-2 infection
Direct SARS-CoV-2 infection of human AM has also been reported [154], with subsequent AM dysfunction, exacerbated pro-inflammatory responses and eventually generalized pulmonary damage [154]. Upon severe coronavirus disease (COVID-19), AM expand into the alveoli along with neutrophils and lymphocytes, and seem to sustain an M1 polarization thereby contributing to SARS-CoV-2-associated “cytokine storm” and acute respiratory distress syndrome [154, 155]. Recently, Grant et al. identified a positive feedback loop between SARS-CoV-2 infected AM and T cells whose continuous interaction perpetuates a spatially restricted alveolitis [156]. Of note, in vitro SARS-CoV-2 infection of monocyte-derived macrophages and dendritic cells led to production of cytokines, such as IFN-α, IFN-β, TNF, IL-1β, IL-6, IL-10, and CXCL10 and also resulted in type I IFN-mediated cell death, despite its abortive character and the fact that the virus failed to replicate efficiently in these cells [157]. However, SARS-CoV-2-induced IFN production was weaker compared to other respiratory viruses [158], a finding that may be attributed to newly identified SARS-CoV-2-encoded genes limiting IFN production [159] as well as a poor ability of AM to sense the invading virus and mount a robust IFN response [160].
Upon SARS-CoV-2 encounter, DCs seem to be unexpectedly affected. Interestingly, a lower DC number, impaired antigen-presenting function and decreased type I IFN production by DCs have been described in patients with severe COVID-19 [161,162,163]. Among cDC1s, cDC2s, and pDCs, only cDC2s accumulate in SARS-CoV-2-infected human lungs [162], while pDC apoptosis is increased [162, 164]. The preferential accumulation of cDC2s in SARS-CoV-2-infected lungs may be explained by the fact that cDC2s can enhance the function of CD4+ T cells and trigger follicular helper T cells, which both contribute to effective antiviral humoral immune responses, as shown in the case of human immunodeficiency virus-1 infection [165]. Of note, upon acute SARS-CoV-2 infection, DCs failed to induce robust T cell responses, thus suggesting an impaired antigen-presenting capacity [163, 164]. A similar DC dysfunction could be seen in SARS-CoV-infected rhesus macaques. In that case, the low rate of viral replication in all infected APCs and the consequently low expression of viral proteins impaired viral sensing from Toll-like receptors (TLRs) thereby facilitating viral escape from mucosal innate immunity and subsequent systemic dissemination using APCs, including DCs, as vehicles [166]. Additionally, SARS-CoV-2 preferentially inhibits pDCs from producing type I IFN and can thus escape immune recognition [163, 167].
Our understanding of ILC implication in COVID-19 is limited to changes observed in peripheral blood and thus, insights into lung-resident ILCs are sparse. Higher expression of activation and homing markers in ILCs from infected individuals was associated with severe infection [168]. This finding together with decreased NK, ILC1 and ILC2 numbers in the blood of COVID-19 patients [169] suggest that ILCs migrate and persist in the lung upon severe infection.
SARS-CoV-2 infection also induces development of lung-resident adaptive immune memory [170, 171]. Human studies report a long-term persistence of virus-specific lung TRM cells up to 10 months after primary infection [170]. Importantly, cross-reactive immunity has also been described in the case of SARS-CoV-2, as, due to its homology with the human coronaviruses OC43 and 229E, cross-reactive CD4+ and CD8+ T cells have been detected in the absence of a prior SARS-CoV-2 infection [172,173,174]. Although both in the case of influenza or SARS-CoV-2 infection, virus-specific lung TRM cells seem to be generated and cross-react with heterologous virus strains, whether these cells confer cross-protection against other antigens remains unclear. Of note, pre-existing immunity to influenza, either due to prior infection or vaccination, seems indeed to impact SARS-CoV-2-specific T-cell immune responses, since it has been associated with enhanced SARS-CoV-2-specific CD4+ T cell immunity [175] and reduced rate of COVID-19 [176]. Such cross-immunity can be attributed to the structural similarities of influenza virus and SARS-CoV-2 [177] and thus, lung TRM cells may be key players in this context, a hypothesis warranting further investigation. However, little is known about the ability of SARS-CoV-2-specific lung TRM cells to sufficiently protect against re-infection. Of note, a sublethal mouse SARS-CoV-2 infection induced pulmonary resident CD4+ and CD8+ T effector cells expressing TRM-related markers such as CD69 and CD103, but, when adoptively transferred to a naïve host, these cells failed to independently protect against a lethal SARS-CoV-2 infection [178]. This finding is in contrast to other studies showing that pulmonary resident T cells are essential for vaccine-induced protection against coronaviruses [179]. Taken together, these observations may indicate that, unlike a SARS-CoV-2 infection, a vaccination may induce the generation of lung-resident T cells with enhanced protective capacity against infection. Another explanation for the inability of transferred pre-trained lung-resident T cells to independently protect against infection may be that immune responses commonly rely on well-orchestrated cellular events with immune cells acting as a network. Thus, an independent protective function of one immune cell population may not be sufficient to confer immunity in this case. Interestingly, a recent study by Zhao et al. uncovered a pathogenic role for tissue-resident memory-like Th17 (TRM17) cells as potential drivers of aberrant inflammation in severe COVID-19 pneumonia [180]. Specifically, COVID-19 severity and inflammation-induced lung injury were associated with the interplay among TRM17 cells, macrophages and CD8+ T cells in the infected lungs [180]. Of note, patients with severe disease exhibited increased serum IL-17A and GM-CSF levels [180]. However, a potential protective function of TRM17 cells at an early infection stage or in asymptomatic SARS-CoV-2 infected individuals could not be ruled out. Therefore, further studies are required to fully decipher the emergence and origin as well as the role of these cells throughout the infection course.
Studies focusing on γδ-T cell responses upon SARS-CoV-2 infection deal mainly with circulating γδ-T cells and largely neglect the lung-resident subtypes. Of note, decreased circulating γδ-T cells are found in the blood of patients with severe COVID-19 and related general lymphopenia [181]. Additionally, recovery from COVID-19 has been associated with a shift of γδ-T cells towards an effector-like phenotype with enhanced tissue infiltration capacity [182]. Taken together, these findings might indeed suggest a recruitment and retention of γδ-T cells in the infected lung [158, 183].
Respiratory bacterial infections
Similar to respiratory viral infections, lung-resident immunity is essentially involved in protection against invading bacteria, such as S. pneumoniae and Mycobacterium tuberculosis (M. tuberculosis).
S. pneumoniae infection
AM are among the first responders to invading bacteria. Upon S. pneumoniae infection, TLRs of AMs can synergistically prevent pneumococci from escaping immune recognition [184]. Following phagocytosis, pathogen elimination can be achieved through AM apoptosis [185]. Nevertheless, the elimination of phagocytized pathogens, such as S. pneumoniae, most commonly relies on the acidity of the phagosome and AM-produced reactive oxygen and nitrogen species [186]. Gut microbiota-derived acetate was found to boost AM bactericidal activity by inducing production of IL-1β and in turn nitric oxide by AM [187]. AM are also critically involved in inflammation resolution and tissue repair. After infection control, reprogramming of AM towards the M2 activation state results in the secretion of anti-inflammatory cytokines, such as IL-10 and IL-1ra thereby controlling inflammation and boosting tissue repair [188]. Additionally, through AM-mediated efferocytosis, dead cells and their intracellular inflammatory residues are also removed [183, 188]. Regarding IM, several subtypes with distinct transcriptional profiles can be found during a bacterial infection [189] and they all express MHCII, a trait likely indicating an antigen presenting role [190]. Additionally, IM population is known to expand significantly after exposure to bacterial unmethylated CpG DNA and can subsequently prevent asthma development through IL-10 production [191]. Unlike AM, insights into the role of lung-resident IM in respiratory bacterial infections remain sparse and thus, further studies focusing on this topic are needed.
Although the role of DCs in antiviral immunity is well-described, their response to bacterial lung infections is less understood. As first-line defenders, DCs exert their antigen presenting activity also upon bacterial infections [192]. On the other hand, DC deficiency in S. pneumoniae-infected mice was associated with reduced systemic bacterial spread and thus, lower systemic inflammation, indicating the ability of pneumococci to exploit DC-mediated proteolysis in order to spread outside the lung [193]. Interestingly, DC depletion was not accompanied by an increased recruitment of another immune cell type [193], and thus, one can only assume that other antigen presenting cells, such as AM, monocytes and epithelial cells undertake the role of DCs in this context [194].
ILCs play also an important role in host responses to bacterial lung infections, with ILC3s being the most important ILC population in this context [195]. Upon infection, pro-inflammatory factors such as IL-1β and IL-23, trigger ILC3s to secrete IL-17 and IL-22 [195], which subsequently enhance airway epithelial barrier function and promote immune responses against S. pneumoniae [196]. Apart from ILC3s, NK cells contribute also to bacterial clearance, likely due to their ability to produce IL-22 and IL-15 [197]. On the other hand, ILC2s seem to indirectly impede immune response to S. pneumoniae by skewing AM phenotype and function towards an M2 activation state thereby favoring immune quiescence in homeostasis [44].
TRM and BRM cells are also integral parts of immune defense against bacterial pneumonia. Bacterial respiratory infections lead to IL-12/IL-18-mediated bystander activation of virus-specific lung CD8+ TRM cells, which in turn secrete IFN-γ and attract neutrophils into the lung thereby reducing disease severity [198]. Importantly, neutrophil recruitment in S. pneumoniae-infected lungs can also be boosted by CD4+ TRM cells, which, in response to pneumococcal antigens, produce IL-17 and reprogram lung epithelial transcriptome to accelerate antimicrobial responses [199]. Interestingly, treatment with antibiotics such as clarithromycin was associated with a reduced lung CD4+ TRM cell number and thus impaired host response to a S. pneumoniae re-infection. Such an impairment of tissue-resident immune memory cells may be a mechanism, underlying the long-term ability of extensive antibiotic use to dysregulate immune responses and weaken the host’s defense against a re-infection [200]. BRM cells are also elicited in human and mouse lungs after pneumococcal infection and their depletion from experienced mouse lungs prior to a re-infection has been associated with increased disease severity [201].
As IL-17 sources, lung resident γδ-T cells are essentially involved in host immune defense against bacterial infections. Additionally, γδ-T cells contribute to the resolution of pneumococcal inflammation, since they can expand and drastically reduce the number of lung DCs and AM following S. pneumoniae clearance [202].
M. tuberculosis infection
As in the case of pneumococcal infection, AM are essentially involved in the elimination of M. tuberculosis upon infection. The central TLR adapter protein myeloid differentiation factor 88 (MyD88) and the macrophage receptor with collagenous structure (MARCO) are essential for elimination of both S. pneumoniae and M. tuberculosis [203,204,205], by, among others, mediating AM interaction with another integral sensor of tissue-specific defense, namely the lung epithelium [204]. Invading mycobacteria are easily recognized and phagocytosed by AM, which subsequently secrete pro-inflammatory cytokines and exert bactericidal effects by highly expressing inducible nitric oxide synthase and antimicrobial agents [206]. Despite their –largely- protective role in bacterial respiratory infections, upon a M. tuberculosis infection, IL-1-mediated interplay between infected AM and non-hematopoietic cells facilitates the migration of the former from the alveolar space to the lung interstitium thereby favoring tuberculosis progression [207]. AM ability to support M. tuberculosis intrapulmonary dissemination can be explained by the fact that M. tuberculosis bacilli can survive in the phagosomes of AM, and thus AM depletion in M. tuberculosis-infected mice enhances bacterial clearance and ameliorates disease outcome [208].
DCs are also critically involved in host defense against M. tuberculosis. Bacillus phagocytosis and induced cytokine production by DCs control the infection and granuloma formation [209]. Infected DCs migrate to draining lymph nodes and trigger adaptive immune responses [210]. Mycobacterium-specific Th1 responses are orchestrated by CD11b+ cDCs, while at the same time being suppressed by CD103+ cDCs via IL-10 production [211]. Additionally, moDCs highly express pro-inflammatory cytokines to support mycobacterium elimination and enhance Th2 and Th17 immune responses [212]. However, little is known about the role of pDCs in tuberculosis, although their number increases in lung-draining lymph nodes upon mycobacterial infection [213].
Recent evidence has uncovered a critical implication of ILCs in antimycobacterial immunity. Studies in mice demonstrate a protective role of lung-resident IFN-γ-producing NK cells in M. tuberculosis infection, which can bind on the pathogen and exert cytotoxic effects [214]. Additionally, early innate immune responses to M. tuberculosis are further facilitated by ILC3s via IL-17 and IL-22 production [214, 215].
The presence of antigen-specific IL-17-producing CD4+ and CD8+TRM cells in M. tuberculosis-infected human lungs has also been recently reported [216], an intriguing finding raising discussions about the efficiency of tissue vaccines against tuberculosis, in an effort to boost TRM cell presence in the passively immunized lung [217]. Finally, through IL-17 production, lung γδ-T cells also play an important role in host immune response against M. tuberculosis, including granuloma formation [218, 219].
Asthma
Asthma is one of the most widespread respiratory conditions worldwide. Its pathogenesis is driven by chronic airway inflammation, AHR and remodeling which overall result in episodic manifestation of respiratory symptoms including wheezing, coughing, and dyspnea. Due to improved understanding of the disease, asthma is now considered as a heterogeneous entity consisting of distinct disease endotypes and phenotypes [220]. Asthma endotypes refer to underlying inflammatory pathways that result in the clinical features of the disease, namely the asthmatic phenotypes [220, 221]. The most well-recognized and defined endotypes of asthma are the type 2-high and type 2-low endotypes, characterized by eosinophilic or neutrophilic/paucigranulocytic airway inflammation, respectively [220,221,222]. Patients with eosinophilic or neutrophilic asthma share similar clinical symptomatology but different endotypes need to be taken into account for optimal selection of treatment strategies [221, 222]. Although different molecular and immune cell pathways predominate distinct disease endotypes, lung-resident immunity is an essential part of all asthma-related immune responses.
AM seem to play a rather dichotomous role in asthma pathogenesis. Although AM depletion exacerbated allergic airway inflammation (AAI) in mice [223], AM-produced mediators, including CCL17, CCL8, and CCL24, enhanced type 2 immunity and eosinophilia in asthmatic patients and mice [224]. Additionally, AM from patients with asthma secrete more pro-inflammatory mediators, such as TNF-a, IL-1β, IL-6, IL-8, and IL-17, which promote AAI [225]. In asthmatic patients, the dysfunctional AM phagocytotic and efferocytotic capacity perpetuate inflammation and tissue remodelling and trigger disease exacerbations [226,227,228]. Of note, an impaired phagocytic capacity is not a general feature of asthma as it correlates with disease severity and depends on the type of product to be phagocytosed [229]. Additionally, AM from patients with severe—but not mild- asthma exhibit a defective efferocytotic capacity, which in turn is associated with an impaired anti-inflammatory function [228, 230]. On the contrary, apoptotic cell efferocytosis by monocyte-derived macrophages from asthmatic patients remains intact [231], a finding that may be attributed to the different cellular origin of lung-resident macrophages [10]. Similarly, lung-resident macrophages exhibit an impaired anti-inflammatory behavior in asthma. Reduced IL-10+ macrophages were observed in asthmatic patients [232], while an increase of IL-10+ IM could dampen airway inflammation or prevent neutrophilic asthma in mice [233]. However, the exact mechanisms underlying macrophage dysfunction and its impact on asthma manifestation remain elusive and thus, further studies in this field are warranted.
Similar to AM, DCs also contribute to asthma-related immunity. Upon allergen encounter, DCs trigger naïve Th cell differentiation and thus Th2 and ILC2 accumulation with type 2 cytokine production [234]. Both lung-resident cDC1s and cDC2s are able to elicit a strong Th2 response upon house dust mite (HDM) exposure [235,236,237], while CD103+ cDC1-deficient mice exhibit attenuated AHR and eosinophilia upon ovalbumin-induced asthma [238]. Of note, pDCs can be divided into distinct subpopulations based on the expression of the surface markers CD8α or CD8β, which were shown to either prevent or enhance asthma manifestation in mice [239]. Interestingly, expression of these markers can be observed after TLR agonist-mediated stimulation and has been linked with distinct cytokine expression profiles and a tolerogenic DC phenotype [239, 240]. Importantly, DCs can not only trigger and sustain but also control and limit allergic Th2 immune responses [241, 242]. For example, CD103+ DCs seem indeed to play an important role in pulmonary tolerance, since they are able to induce Treg cell generation in ovalbumin-challenged mice [241, 242], while their absence has been linked with exacerbated AAI and eosinophilia in both ovalbumin- and HDM-induced asthma [241,242,243]. Interestingly, regular administration of Helicobacter pylori extract to ovalbumin-treated mice attenuated AAI, eosinophilia and AHR, with the success of the treatment relying mainly on CD103+ DCs and their intrinsic IL-10 production [244]. Treg cell generation can also be induced by pDCs, which seem to mostly attenuate AAI and promote pulmonary tolerance to harmless inhaled antigens [245, 246].
As well-known type 2 cytokine producers, ILC2s are highly involved in asthma pathogenesis [56]. Indeed, increased blood circulating and lung-resident ILC2s can be found in asthmatic patients [56, 247]. Interestingly, this increase applies mainly to eosinophilic asthma, whereas neutrophilic asthma has been associated with elevated ILC1s and ILC3s [248]. ILC2s boost airway eosinophilia via IL-5 production [249] and enhance AHR, goblet cell hyperplasia and Th2-mediated AAI through IL-13 secretion [37, 250]. Additionally, increased bronchial epithelial barrier permeability and disrupted tight junction integrity have been associated with ILC2s and IL-13 production [251]. A recent study identified the neuropeptide neuromedin U as a powerful effector of AAI through ILC2 activation [252]. On the other hand, several mediators, including IFN-γ, IL-1β, and Nrf2, have been found to tightly control ILC2-mediated immunity thereby preventing or suppressing AAI in mice [250, 253, 254]. Although little is known about the role of ILC1s in asthma, NK cells can be both protective and pathogenic in this context. On one hand, NK cells seem to restrict AAI by promoting eosinophilic apoptosis and impeding viral-induced allergic immune responses [143, 255]. Of note, severe asthma has been associated with decreased lipoxin A4 and thus, impaired ability of NK cells to trigger eosinophil apoptosis [256]. In mice, IFN-γ-producing NK cells and ILC1s can suppress ILC2 expansion and activation thereby contributing to type 2 inflammation resolution [257]. Consistently, NK cell depletion during the early phase of papain-induced lung inflammation in mice led to ILC2 expansion, increased type 2 cytokine production and thus, aggravated asthma manifestation [258]. On the other hand, NK cells can also promote type 2 cytokine production and thus, trigger AAI [259]. Overall, NK cells are critically involved in the phase of sensitization upon allergen encounter, facilitate the balance between Th1 and Th2 inflammation and finally contribute to the resolution of allergic inflammation. The impact of NK cells on these steps depends largely on their activation status and subtype. Of note, the result of NK activation can be influenced by the type of environmental stimulus, the cytokine and inflammatory milieu, the interaction with other parenchymal and immune cells, the differentiation status of the cell and the developmental phase of the host individual [260]. Since immune cells, and in this case NK cells, exert their functions within an interacting network, a modulation of this cross-talk in favor of NK activation or inhibition may be a promising immunotherapeutic approach. A better understanding of the implication of NK cells in allergic asthma including the crucial elements that govern their dichotomous mechanisms of action is thus required. Finally, although insights into the role of group 3 ILCs in asthma are sparse, IL-17-producing ILC3s may be involved, since IL-17 has been associated with neutrophilic asthma in both humans and mice [261].
TRM and BRM cells are also implicated in the pathogenesis of asthma [73, 262]. Specifically, Th2-TRM cells secrete cytokines that promote and sustain airway eosinophilia [72] [262]. Upon HDM exposure, IL-2 signalling enables the generation, migration and retention of allergen-specific Th2-TRM cells in the lungs, where they drive AAI [72]. Of note, these long-lived pathogenic cells remain in the mouse lung for its entire lifetime as safekeepers of allergic memory [71]. Therefore, allergen re-challenge induces rapid Th2-TRM cell proliferation and type 2 cytokine secretion with subsequent exacerbated clinical features of asthma [73, 74]. BRM cells could also be identified in the lungs of HDM-sensitized and challenged mice and were implicated in the allergic response, while HDM re-challenge resulted in their activation [263].
Lung-resident γδ-T cells seem to play a dichotomous role in allergen-induced Th2 immunity. In ovalbumin-induced asthma, γδ-T cell-deficient mice had reduced AHR, airway eosinophilia, peribronchial lymphocytic infiltration as well as lower serum IgE and lung IL-5 levels compared to wild-type mice [264, 265]. Besides promoting airway eosinophilia and regulating IgE production, Vγ1+ γδ-T cells boost AHR by suppressing IL-10-producing Treg cells in the lung of ovalbumin-treated mice [266]. However, IL-17-producing γδ-T cells have been identified as key regulators of pulmonary allergic responses, as they could ameliorate AHR thereby enhancing resolution of eosinophilic and Th2-mediated AAI [110, 267]. Indeed, activation of Th17-like γδ-T cells has been associated with lower AHR [268], attenuated airway eosinophilia as well as increased neutrophil airway recruitment [268] and higher AM number [110].
Tissue-resident immunity in lung cancer and metastasis
Lung cancer is the most frequently occurring type of cancer and the leading cause of cancer-related death in men worldwide, while it has the third highest incidence and second highest cancer-related mortality in women [269]. Apart from primary tumor growth, the lungs are frequently targeted by metastatic tumor cells originating from primary tumors located at other parts of the lung itself or distant sites, such as the breast, colon or the skin [270]. Similar to lung cancer, lung metastasis is a major health burden worldwide and a common cause of cancer-related death [271]. Although the pathogenesis of lung cancer and metastasis has not been fully elucidated yet, mounting evidence identifies the lung microenvironment and the crosstalk between cancer and immune cells as key players in this process [272].
Tissue-resident immune cells can play a critical role in tumor surveillance and immunity. Despite the multifaceted nature of lung-resident immunity, most studies focus on the role of TRM or –to a smaller extent- of tissue-resident macrophages in lung tumorigenesis and metastasis formation, while insights into the potential involvement of other resident immune cell subsets are largely missing.
During tumor or metastasis formation, lung-resident macrophages are among the first cells to interact with tumor cells and may thus play a critical role in this process. Th2-driven inflammation can be typically found at tumor sites and tumor microenvironment (TME) [273], and evidence demonstrates that IL-4 boosts tissue-resident macrophage proliferation [274]. In the mouse lung, monocyte-derived macrophages were shown to induce metastasis, while tissue-resident macrophages were associated with primary tumor growth [275]. Additionally, tumor-associated macrophages (TAMs) in mouse lungs were shown to be tissue-resident IM and CCR2-dependent recruited macrophages, with the former mainly promoting tumor growth in—among others- an IL-9-dependent manner [276], and the latter facilitating tumor cell dissemination [275]. In accordance to these findings, intratracheal L-Clodronate-mediated AM depletion did not affect lung metastasis of mammary carcinoma-derived Met-1 cells [277]. However, a role of AM in metastasis cannot be ruled out, since in a mouse model of breast cancer, AM were found to enhance lung metastasis by impeding antitumor T cell activity in the lung and thus, could be identified as an important resident of the pre-metastatic niche and a potent target of future cancer immunotherapies [278].
Recent findings suggested that tumor-associated lymphocytes in non-small cell lung cancer (NSCLC) exhibit TRM cell function [279]. In TME, CD8+ TRM cells comprise a homogeneous CD103+ CD49+CD69+ population characterized by T-bet, porylated (p)STAT-3, and Aiolos transcription factor expression, while a small subset produces IFN-γ and IL-17 [279].
In human NSCLC, cytotoxic CD8+ T cells with high CD103 expression exhibit increased cytotoxicity, are highly proliferative, and can thus contribute to robust antitumor immunity [280]. In early-stage NSCLC, effector memory T cells face tumor-related antigens and transform into CD103+ TRM cells with antitumor activity [281]. Of note, the function of CD8+ tumor-infiltrating lymphocytes (TIL) in this stage seems to be determined by a balance between an antitumor CD103+ TRM program and an exhaustion program driven by Eomes expression. During tumor growth, the exhaustion program may prevail, thus reducing TIL function [281].
Furthermore, transcription factors may be also essentially involved in this process. A gradual reduction of TILs as well as progressively reducing nuclear factor of activated T cells (NFATc1) expression in cancer cells have been described in patients with advanced-stage NSCLC [282]. Interestingly, enhanced tumor growth together with decreased effector memory and CD103+ TRM cells were observed in tumor-bearing lungs of mice with T-cell-specific NFATc1 inactivation, thus highlighting the role of this transcription factor in cytotoxic T-cell immunity and TRM cell tissue retention [282, 283]. Additionally, CXCR6 surface expression on mouse and human lung TRM cells upon tumor antigen encounter facilitates their migration and maintenance in lung TME [284]. Moreover, CD103+ TRM cell-derived granzyme B and IFN-γ in humans control tumor formation and metastasis through fibronectin secretion, facilitate the priming of newly generated tumor-specific T cells, and boost immune cell recruitment in the tumor [285]. Interestingly, enhanced proliferation of CD103+ T cells in tumors with high CD8+ T cell numbers was associated with a prolonged survival, a finding likely identifying CD8+CD103+ TRM cell infiltration as a positive prognostic factor [70, 286].
Although several studies report an association of CD8+ TRM cells in human NSCLC with a good disease prognosis [283], little is known about the role of CD4+ TRM-like TILs in this context. CD4+ TRM cells are a phenotypically and functional heterogeneous population and thus, multiple, even contradictory, functions in TME are to be expected. Of note, CD8+ T cell cytotoxicity largely depends on CD4+ TRM cells, which can also hinder tumor growth via IFN-γ production or by tumor cell elimination [287].
Regarding the implication of TRM cells in lung metastasis, Christian et al. recently demonstrated that TRM cells develop in the tumor, the contralateral mammary mucosa, and the pre-metastatic lung. In a functional level, CXCR6 is critically involved in TRM retention in the primary tumor. This amplifies tumor-derived TRM cells in the lung and induces protection against metastasis, thus, overall suggesting a potential strategical approach to prevent metastasis [288].
TRM cells are currently considered a valuable tool in tumor immunotherapy. For instance, checkpoint therapy enhanced TRM formation in mice with melanoma [289]. Specifically, programmed cell death protein (PD)-1 inhibition in combination with central memory T cell transfer induced TRM infiltration and subsequently inhibited tumor growth [289]. Similarly, PD-1 inhibition in human NSCLC-derived TRM cells boosted their cytotoxic capacity against autologous tumor cells ex vivo [70]. Additionally, PD-1 blockade enhanced CD8+CD103+ TRM cell proliferation in patients with melanoma, with higher cell numbers correlating with longer survival [290]. Hence, TRM modulation appears to be a potent future approach to enhance cancer therapy efficacy.
Research approaches for lung tissue-resident immunity assessment
Since the first identification of tissue-resident immunity, its assessment has been a challenge and calls for constant optimization of respective research approaches.
A first and likely the simplest method to address tissue-residence is the in vivo immune cell labelling (Fig. 4a). Intravenous staining distinguishes between immune cells in vasculature and those outside intact endothelium, e.g., in the lung parenchyma [291].
Another technique to label cells in the lung and track their movement in vivo is the photoconversion of one lung after thoracotomy (Fig. 4b). This method is possible for mice carrying the Kaede protein, a coral-derived fluorescent protein whose emission alters from green to red fluorescence after exposure to violet light. Photoconversion allows in situ labeling of Kaede protein-expressing cells in the lung [292].
Assessment of lung-resident immunity can also be achieved with parabiosis (Fig. 4c). Surgical generation of parabiotic pairs is the conjoining of two congenic mice, which share their blood circulation through newly developed vasculature within approximately one week. Through the blood circulation, recirculating lymphocyte populations equilibrate between the mice of the pair. Failure of an immune cell population to equilibrate between tissues in each mouse of the parabiotic pair indicates residence [293].
Finally, another powerful method to track tissue-resident immune cells is orthotopic lung transplantation, allowing both cell ingress and egress assessment (Fig. 4d). Tissue-resident cells are transplanted together with the organ in the recipient congenic mouse, thus, allowing one to discriminate between tissue-resident and circulating immune cells by the analysis of the congene in the transplant organ and in the periphery [66, 294].
Sex-specific differences in tissue resident immunity
Sex differences in both innate and adaptive immunity are well documented in humans and mice [295]. However, insights into a potential sexual dimorphism of tissue-resident immunity remain sparse. Of note, sex differences in tissue-resident immune cells can be tissue- or organ-specific. For example, female mice have higher number of tissue-resident macrophages, T and B cells in naïve peritoneal and pleural cavities, compared to male ones [296]. Within macrophages, upregulated expression of TLRs, especially TLR2 and TLR4, was demonstrated and associated with a higher phagocytotic capacity in female mice. Among T cell subpopulations, CD4+ and CD8+ cells, but not Treg or γδ-T cells, were significantly increased in females compared to males. Additionally, increased lung-resident ILCs were observed in female compared to male mice in steady-state conditions [297, 298].
Further phenotypic and functional characterization of sex-specific differences in tissue-resident immunity is warranted to elucidate the mechanisms underlying the sex-specific manifestation of several respiratory immune diseases. For example, a sex bias in the incidence and severity of allergic asthma is well-established, with male and female predominance in childhood and adulthood, respectively [299]. Given the aforementioned role of tissue-resident immunity in asthma pathogenesis, sex-specific differences in tissue-resident immune cells and responses may – at least to some extent- account for this sex bias. Although the mechanisms driving the observed sex disparity in relation to asthma in general remain unclear, a crosstalk between immune cells and sex hormones seems to play a key role in this context and could likely apply in the case of tissue-resident immunity. Indeed, androgen signaling impacts ILC2 responsiveness while estrogen-mediated signaling influences macrophage polarization and therefore contribute to sex differences in allergic asthma [300, 301]. Additionally, estrogens promote mast cell degranulation thereby exacerbating asthma severity [297].
In conclusion, more studies are required in order to thoroughly characterize a potential sexual dimorphism in lung-resident immunity and uncover its role in respiratory health and disease. If obtained, such insights would pave the way for targeted optimized therapeutic approaches in a sex-dependent, highly personalized manner.
References
Hewitt RJ, Lloyd CM (2021) Regulation of immune responses by the airway epithelial cell landscape. Nat Rev Immunol 21:347–362
Sun H, Sun C, Xiao W, Sun R (2019) Tissue-resident lymphocytes: from adaptive to innate immunity. Cell Mol Immunol 16:205–215
Allie SR, Bradley JE, Mudunuru U, Schultz MD, Graf BA, Lund FE, Randall TD (2019) The establishment of resident memory B cells in the lung requires local antigen encounter. Nat Immunol 20:97–108
Yuan R, Yu J, Jiao Z, Li J, Wu F, Yan R, Huang X, Chen C (2021) The roles of tissue-resident memory T cells in lung diseases. Front Immunol 12:710375
Ardain A, Marakalala MJ, Leslie A (2020) Tissue-resident innate immunity in the lung. Immunology 159:245–256
Evren E, Ringqvist E, Willinger T (2020) Origin and ontogeny of lung macrophages: from mice to humans. Immunology 160:126–138
Kopf M, Schneider C, Nobs SP (2015) The development and function of lung-resident macrophages and dendritic cells. Nat Immunol 16:36–44
Ginhoux F, Guilliams M (2016) Tissue-resident macrophage ontogeny and homeostasis. Immunity 44:439–449
Perdiguero EG, Geissmann F (2016) The development and maintenance of resident macrophages. Nat Immunol 17:2–8
Guilliams M, De Kleer I, Henri S, Post S, Vanhoutte L, De Prijck S, Deswarte K, Malissen B, Hammad H, Lambrecht BN (2013) Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J Exp Med 210:1977–1992
Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, Becker CD, See P, Price J, Lucas D, Greter M, Mortha A, Boyer SW, Forsberg EC, Tanaka M, van Rooijen N, García-Sastre A, Stanley ER, Ginhoux F, Frenette PS, Merad M (2013) Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 38:792–804
Schneider C, Nobs SP, Kurrer M, Rehrauer H, Thiele C, Kopf M (2014) Induction of the nuclear receptor PPAR-γ by the cytokine GM-CSF is critical for the differentiation of fetal monocytes into alveolar macrophages. Nat Immunol 15:1026–1037
Aegerter H, Kulikauskaite J, Crotta S, Patel H, Kelly G, Hessel EM, Mack M, Beinke S, Wack A (2020) Influenza-induced monocyte-derived alveolar macrophages confer prolonged antibacterial protection. Nat Immunol 21:145–157
Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A, Chak S, Naikawadi RP, Wolters PJ, Abate AR, Butte AJ, Bhattacharya M (2019) Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol 20:163–172
Lechner AJ, Driver IH, Lee J, Conroy CM, Nagle A, Locksley RM, Rock JR (2017) Recruited monocytes and type 2 immunity promote lung regeneration following pneumonectomy. Cell Stem Cell 21:120–34.e7
McQuattie-Pimentel AC, Ren Z, Joshi N, Watanabe S, Stoeger T, Chi M, Lu Z, Sichizya L, Aillon RP, Chen CI, Soberanes S, Chen Z, Reyfman PA, Walter JM, Anekalla KR, Davis JM, Helmin KA, Runyan CE, Abdala-Valencia H et al (2021) The lung microenvironment shapes a dysfunctional response of alveolar macrophages in aging. J Clin Invest 131:e140299
Misharin AV, Morales-Nebreda L, Reyfman PA, Cuda CM, Walter JM, McQuattie-Pimentel AC, Chen CI, Anekalla KR, Joshi N, Williams KJN, Abdala-Valencia H, Yacoub TJ, Chi M, Chiu S, Gonzalez-Gonzalez FJ, Gates K, Lam AP, Nicholson TT, Homan PJ, Soberanes S, Dominguez S, Morgan VK, Saber R, Shaffer A, Hinchcliff M, Marshall SA, Bharat A, Berdnikovs S, Bhorade SM, Bartom ET, Morimoto RI, Balch WE, Sznajder JI, Chandel NS, Mutlu GM, Jain M, Gottardi CJ, Singer BD, Ridge KM, Bagheri N, Shilatifard A, Budinger GRS, Perlman H (2017) Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J Exp Med 214:2387–2404
Tan SYS, Krasnow MA (2016) Developmental origin of lung macrophage diversity. Development 143:1318–1327
Nakata K, Gotoh H, Watanabe J, Uetake T, Komuro I, Yuasa K, Watanabe S, Ieki R, Sakamaki H, Akiyama H, Kudoh S, Naitoh M, Satoh H, Shimada K (1999) Augmented proliferation of human alveolar macrophages after allogeneic bone marrow transplantation. Blood 93:667–673
Wickenhauser C, Thiele J, Pérez F, Varus E, Stoffel MS, Kvasnicka HM, Beelen DW, Schaefer UW (2002) Mixed chimerism of the resident macrophage population after allogeneic bone marrow transplantation for chronic myeloid leukemia. Transplantation 73:104–111
Eguíluz-Gracia I, Schultz HH, Sikkeland LI, Danilova E, Holm AM, Pronk CJ, Agace WW, Iversen M, Andersen C, Jahnsen FL, Baekkevold ES (2016) Long-term persistence of human donor alveolar macrophages in lung transplant recipients. Thorax 71:1006–1011
Nayak DK, Zhou F, Xu M, Huang J, Tsuji M, Hachem R, Mohanakumar T (2016) Long-term persistence of donor alveolar macrophages in human lung transplant recipients that influences donor-specific immune responses. Am J Transplant 16:2300–2311
Byrne AJ, Powell JE, O'Sullivan BJ, Ogger PP, Hoffland A, Cook J, Bonner KL, Hewitt RJ, Wolf S, Ghai P, Walker SA, Lukowski SW, Molyneaux PL, Saglani S, Chambers DC, Maher TM, Lloyd CM (2020) Dynamics of human monocytes and airway macrophages during healthy aging and after transplant. J Exp Med 217:e20191236
Evren E, Ringqvist E, Tripathi KP, Sleiers N, Rives IC, Alisjahbana A, Gao Y, Sarhan D, Halle T, Sorini C, Lepzien R, Marquardt N, Michaelsson J, Smed-Sorensen A, Botling J, Karlsson MCI, Villablanca EJ, Willinger T (2021) Distinct developmental pathways from blood monocytes generate human lung macrophage diversity. Immunity 54(259–75):e7
Bharat A, Bhorade SM, Morales-Nebreda L, McQuattie-Pimentel AC, Soberanes S, Ridge K, DeCamp MM, Mestan KK, Perlman H, Budinger GR, Misharin AV (2016) Flow cytometry reveals similarities between lung macrophages in humans and mice. Am J Respir Cell Mol Biol 54:147–149
Evren E, Ringqvist E, Doisne J-M, Thaller A, Sleiers N, Flavell RA, Di Santo JP, Willinger T (2022) CD116+ fetal precursors migrate to the perinatal lung and give rise to human alveolar macrophages. J Exp Med 219:e20210987
Misharin AV, Morales-Nebreda L, Mutlu GM, Budinger GR, Perlman H (2013) Flow cytometric analysis of macrophages and dendritic cell subsets in the mouse lung. Am J Respir Cell Mol Biol 49:503–510
del Rio ML, Rodriguez-Barbosa JI, Kremmer E, Förster R (2007) CD103- and CD103+ bronchial lymph node dendritic cells are specialized in presenting and cross-presenting innocuous antigen to CD4+ and CD8+ T cells. J Immunol 178:6861–6866
Demedts IK, Brusselle GG, Vermaelen KY, Pauwels RA (2005) Identification and characterization of human pulmonary dendritic cells. Am J Respir Cell Mol Biol 32:177–184
Lambrecht BN, Hammad H (2012) Lung dendritic cells in respiratory viral infection and asthma: from protection to immunopathology. Annu Rev Immunol 30:243–270
Swiecki M, Omattage NS, Brett TJ (2013) BST-2/tetherin: structural biology, viral antagonism, and immunobiology of a potent host antiviral factor. Mol Immunol 54:132–139
Diefenbach A, Colonna M, Koyasu S (2014) Development, differentiation, and diversity of innate lymphoid cells. Immunity 41:354–365
Vivier E, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, Koyasu S, Locksley RM, McKenzie ANJ, Mebius RE, Powrie F, Spits H (2018) Innate Lymphoid Cells: 10 Years On. Cell 174:1054–1066
Monticelli LA, Sonnenberg GF, Abt MC, Alenghat T, Ziegler CG, Doering TA, Angelosanto JM, Laidlaw BJ, Yang CY, Sathaliyawala T, Kubota M, Turner D, Diamond JM, Goldrath AW, Farber DL, Collman RG, Wherry EJ, Artis D (2011) Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat Immunol 12:1045–1054
Sonnenberg GF, Artis D (2015) Innate lymphoid cells in the initiation, regulation and resolution of inflammation. Nat Med 21:698–708
Ebbo M, Crinier A, Vely F, Vivier E (2017) Innate lymphoid cells: major players in inflammatory diseases. Nat Rev Immunol 17:665–678
Halim TY, Steer CA, Matha L, Gold MJ, Martinez-Gonzalez I, McNagny KM, McKenzie AN, Takei F (2014) Group 2 innate lymphoid cells are critical for the initiation of adaptive T helper 2 cell-mediated allergic lung inflammation. Immunity 40:425–435
Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, Koyasu S, Locksley RM, McKenzie AN, Mebius RE, Powrie F, Vivier E (2013) Innate lymphoid cells–a proposal for uniform nomenclature. Nat Rev Immunol 13:145–149
Cherrier DE, Serafini N, Di Santo JP (2018) Innate lymphoid cell development: a T cell perspective. Immunity 48:1091–1103
Robinette ML, Colonna M (2016) Immune modules shared by innate lymphoid cells and T cells. J Allergy Clin Immunol 138:1243–1251
Mebius RE, Rennert P, Weissman IL (1997) Developing lymph nodes collect CD4+CD3- LTbeta+ cells that can differentiate to APC, NK cells, and follicular cells but not T or B cells. Immunity 7:493–504
Constantinides MG, McDonald BD, Verhoef PA, Bendelac A (2014) A committed precursor to innate lymphoid cells. Nature 508:397–401
Gasteiger G, Fan X, Dikiy S, Lee SY, Rudensky AY (2015) Tissue residency of innate lymphoid cells in lymphoid and nonlymphoid organs. Science 350:981–985
Saluzzo S, Gorki AD, Rana BMJ, Martins R, Scanlon S, Starkl P, Lakovits K, Hladik A, Korosec A, Sharif O, Warszawska JM, Jolin H, Mesteri I, McKenzie ANJ, Knapp S (2017) First-Breath-Induced Type 2 Pathways Shape the Lung Immune Environment. Cell Rep 18:1893–1905
Oherle K, Acker E, Bonfield M, Wang T, Gray J, Lang I, Bridges J, Lewkowich I, Xu Y, Ahlfeld S, Zacharias W, Alenghat T, Deshmukh H (2020) Insulin-like growth factor 1 supports a pulmonary niche that promotes type 3 innate lymphoid cell development in newborn lungs. Immunity 52:275–94.e9
Mjosberg J, Spits H (2016) Human innate lymphoid cells. J Allergy Clin Immunol 138:1265–1276
Fuchs A, Vermi W, Lee JS, Lonardi S, Gilfillan S, Newberry RD, Cella M, Colonna M (2013) Intraepithelial type 1 innate lymphoid cells are a unique subset of IL-12- and IL-15-responsive IFN-gamma-producing cells. Immunity 38:769–781
Qi J, Crinier A, Escalière B, Ye Y, Wang Z, Zhang T, Batista L, Liu H, Hong L, Wu N, Zhang M, Chen L, Liu Y, Shen L, Narni-Mancinelli E, Vivier E, Su B (2021) Single-cell transcriptomic landscape reveals tumor specific innate lymphoid cells associated with colorectal cancer progression. Cell Reports Medicine 2:100353
Vashist N, Trittel S, Ebensen T, Chambers BJ, Guzman CA, Riese P (2018) Influenza-activated ILC1s contribute to antiviral immunity partially influenced by differential GITR expression. Front Immunol 9:505
Weizman OE, Adams NM, Schuster IS, Krishna C, Pritykin Y, Lau C, Degli-Esposti MA, Leslie CS, Sun JC, O’Sullivan TE (2017) ILC1 confer early host protection at initial sites of viral infection. Cell 171(795–808):e12
Bernink JH, Peters CP, Munneke M, te Velde AA, Meijer SL, Weijer K, Hreggvidsdottir HS, Heinsbroek SE, Legrand N, Buskens CJ, Bemelman WA, Mjosberg JM, Spits H (2013) Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat Immunol 14:221–229
Cortez VS, Robinette ML, Colonna M (2015) Innate lymphoid cells: new insights into function and development. Curr Opin Immunol 32:71–77
Leyva-Castillo JM, Galand C, Mashiko S, Bissonnette R, McGurk A, Ziegler SF, Dong C, McKenzie ANJ, Sarfati M, Geha RS (2020) ILC2 activation by keratinocyte-derived IL-25 drives IL-13 production at sites of allergic skin inflammation. J Allergy Clin Immunol 145(1606–14):e4
Miller JE, Lingegowda H, Symons LK, Bougie O, Young SL, Lessey BA, Koti M, Tayade C (2021) IL-33 activates group 2 innate lymphoid cell expansion and modulates endometriosis. JCI Insight 6
Bouchery T, Kyle R, Camberis M, Shepherd A, Filbey K, Smith A, Harvie M, Painter G, Johnston K, Ferguson P, Jain R, Roediger B, Delahunt B, Weninger W, Forbes-Blom E, Le Gros G (2015) ILC2s and T cells cooperate to ensure maintenance of M2 macrophages for lung immunity against hookworms. Nat Commun 6:6970
Winkler C, Hochdörfer T, Israelsson E, Hasselberg A, Cavallin A, Thörn K, Muthas D, Shojaee S, Lüer K, Müller M, Mjösberg J, Vaarala O, Hohlfeld J, Pardali K (2019) Activation of group 2 innate lymphoid cells after allergen challenge in asthmatic patients. J Allergy Clin Immunol 144:61–9.e7
Mjosberg J, Bernink J, Golebski K, Karrich JJ, Peters CP, Blom B, te Velde AA, Fokkens WJ, van Drunen CM, Spits H (2012) The transcription factor GATA3 is essential for the function of human type 2 innate lymphoid cells. Immunity 37:649–659
Kasal DN, Liang Z, Hollinger MK, O'Leary CY, Lisicka W, Sperling AI, Bendelac A (2021) A Gata3 enhancer necessary for ILC2 development and function. Proc Natl Acad Sci U S A 118:e2106311118
Ricardo-Gonzalez RR, Van Dyken SJ, Schneider C, Lee J, Nussbaum JC, Liang HE, Vaka D, Eckalbar WL, Molofsky AB, Erle DJ, Locksley RM (2018) Tissue signals imprint ILC2 identity with anticipatory function. Nat Immunol 19:1093–1099
Schneider C, Lee J, Koga S, Ricardo-Gonzalez RR, Nussbaum JC, Smith LK, Villeda SA, Liang HE, Locksley RM (2019) Tissue-resident group 2 innate lymphoid cells differentiate by layered ontogeny and in situ perinatal priming. Immunity 50(1425–38):e5
Drake LY, Kita H (2014) Group 2 innate lymphoid cells in the lung. Adv Immunol 124:1–16
Takatori H, Kanno Y, Watford WT, Tato CM, Weiss G, Ivanov II, Littman DR, O’Shea JJ (2009) Lymphoid tissue inducer-like cells are an innate source of IL-17 and IL-22. J Exp Med 206:35–41
Victor AR, Nalin AP, Dong W, McClory S, Wei M, Mao C, Kladney RD, Youssef Y, Chan WK, Briercheck EL, Hughes T, Scoville SD, Pitarresi JR, Chen C, Manz S, Wu LC, Zhang J, Ostrowski MC, Freud AG, Leone GW, Caligiuri MA, Yu J (2017) IL-18 drives ILC3 proliferation and promotes IL-22 production via NF-κB. J Immunol 199:2333–2342
Zhong C, Cui K, Wilhelm C, Hu G, Mao K, Belkaid Y, Zhao K, Zhu J (2016) Group 3 innate lymphoid cells continuously require the transcription factor GATA-3 after commitment. Nat Immunol 17:169–178
Hirose K, Ito T, Nakajima H (2018) Roles of IL-22 in allergic airway inflammation in mice and humans. Int Immunol 30:413–418
Gebhardt T, Wakim LM, Eidsmo L, Reading PC, Heath WR, Carbone FR (2009) Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat Immunol 10:524–530
Steinert Elizabeth M, Schenkel Jason M, Fraser Kathryn A, Beura Lalit K, Manlove Luke S, Igyártó Botond Z, Southern Peter J, Masopust D (2015) Quantifying memory CD8 T cells reveals regionalization of immunosurveillance. Cell 161:737–749
Masopust D, Soerens AG (2019) Tissue-Resident T Cells and Other Resident Leukocytes. Annu Rev Immunol 37:521–546
McMaster SR, Gabbard JD, Koutsonanos DG, Compans RW, Tripp RA, Tompkins SM, Kohlmeier JE (2015) Memory T cells generated by prior exposure to influenza cross react with the novel H7N9 influenza virus and confer protective heterosubtypic immunity. PLoS ONE 10:e0115725
Djenidi F, Adam J, Goubar A, Durgeau A, Meurice G, de Montpréville V, Validire P, Besse B, Mami-Chouaib F (2015) CD8+CD103+ tumor-infiltrating lymphocytes are tumor-specific tissue-resident memory T cells and a prognostic factor for survival in lung cancer patients. J Immunol 194:3475–3486
Bošnjak B, Kazemi S, Altenburger LM, Mokrović G, Epstein MM (2019) Th2-TRMs maintain life-long allergic memory in experimental asthma in mice. Front Immunol 10:840
Hondowicz BD, An D, Schenkel JM, Kim KS, Steach HR, Krishnamurty AT, Keitany GJ, Garza EN, Fraser KA, Moon JJ, Altemeier WA, Masopust D, Pepper M (2016) Interleukin-2-dependent allergen-specific tissue-resident memory cells drive asthma. Immunity 44:155–166
Rahimi RA, Nepal K, Cetinbas M, Sadreyev RI, Luster AD (2020) Distinct functions of tissue-resident and circulating memory Th2 cells in allergic airway disease. J Exp Med 217:e20190865
Turner DL, Goldklang M, Cvetkovski F, Paik D, Trischler J, Barahona J, Cao M, Dave R, Tanna N, D’Armiento JM, Farber DL (2018) Biased generation and in situ activation of lung tissue-resident memory CD4 T cells in the pathogenesis of allergic asthma. J Immunol 200:1561–1569
Mackay LK, Wynne-Jones E, Freestone D, Pellicci DG, Mielke LA, Newman DM, Braun A, Masson F, Kallies A, Belz GT, Carbone FR (2015) T-box transcription factors combine with the cytokines TGF-β and IL-15 to control tissue-resident memory T cell fate. Immunity 43:1101–1111
Skon CN, Lee JY, Anderson KG, Masopust D, Hogquist KA, Jameson SC (2013) Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat Immunol 14:1285–1293
Kumar BV, Ma W, Miron M, Granot T, Guyer RS, Carpenter DJ, Senda T, Sun X, Ho SH, Lerner H, Friedman AL, Shen Y, Farber DL (2017) Human tissue-resident memory T cells are defined by core transcriptional and functional signatures in lymphoid and mucosal sites. Cell Rep 20:2921–2934
Reilly EC, Emo KL, Buckley PM, Reilly NS, Smith I, Chaves FA, Yang H, Oakes PW, Topham DJ (2020) T<sub>RM</sub> integrins CD103 and CD49a differentially support adherence and motility after resolution of influenza virus infection. Proc Natl Acad Sci 117:12306–12314
Kim TS, Braciale TJ (2009) Respiratory dendritic cell subsets differ in their capacity to support the induction of virus-specific cytotoxic CD8+ T cell responses. PLoS ONE 4:e4204
Mikhak Z, Strassner JP, Luster AD (2013) Lung dendritic cells imprint T cell lung homing and promote lung immunity through the chemokine receptor CCR4. J Exp Med 210:1855–1869
Takamura S (2017) Persistence in temporary lung niches: a survival strategy of lung-resident memory CD8(+) T cells. Viral Immunol 30:438–450
Takamura S, Yagi H, Hakata Y, Motozono C, McMaster SR, Masumoto T, Fujisawa M, Chikaishi T, Komeda J, Itoh J, Umemura M, Kyusai A, Tomura M, Nakayama T, Woodland DL, Kohlmeier JE, Miyazawa M (2016) Specific niches for lung-resident memory CD8+ T cells at the site of tissue regeneration enable CD69-independent maintenance. J Exp Med 213:3057–3073
Dunbar PR, Cartwright EK, Wein AN, Tsukamoto T, Tiger Li Z-R, Kumar N, Uddbäck IE, Hayward SL, Ueha S, Takamura S, Kohlmeier JE (2020) Pulmonary monocytes interact with effector T cells in the lung tissue to drive T(RM) differentiation following viral infection. Mucosal Immunol 13:161–171
Ferreira C, Barros L, Baptista M, Blankenhaus B, Barros A, Figueiredo-Campos P, Konjar Š, Lainé A, Kamenjarin N, Stojanovic A, Cerwenka A, Probst HC, Marie JC, Veldhoen M (2020) Type 1 Treg cells promote the generation of CD8+ tissue-resident memory T cells. Nat Immunol 21:766–776
Mackay LK, Stock AT, Ma JZ, Jones CM, Kent SJ, Mueller SN, Heath WR, Carbone FR, Gebhardt T (2012) Long-lived epithelial immunity by tissue-resident memory T (TRM) cells in the absence of persisting local antigen presentation. Proc Natl Acad Sci U S A 109:7037–7042
McMaster SR, Wein AN, Dunbar PR, Hayward SL, Cartwright EK, Denning TL, Kohlmeier JE (2018) Pulmonary antigen encounter regulates the establishment of tissue-resident CD8 memory T cells in the lung airways and parenchyma. Mucosal Immunol 11:1071–1078
Mackay LK, Minnich M, Kragten NAM, Liao Y, Nota B, Seillet C, Zaid A, Man K, Preston S, Freestone D, Braun A, Wynne-Jones E, Behr FM, Stark R, Pellicci DG, Godfrey DI, Belz GT, Pellegrini M, Gebhardt T, Busslinger M, Shi W, CarboneLier FRRAWV, Kallies A, Gisbergen KPJMV (2016) Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science 352:459–463
Behr FM, Kragten NAM, Wesselink TH, Nota B, van Lier RAW, Amsen D, Stark R, Hombrink P, van Gisbergen KPJM (2019) Blimp-1 rather than hobit drives the formation of tissue-resident memory CD8+ T cells in the lungs. Front Immunol 10:400
Teijaro JR, Turner D, Pham Q, Wherry EJ, Lefrancois L, Farber DL (2011) Cutting edge: Tissue-retentive lung memory CD4 T cells mediate optimal protection to respiratory virus infection. J Immunol 187:5510–5514
Turner DL, Bickham KL, Thome JJ, Kim CY, D’Ovidio F, Wherry EJ, Farber DL (2014) Lung niches for the generation and maintenance of tissue-resident memory T cells. Mucosal Immunol 7:501–510
Schreiner D, King CG (2018) CD4+ Memory T cells at home in the tissue: mechanisms for health and disease. Front Immunol 9:2394
Strutt TM, Dhume K, Finn CM, Hwang JH, Castonguay C, Swain SL, McKinstry KK (2018) IL-15 supports the generation of protective lung-resident memory CD4 T cells. Mucosal Immunol 11:668–680
Hwang JY, Randall TD, Silva-Sanchez A (2016) Inducible bronchus-associated lymphoid tissue: taming inflammation in the lung. Front Immunol 7:258
Ely KH, Cookenham T, Roberts AD, Woodland DL (2006) Memory T cell populations in the lung airways are maintained by continual recruitment. J Immunol 176:537–543
Allie SR, Randall TD (2020) Resident Memory B Cells. Viral Immunol 33:282–293
Tomayko MM, Steinel NC, Anderson SM, Shlomchik MJ (2010) Cutting edge: hierarchy of maturity of murine memory B cell subsets. J Immunol 185:7146–7150
Weisel F, Shlomchik M (2017) Memory B cells of mice and humans. Annu Rev Immunol 35:255–284
Tan H-X, Juno JA, Esterbauer R, Kelly HG, Wragg KM, Konstandopoulos P, Alcantara S, Alvarado C, Jones R, Starkey G, Wang BZ, Yoshino O, Tiang T, Grayson ML, Opdam H, D’Costa R, Vago A, Mackay LK, Gordon CL, Masopust D, Groom JR, Kent SJ, Wheatley AK (2022) Lung-resident memory B cells established after pulmonary influenza infection display distinct transcriptional and phenotypic profiles. Sci Immunol 7:eabf5314
Kato A, Hulse KE, Tan BK, Schleimer RP (2013) B-lymphocyte lineage cells and the respiratory system. J Allergy Clin Immunol 131: 933–57; quiz 58
Moyron-Quiroz JE, Rangel-Moreno J, Kusser K, Hartson L, Sprague F, Goodrich S, Woodland DL, Lund FE, Randall TD (2004) Role of inducible bronchus associated lymphoid tissue (iBALT) in respiratory immunity. Nat Med 10:927–934
Tan HX, Juno JA, Esterbauer R, Kelly HG, Wragg KM, Konstandopoulos P, Alcantara S, Alvarado C, Jones R, Starkey G, Wang BZ, Yoshino O, Tiang T, Grayson ML, Opdam H, D’Costa R, Vago A, Mackay LK, Gordon CL, Masopust D, Groom JR, Kent SJ, Wheatley AK (2022) Lung-resident memory B cells established after pulmonary influenza infection display distinct transcriptional and phenotypic profiles. Sci Immunol 7:eabf5314
Vantourout P, Hayday A (2013) Six-of-the-best: unique contributions of γδ T cells to immunology. Nat Rev Immunol 13:88–100
Cheng M, Hu S (2017) Lung-resident γδ T cells and their roles in lung diseases. Immunology 151:375–384
Augustin A, Kubo RT, Sim G-K (1989) Resident pulmonary lymphocytes expressing the γ/δ T-cell receptor. Nature 340:239–241
Itohara S, Farr AG, Lafaille JJ, Bonneville M, Takagaki Y, Haas W, Tonegawa S (1990) Homing of a γδ thymocyte subset with homogeneous T-cell receptors to mucosal epithelia. Nature 343:754–757
Sim G-K, Rajaserkar R, Dessing M, Augustin A (1994) Homing and in situ differentiation of resident pulmonary lymphocytes. Int Immunol 6:1287–1295
Wands JM, Roark CL, Aydintug MK, Jin N, Hahn YS, Cook L, Yin X, Dal Porto J, Lahn M, Hyde DM, Gelfand EW, Mason RJ, O’Brien RL, Born WK (2005) Distribution and leukocyte contacts of gammadelta T cells in the lung. J Leukoc Biol 78:1086–1096
Simonian PL, Roark CL, Wehrmann F, Lanham AM, Born WK, O'Brien RL, Fontenot AP (2009) IL-17A-expressing T cells are essential for bacterial clearance in a murine model of hypersensitivity pneumonitis. J Immunol (Baltimore, Md. : 1950) 182: 6540–9
Simonian PL, Wehrmann F, Roark CL, Born WK, O’Brien RL, Fontenot AP (2010) γδ T cells protect against lung fibrosis via IL-22. J Exp Med 207:2239–2253
Murdoch JR, Lloyd CM (2010) Resolution of allergic airway inflammation and airway hyperreactivity is mediated by IL-17-producing {gamma}{delta}T cells. Am J Respir Crit Care Med 182:464–476
van Riel D, Leijten LME, van der Eerden M, Hoogsteden HC, Boven LA, Lambrecht BN, Osterhaus ADME, Kuiken T (2011) Highly pathogenic avian influenza virus H5N1 infects alveolar macrophages without virus production or excessive TNF-alpha induction. PLoS Pathog 7:e1002099
Purnama C, Ng SL, Tetlak P, Setiagani YA, Kandasamy M, Baalasubramanian S, Karjalainen K, Ruedl C (2014) Transient ablation of alveolar macrophages leads to massive pathology of influenza infection without affecting cellular adaptive immunity. Eur J Immunol 44:2003–2012
Kumagai Y, Takeuchi O, Kato H, Kumar H, Matsui K, Morii E, Aozasa K, Kawai T, Akira S (2007) Alveolar macrophages are the primary interferon-alpha producer in pulmonary infection with RNA viruses. Immunity 27:240–252
Guarda G, Braun M, Staehli F, Tardivel A, Mattmann C, Förster I, Farlik M, Decker T, Du Pasquier RA, Romero P, Tschopp J (2011) Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity 34:213–223
Mayer-Barber KD, Andrade BB, Barber DL, Hieny S, Feng CG, Caspar P, Oland S, Gordon S, Sher A (2011) Innate and adaptive interferons suppress IL-1α and IL-1β production by distinct pulmonary myeloid subsets during Mycobacterium tuberculosis infection. Immunity 35:1023–1034
Neupane AS, Willson M, Chojnacki AK, Vargas ESCF, Morehouse C, Carestia A, Keller AE, Peiseler M, DiGiandomenico A, Kelly MM, Amrein M, Jenne C, Thanabalasuriar A, Kubes P (2020) Patrolling alveolar macrophages conceal bacteria from the immune system to maintain homeostasis. Cell 183:110–25.e11
Corry J, Kettenburg G, Upadhyay AA, Wallace M, Marti MM, Wonderlich ER, Bissel SJ, Goss K, Sturgeon TJ, Watkins SC, Reed DS, Bosinger SE, Barratt-Boyes SM (2022) Infiltration of inflammatory macrophages and neutrophils and widespread pyroptosis in lung drive influenza lethality in nonhuman primates. PLoS Pathog 18:e1010395
Hargadon KM, Zhou H, Albrecht RA, Dodd HA, García-Sastre A, Braciale TJ (2011) Major histocompatibility complex class II expression and hemagglutinin subtype influence the infectivity of type A influenza virus for respiratory dendritic cells. J Virol 85:11955–11963
Li Q, Spriggs MK, Kovats S, Turk SM, Comeau MR, Nepom B, Hutt-Fletcher LM (1997) Epstein-Barr virus uses HLA class II as a cofactor for infection of B lymphocytes. J Virol 71:4657–4662
GeurtsvanKessel CH, Willart MAM, van Rijt LS, Muskens F, Kool M, Baas C, Thielemans K, Bennett C, Clausen BE, Hoogsteden HC, Osterhaus ADME, Rimmelzwaan GF, Lambrecht BN (2008) Clearance of influenza virus from the lung depends on migratory langerin+CD11b- but not plasmacytoid dendritic cells. J Exp Med 205:1621–1634
Ng SL, Teo YJ, Setiagani YA, Karjalainen K, Ruedl C (2018) Type 1 conventional CD103+ dendritic cells control effector CD8+ T cell migration, survival, and memory responses during influenza infection. Front Immunol 9
Ballesteros-Tato A, León B, Lund FE, Randall TD (2010) Temporal changes in dendritic cell subsets, cross-priming and costimulation via CD70 control CD8(+) T cell responses to influenza. Nat Immunol 11:216–224
Bosteels C, Neyt K, Vanheerswynghels M, van Helden MJ, Sichien D, Debeuf N, De Prijck S, Bosteels V, Vandamme N, Martens L, Saeys Y, Louagie E, Lesage M, Williams DL, Tang S-C, Mayer JU, Ronchese F, Scott CL, Hammad H, Guilliams M, Lambrecht BN (2020) Inflammatory Type 2 cDCs Acquire Features of cDC1s and Macrophages to Orchestrate Immunity to Respiratory Virus Infection. Immunity 52:1039–56.e9
Li T, Wang J, Wang Y, Chen Y, Wei H, Sun R, Tian Z (2017) Respiratory influenza virus infection induces memory-like liver NK cells in mice. J Immunol 198:1242–1252
Nogusa S, Ritz BW, Kassim SH, Jennings SR, Gardner EM (2008) Characterization of age-related changes in natural killer cells during primary influenza infection in mice. Mech Ageing Dev 129:223–230
Zhou G, Juang SW, Kane KP (2013) NK cells exacerbate the pathology of influenza virus infection in mice. Eur J Immunol 43:929–938
Pociask DA, Scheller EV, Mandalapu S, McHugh KJ, Enelow RI, Fattman CL, Kolls JK, Alcorn JF (2013) IL-22 is essential for lung epithelial repair following influenza infection. Am J Pathol 182:1286–1296
Ivanov S, Renneson J, Fontaine J, Barthelemy A, Paget C, Fernandez EM, Blanc F, De Trez C, Van Maele L, Dumoutier L, Huerre M-R, Eberl G, Si-Tahar M, Gosset P, Renauld JC, Sirard JC, Faveeuw C, Trottein F (2013) Interleukin-22 reduces lung inflammation during influenza A virus infection and protects against secondary bacterial infection. J Virol 87:6911–6924
Koutsakos M, Illing PT, Nguyen THO, Mifsud NA, Crawford JC, Rizzetto S, Eltahla AA, Clemens EB, Sant S, Chua BY, Wong CY, Allen EK, Teng D, Dash P, Boyd DF, Grzelak L, Zeng W, Hurt AC, Barr I, Rockman S, Jackson DC, Kotsimbos TC, Cheng AC, Richards M, Westall GP, Loudovaris T, Mannering SI, Elliott M, Tangye SG, Wakim LM, Rossjohn J, Vijaykrishna D, Luciani F, Thomas PG, Gras S, Purcell AW, Kedzierska K (2019) Human CD8+ T cell cross-reactivity across influenza A, B and C viruses. Nat Immunol 20:613–625
Pizzolla A, Nguyen TH, Sant S, Jaffar J, Loudovaris T, Mannering SI, Thomas PG, Westall GP, Kedzierska K, Wakim LM (2018) Influenza-specific lung-resident memory T cells are proliferative and polyfunctional and maintain diverse TCR profiles. J Clin Invest 128:721–733
Pizzolla A, Wakim LM (2019) Memory T Cell Dynamics in the Lung during Influenza Virus Infection. J Immunol 202:374
Zens KD, Chen JK, Farber DL (2016) Vaccine-generated lung tissue-resident memory T cells provide heterosubtypic protection to influenza infection. JCI Insight 1:e85832
Son YM, Cheon IS, Wu Y, Li C, Wang Z, Gao X, Chen Y, Takahashi Y, Fu YX, Dent AL, Kaplan MH, Taylor JJ, Cui W, Sun J (2021) Tissue-resident CD4(+) T helper cells assist the development of protective respiratory B and CD8(+) T cell memory responses. Sci Immunol 6:eabb6852
MacLean AJ, Richmond N, Koneva L, Attar M, Medina CAP, Thornton EE, Gomes AC, El-Turabi A, Bachmann MF, Rijal P, Tan TK, Townsend A, Sansom SN, Bannard O, Arnon TI (2022) Secondary influenza challenge triggers resident memory B cell migration and rapid relocation to boost antibody secretion at infected sites. Immunity 55:718–33.e8
Guo XJ, Dash P, Crawford JC, Allen EK, Zamora AE, Boyd DF, Duan S, Bajracharya R, Awad WA, Apiwattanakul N, Vogel P, Kanneganti TD, Thomas PG (2018) Lung γδ T Cells Mediate Protective Responses during Neonatal Influenza Infection that Are Associated with Type 2 Immunity. Immunity 49:531–44.e6
Sant S, Jenkins MR, Dash P, Watson KA, Wang Z, Pizzolla A, Koutsakos M, Nguyen TH, Lappas M, Crowe J, Loudovaris T, Mannering SI, Westall GP, Kotsimbos TC, Cheng AC, Wakim L, Doherty PC, Thomas PG, Loh L, Kedzierska K (2019) Human γδ T-cell receptor repertoire is shaped by influenza viruses, age and tissue compartmentalisation. Clin Transl Immunology 8:e1079
Li H, Xiang Z, Feng T, Li J, Liu Y, Fan Y, Lu Q, Yin Z, Yu M, Shen C, Tu W (2013) Human Vγ9Vδ2-T cells efficiently kill influenza virus-infected lung alveolar epithelial cells. Cell Mol Immunol 10:159–164
Tu W, Zheng J, Liu Y, Sia SF, Liu M, Qin G, Ng IHY, Xiang Z, Lam K-T, Peiris JSM, Lau Y-L (2011) The aminobisphosphonate pamidronate controls influenza pathogenesis by expanding a gammadelta T cell population in humanized mice. J Exp Med 208:1511–1522
Kolli D, Gupta MR, Sbrana E, Velayutham TS, Chao H, Casola A, Garofalo RP (2014) Alveolar macrophages contribute to the pathogenesis of human metapneumovirus infection while protecting against respiratory syncytial virus infection. Am J Respir Cell Mol Biol 51:502–515
Smit JJ, Rudd BD, Lukacs NW (2006) Plasmacytoid dendritic cells inhibit pulmonary immunopathology and promote clearance of respiratory syncytial virus. J Exp Med 203:1153–1159
Goritzka M, Makris S, Kausar F, Durant LR, Pereira C, Kumagai Y, Culley FJ, Mack M, Akira S, Johansson C (2015) Alveolar macrophage-derived type I interferons orchestrate innate immunity to RSV through recruitment of antiviral monocytes. J Exp Med 212:699–714
Makris S, Bajorek M, Culley FJ, Goritzka M, Johansson C (2016) Alveolar macrophages can control respiratory syncytial virus infection in the absence of type I interferons. J Innate Immun 8:452–463
Kaiko GE, Phipps S, Angkasekwinai P, Dong C, Foster PS (2010) NK cell deficiency predisposes to viral-induced Th2-type allergic inflammation via epithelial-derived IL-25. J Immunol 185:4681–4690
Stier MT, Bloodworth MH, Toki S, Newcomb DC, Goleniewska K, Boyd KL, Quitalig M, Hotard AL, Moore ML, Hartert TV, Zhou B, McKenzie AN, Peebles RS Jr (2016) Respiratory syncytial virus infection activates IL-13–producing group 2 innate lymphoid cells through thymic stromal lymphopoietin. J Allergy Clin Immunol 138:814–24.e11
Vu LD, Siefker D, Jones TL, You D, Taylor R, DeVincenzo J, Cormier SA (2019) Elevated Levels of Type 2 Respiratory Innate Lymphoid Cells in Human Infants with Severe Respiratory Syncytial Virus Bronchiolitis. Am J Respir Crit Care Med 200:1414–1423
Wu YH, Lai AC, Chi PY, Thio CL, Chen WY, Tsai CH, Lee YL, Lukacs NW, Chang YJ (2020) Pulmonary IL-33 orchestrates innate immune cells to mediate respiratory syncytial virus-evoked airway hyperreactivity and eosinophilia. Allergy 75:818–830
Malinczak CA, Fonseca W, Rasky AJ, Ptaschinski C, Morris S, Ziegler SF, Lukacs NW (2019) Sex-associated TSLP-induced immune alterations following early-life RSV infection leads to enhanced allergic disease. Mucosal Immunol 12:969–979
Beigelman A, Bacharier LB (2016) Early-life respiratory infections and asthma development: role in disease pathogenesis and potential targets for disease prevention. Curr Opin Allergy Clin Immunol 16:172–178
Jozwik A, Habibi MS, Paras A, Zhu J, Guvenel A, Dhariwal J, Almond M, Wong EHC, Sykes A, Maybeno M, Del Rosario J, Trujillo-Torralbo MB, Mallia P, Sidney J, Peters B, Kon OM, Sette A, Johnston SL, Openshaw PJ, Chiu C (2015) RSV-specific airway resident memory CD8+ T cells and differential disease severity after experimental human infection. Nat Commun 6:10224
Luangrath MA, Schmidt ME, Hartwig SM, Varga SM (2021) Tissue-resident memory T cells in the lungs protect against acute respiratory syncytial virus infection. Immunohorizons 5:59–69
Guvenel A, Jozwik A, Ascough S, Ung SK, Paterson S, Kalyan M, Gardener Z, Bergstrom E, Kar S, Habibi MS, Paras A, Zhu J, Park M, Dhariwal J, Almond M, Wong EHC, Sykes A, Del Rosario J, Trujillo-Torralbo M-B, Mallia P, Sidney J, Peters B, Kon OM, Sette A, Johnston SL, Openshaw PJ, Chiu C (2020) Epitope-specific airway-resident CD4+ T cell dynamics during experimental human RSV infection. J Clin Investig 130:523–538
Dodd J, Riffault S, Kodituwakku JS, Hayday AC, Openshaw PJM (2009) Pulmonary V gamma 4+ gamma delta T cells have proinflammatory and antiviral effects in viral lung disease. Journal of immunology (Baltimore, Md. : 1950) 182: 1174–81
Huang H, Saravia J, You D, Shaw AJ, Cormier SA (2015) Impaired gamma delta T cell-derived IL-17A and inflammasome activation during early respiratory syncytial virus infection in infants. Immunol Cell Biol 93:126–135
Wang C, Xie J, Zhao L, Fei X, Zhang H, Tan Y, Nie X, Zhou L, Liu Z, Ren Y, Yuan L, Zhang Y, Zhang J, Liang L, Chen X, Liu X, Wang P, Han X, Weng X et al (2020) Alveolar macrophage dysfunction and cytokine storm in the pathogenesis of two severe COVID-19 patients. eBioMedicine 57:102833
Kosyreva A, Dzhalilova D, Lokhonina A, Vishnyakova P, Fatkhudinov T (2021) The Role of Macrophages in the pathogenesis of SARS-CoV-2-associated acute respiratory distress syndrome. Front Immunol 12:682871
Grant RA, Morales-Nebreda L, Markov NS, Swaminathan S, Querrey M et al (2021) Circuits between infected macrophages and T cells in SARS-CoV-2 pneumonia. Nature 590:635–641
Zheng J, Wang Y, Li K, Meyerholz DK, Allamargot C, Perlman S (2021) Severe acute respiratory syndrome coronavirus 2-induced immune activation and death of monocyte-derived human macrophages and dendritic cells. J Infect Dis 223:785–795
Blanco-Melo D, Nilsson-Payant BE, Liu WC, Uhl S, Hoagland D, Møller R, Jordan TX, Oishi K, Panis M, Sachs D, Wang TT, Schwartz RE, Lim JK, Albrecht RA, tenOever BR (2020) Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell 181:1036–45.e9
Lei X, Dong X, Ma R, Wang W, Xiao X, Tian Z, Wang C, Wang Y, Li L, Ren L, Guo F, Zhao Z, Zhou Z, Xiang Z, Wang J (2020) Activation and evasion of type I interferon responses by SARS-CoV-2. Nat Commun 11:3810
Dalskov L, Møhlenberg M, Thyrsted J, Blay-Cadanet J, Poulsen ET, Folkersen BH, Skaarup SH, Olagnier D, Reinert L, Enghild JJ, Hoffmann HJ, Holm CK, Hartmann R (2020) SARS-CoV-2 evades immune detection in alveolar macrophages. EMBO reports 21: e51252-e
Chang T, Yang J, Deng H, Chen D, Yang X, Tang ZH (2022) Depletion and dysfunction of dendritic cells: understanding SARS-CoV-2 infection. Front Immunol 13:843342
Sánchez-Cerrillo I, Landete P, Aldave B, Sánchez-Alonso S, Sánchez-Azofra A, Marcos-Jiménez A, Ávalos E, Alcaraz-Serna A, de Los SI, Mateu-Albero T, Esparcia L, López-Sanz C, Martínez-Fleta P, Gabrie L, Del Campo GL, de la Fuente H, Calzada MJ, González-Álvaro I, Alfranca A, Sánchez-Madrid F, Muñoz-Calleja C, Soriano JB, Ancochea J, Martín-Gayo E (2020) COVID-19 severity associates with pulmonary redistribution of CD1c+ DCs and inflammatory transitional and nonclassical monocytes. J Clin Invest 130:6290–6300
Zhou R, To KK-W, Wong Y-C, Liu L, Zhou B, Li X, Huang H, Mo Y, Luk T-Y, Lau TT-K, Yeung P, Chan W-M, Wu AK-L, Lung K-C, Tsang OT-Y, Leung W-S, Hung IF-N, Yuen K-Y, Chen Z (2020) Acute SARS-CoV-2 infection impairs dendritic cell and T cell responses. Immunity 53:864–77.e5
Saichi M, Ladjemi MZ, Korniotis S, Rousseau C, Ait Hamou Z, Massenet-Regad L, Amblard E, Noel F, Marie Y, Bouteiller D, Medvedovic J, Pène F, Soumelis V (2021) Single-cell RNA sequencing of blood antigen-presenting cells in severe COVID-19 reveals multi-process defects in antiviral immunity. Nat Cell Biol 23:538–551
Martin-Gayo E, Gao C, Chen HR, Ouyang Z, Kim D, Kolb KE, Shalek AK, Walker BD, Lichterfeld M, Yu XG (2020) Immunological fingerprints of controllers developing neutralizing HIV-1 antibodies. Cell Rep 30(984–96):e4
Liu L, Wei Q, Nishiura K, Peng J, Wang H, Midkiff C, Alvarez X, Qin C, Lackner A, Chen Z (2016) Spatiotemporal interplay of severe acute respiratory syndrome coronavirus and respiratory mucosal cells drives viral dissemination in rhesus macaques. Mucosal Immunol 9:1089–1101
Parackova Z, Zentsova I, Bloomfield M, Vrabcova P, Smetanova J, Klocperk A, Mesežnikov G, Casas Mendez LF, Vymazal T, Sediva A (2020) Disharmonic inflammatory signatures in COVID-19: augmented neutrophils’ but impaired monocytes’ and dendritic cells’ Responsiveness. Cells 9:2206
García M, Kokkinou E, Carrasco García A, Parrot T, Palma Medina LM, Maleki KT, Christ W, Varnaitė R, Filipovic I, Ljunggren HG, Björkström NK, Folkesson E, Rooyackers O, Eriksson LI, Sönnerborg A, Aleman S, Strålin K, Gredmark-Russ S, Klingström J, Mjösberg J (2020) Innate lymphoid cell composition associates with COVID-19 disease severity. Clin Transl Immunol 9:e1224
Silverstein NJ, Wang Y, Manickas-Hill Z, Carbone C, Dauphin A, Boribong BP, Loiselle M, Davis J, Leonard MM, Kuri-Cervantes L, Meyer NJ, Betts MR, Li JZ, Walker BD, Yu XG, Yonker LM, Luban J (2022) Innate lymphoid cells and COVID-19 severity in SARS-CoV-2 infection. Elife 11
Grau-Exposito J, Sanchez-Gaona N, Massana N, Suppi M, Astorga-Gamaza A, Perea D, Rosado J, Falco A, Kirkegaard C, Torrella A, Planas B, Navarro J, Suanzes P, Alvarez-Sierra D, Ayora A, Sansano I, Esperalba J, Andres C, Anton A, Ramon YCS, Almirante B, Pujol-Borrell R, Falco V, Burgos J, Buzon MJ, Genesca M (2021) Peripheral and lung resident memory T cell responses against SARS-CoV-2. Nat Commun 12:3010
Poon MML, Rybkina K, Kato Y, Kubota M, Matsumoto R, Bloom NI, Zhang Z, Hastie KM, Grifoni A, Weiskopf D, Wells SB, Ural BB, Lam N, Szabo PA, Dogra P, Lee YS, Gray JI, Bradley MC, Brusko MA, Brusko TM, Saphire EO, Connors TJ, Sette A, Crotty S, Farber DL (2021) SARS-CoV-2 infection generates tissue-localized immunological memory in humans. Sci Immunol 6:eabl9105
Braun J, Loyal L, Frentsch M, Wendisch D, Georg P, Kurth F, Hippenstiel S, Dingeldey M, Kruse B, Fauchere F, Baysal E, Mangold M, Henze L, Lauster R, Mall MA, Beyer K, Röhmel J, Voigt S, Schmitz J, Miltenyi S, Demuth I, Müller MA, Hocke A, Witzenrath M, Suttorp N, Kern F, Reimer U, Wenschuh H, Drosten C, Corman VM, Giesecke-Thiel C, Sander LE, Thiel A (2020) SARS-CoV-2-reactive T cells in healthy donors and patients with COVID-19. Nature 587:270–274
Mateus J, Grifoni A, Tarke A, Sidney J, Ramirez SI, Dan JM, Burger ZC, Rawlings SA, Smith DM, Phillips E, Mallal S, Lammers M, Rubiro P, Quiambao L, Sutherland A, Yu ED, da Silva AR, Greenbaum J, Frazier A, Markmann AJ, Premkumar L, de Silva A, Peters B, Crotty S, Sette A, Weiskopf D (2020) Selective and cross-reactive SARS-CoV-2 T cell epitopes in unexposed humans. Science 370:89–94
Kundu R, Narean JS, Wang L, Fenn J, Pillay T, Fernandez ND, Conibear E, Koycheva A, Davies M, Tolosa-Wright M, Hakki S, Varro R, McDermott E, Hammett S, Cutajar J, Thwaites RS, Parker E, Rosadas C, McClure M, Tedder R, Taylor GP, Dunning J, Lalvani A (2022) Cross-reactive memory T cells associate with protection against SARS-CoV-2 infection in COVID-19 contacts. Nat Commun 13:80
Pallikkuth S, Williams E, Pahwa R, Hoffer M, Pahwa S (2021) Association of Flu specific and SARS-CoV-2 specific CD4 T cell responses in SARS-CoV-2 infected asymptomatic heath care workers. Vaccine 39:6019–6024
Green I, Ashkenazi S, Merzon E, Vinker S, Golan-Cohen A (2021) The association of previous influenza vaccination and coronavirus disease-2019. Hum Vaccin Immunother 17:2169–2175
Zeng Q, Langereis MA, van Vliet ALW, Huizinga EG, de Groot RJ (2008) Structure of coronavirus hemagglutinin-esterase offers insight into corona and influenza virus evolution. Proc Natl Acad Sci 105:9065–9069
Roberts LM, Jessop F, Wehrly TD, Bosio CM (2021) Cutting edge: lung-resident T cells elicited by SARS-CoV-2 do not mediate protection against secondary infection. J Immunol 207:2399–2404
Zhao J, Zhao J, Mangalam AK, Channappanavar R, Fett C, Meyerholz DK, Agnihothram S, Baric RS, David CS, Perlman S (2016) Airway memory CD4(+) T cells mediate protective immunity against emerging respiratory coronaviruses. Immunity 44:1379–1391
Zhao Y, Kilian C, Turner J-E, Bosurgi L, Roedl K, Bartsch P, Gnirck A-C, Cortesi F, Schultheiß C, Hellmig M, Enk LUB, Hausmann F, Borchers A, Wong MN, Paust H-J, Siracusa F, Scheibel N, Herrmann M, Rosati E, Bacher P, Kylies D, Jarczak D, Lütgehetmann M, Pfefferle S, Steurer S, Wiesch JSZ, Puelles VG, Sperhake J-P, Addo MM, Lohse AW, Binder M, Huber S, Huber TB, Kluge S, Bonn S, Panzer U, Gagliani N, Krebs CF (2021) Clonal expansion and activation of tissue-resident memory-like TH17 cells expressing GM-CSF in the lungs of patients with severe COVID-19. Sci Immunol 6:eabf6692
Wilk AJ, Rustagi A, Zhao NQ, Roque J, Martínez-Colón GJ, McKechnie JL, Ivison GT, Ranganath T, Vergara R, Hollis T, Simpson LJ, Grant P, Subramanian A, Rogers AJ, Blish CA (2020) A single-cell atlas of the peripheral immune response in patients with severe COVID-19. Nat Med 26:1070–1076
Odak I, Barros-Martins J, Bošnjak B, Stahl K, David S, Wiesner O, Busch M, Hoeper MM, Pink I, Welte T, Cornberg M, Stoll M, Goudeva L, Blasczyk R, Ganser A, Prinz I, Förster R, Koenecke C, Schultze-Florey CR (2020) Reappearance of effector T cells is associated with recovery from COVID-19. EBioMedicine 57:102885
Boada-Romero E, Martinez J, Heckmann BL, Green DR (2020) The clearance of dead cells by efferocytosis. Nat Rev Mol Cell Biol 21:398–414
Famà A, Midiri A, Mancuso G, Biondo C, Lentini G, Galbo R, Giardina MM, De Gaetano GV, Romeo L, Teti G, Beninati C (2020) Nucleic acid-sensing Toll-like receptors play a dominant role in innate immune recognition of pneumococci. mBio 11:e00415–e00420
Bewley MA, Marriott HM, Tulone C, Francis SE, Mitchell TJ, Read RC, Chain B, Kroemer G, Whyte MKB, Dockrell DH (2011) A cardinal role for cathepsin D in co-ordinating the host-mediated apoptosis of macrophages and killing of pneumococci. PLoS Pathog 7:e1001262
Marriott HM, Hellewell PG, Whyte MKB, Dockrell DH (2007) Contrasting roles for reactive oxygen species and nitric oxide in the innate response to pulmonary infection with Streptococcus pneumoniae. Vaccine 25:2485–2490
Machado MG, Patente TA, Rouillé Y, Heumel S, Melo EM, Deruyter L, Pourcet B, Sencio V, Teixeira MM, Trottein F (2022) Acetate improves the killing of Streptococcus pneumoniae by alveolar macrophages via NLRP3 inflammasome and glycolysis-HIF-1a axis. Front Immunol 13:773261
Aggarwal NR, King LS, D’Alessio FR (2014) Diverse macrophage populations mediate acute lung inflammation and resolution. Am J Physiol-Lung Cell Mol Physiol 306:L709–L725
Pisu D, Huang L, Narang V, Theriault M, Lê-Bury G, Lee B, Lakudzala AE, Mzinza DT, Mhango DV, Mitini-Nkhoma SC, Jambo KC, Singhal A, Mwandumba HC, Russell DG (2021) Single cell analysis of M. tuberculosis phenotype and macrophage lineages in the infected lung. J Exp Med 218:e20210615
Schyns J, Bureau F, Marichal T (2018) Lung interstitial macrophages: past, present, and future. J Immunol Res 2018:5160794
Sabatel C, Radermecker C, Fievez L, Paulissen G, Chakarov S, Fernandes C, Olivier S, Toussaint M, Pirottin D, Xiao X, Quatresooz P, Sirard JC, Cataldo D, Gillet L, Bouabe H, Desmet CJ, Ginhoux F, Marichal T, Bureau F (2017) Exposure to bacterial CpG DNA protects from airway allergic inflammation by expanding regulatory lung interstitial macrophages. Immunity 46:457–473
Sun X, Jones HP, Dobbs N, Bodhankar S, Simecka JW (2013) Dendritic cells are the major antigen presenting cells in inflammatory lesions of murine mycoplasma respiratory disease. PLoS ONE 8:e55984
Rosendahl A, Bergmann S, Hammerschmidt S, Goldmann O, Medina E (2013) Lung dendritic cells facilitate extrapulmonary bacterial dissemination during pneumococcal pneumonia. Front Cell Infect Microbiol 3:21
Kawasaki T, Ikegawa M, Kawai T (2022) Antigen presentation in the lung. Front Immunol 13:860915
Hoffmann JP, Kolls JK, McCombs JE (2021) Regulation and function of ILC3s in pulmonary infections. Front Immunol 12:672523
Van Maele L, Carnoy C, Cayet D, Ivanov S, Porte R, Deruy E, Chabalgoity JA, Renauld JC, Eberl G, Benecke AG, Trottein F, Faveeuw C, Sirard JC (2014) Activation of type 3 innate lymphoid cells and interleukin 22 secretion in the lungs during Streptococcus pneumoniae infection. J Infect Dis 210:493–503
Elhaik-Goldman S, Kafka D, Yossef R, Hadad U, Elkabets M, Vallon-Eberhard A, Hulihel L, Jung S, Ghadially H, Braiman A, Apte RN, Mandelboim O, Dagan R, Mizrachi-Nebenzahl Y, Porgador A (2011) The natural cytotoxicity receptor 1 contribution to early clearance of Streptococcus pneumoniae and to natural killer-macrophage cross talk. PLoS ONE 6:e23472
Ge C, Monk IR, Pizzolla A, Wang N, Bedford JG, Stinear TP, Westall GP, Wakim LM (2019) Bystander activation of pulmonary Trm cells attenuates the severity of bacterial pneumonia by enhancing neutrophil recruitment. Cell Rep 29:4236–44.e3
Shenoy AT, Wasserman GA, Arafa EI, Wooten AK, Smith NMS, Martin IMC, Jones MR, Quinton LJ, Mizgerd JP (2020) Lung CD4(+) resident memory T cells remodel epithelial responses to accelerate neutrophil recruitment during pneumonia. Mucosal Immunol 13:334–343
Lindenberg M, Almeida L, Dhillon-LaBrooy A, Siegel E, Henriques-Normark B, Sparwasser T (2021) Clarithromycin impairs tissue-resident memory and Th17 responses to macrolide-resistant Streptococcus pneumoniae infections. J Mol Med (Berl) 99:817–829
Barker KA, Etesami NS, Shenoy AT, Arafa EI, Lyon de Ana C, Smith NM, Martin IM, Goltry WN, Barron AM, Browning JL, Kathuria H, Belkina AC, Guillon A, Zhong X, Crossland NA, Jones MR, Quinton LJ, Mizgerd JP (2021) Lung-resident memory B cells protect against bacterial pneumonia. J Clin Invest 131:e141810
Kirby AC, Newton DJ, Carding SR, Kaye PM (2007) Evidence for the involvement of lung-specific gammadelta T cell subsets in local responses to Streptococcus pneumoniae infection. Eur J Immunol 37:3404–3413
Arredouani M, Yang Z, Ning Y, Qin G, Soininen R, Tryggvason K, Kobzik L (2004) The scavenger receptor MARCO is required for lung defense against pneumococcal pneumonia and inhaled particles. J Exp Med 200:267–272
Dudek M, Puttur F, Arnold-Schrauf C, Kühl AA, Holzmann B, Henriques-Normark B, Berod L, Sparwasser T (2016) Lung epithelium and myeloid cells cooperate to clear acute pneumococcal infection. Mucosal Immunol 9:1288–1302
Thuong NT, Tram TT, Dinh TD, Thai PV, Heemskerk D, Bang ND, Chau TT, Russell DG, Thwaites GE, Hawn TR, Caws M, Dunstan SJ (2016) MARCO variants are associated with phagocytosis, pulmonary tuberculosis susceptibility and Beijing lineage. Genes Immun 17:419–425
Nicholson S, Bonecini-Almeida Mda G, Lapa e Silva JR, Nathan C, Xie QW, Mumford R, Weidner JR, Calaycay J, Geng J, Boechat N, Linhares C, Rom W, Ho JL (1996) Inducible nitric oxide synthase in pulmonary alveolar macrophages from patients with tuberculosis. J Exp Med 183:2293–302
Cohen SB, Gern BH, Delahaye JL, Adams KN, Plumlee CR, Winkler JK, Sherman DR, Gerner MY, Urdahl KB (2018) Alveolar macrophages provide an early Mycobacterium tuberculosis niche and initiate dissemination. Cell Host Microbe 24:439–46.e4
Leemans JC, Juffermans NP, Florquin S, van Rooijen N, Vervoordeldonk MJ, Verbon A, van Deventer SJH, van der Poll T (2001) Depletion of alveolar macrophages exerts protective effects in pulmonary tuberculosis in mice. J Immunol 166:4604–4611
Takeda K, Akira S (2005) Toll-like receptors in innate immunity. Int Immunol 17:1–14
Khader SA, Partida-Sanchez S, Bell G, Jelley-Gibbs DM, Swain S, Pearl JE, Ghilardi N, Desauvage FJ, Lund FE, Cooper AM (2006) Interleukin 12p40 is required for dendritic cell migration and T cell priming after Mycobacterium tuberculosis infection. J Exp Med 203:1805–1815
Lai R, Jeyanathan M, Afkhami S, Zganiacz A, Hammill JA, Yao Y, Kaushic C, Xing Z (2018) CD11b<sup>+</sup> dendritic cell–mediated anti–<em>Mycobacterium tuberculosis</em> Th1 activation is counterregulated by CD103<sup>+</sup> dendritic cells via IL-10. J Immunol 200:1746–1760
Balboa L, Kviatcovsky D, Schierloh P, García M, de la Barrera S, Sasiain MDC (2016) Monocyte-derived dendritic cells early exposed to Mycobacterium tuberculosis induce an enhanced T helper 17 response and transfer mycobacterial antigens. Int J Med Microbiol 306:541–553
Ovchinnikova OA, Berge N, Kang C, Urien C, Ketelhuth DF, Pottier J, Drouet L, Hansson GK, Marchal G, Bäck M, Schwartz-Cornil I, Lagranderie M (2014) Mycobacterium bovis BCG killed by extended freeze-drying induces an immunoregulatory profile and protects against atherosclerosis. J Intern Med 275:49–58
Feng CG, Kaviratne M, Rothfuchs AG, Cheever A, Hieny S, Young HA, Wynn TA, Sher A (2006) NK cell-derived IFN-γ differentially regulates innate resistance and neutrophil response in T cell-deficient hosts infected with <em>Mycobacterium tuberculosis</em>. J Immunol 177:7086–7093
Ardain A, Domingo-Gonzalez R, Das S, Kazer SW, Howard NC, Singh A, Ahmed M, Nhamoyebonde S, Rangel-Moreno J, Ogongo P, Lu L, Ramsuran D, de la Luz Garcia-Hernandez M, K. Ulland T, Darby M, Park E, Karim F, Melocchi L, Madansein R, Dullabh KJ, Dunlap M, Marin-Agudelo N, Ebihara T, Ndung’u T, Kaushal D, Pym AS, Kolls JK, Steyn A, Zúñiga J, Horsnell W, Yokoyama WM, Shalek AK, Kløverpris HN, Colonna M, Leslie A, Khader SA (2019) Group 3 innate lymphoid cells mediate early protective immunity against tuberculosis. Nature 570:528-32
Ogongo P, Tezera LB, Ardain A, Nhamoyebonde S, Ramsuran D, Singh A, Ng'oepe A, Karim F, Naidoo T, Khan K, Dullabh KJ, Fehlings M, Lee BH, Nardin A, Lindestam Arlehamn CS, Sette A, Behar SM, Steyn AJ, Madansein R et al (2021) Tissue-resident-like CD4+ T cells secreting IL-17 control Mycobacterium tuberculosis in the human lung. J Clin Invest 131:e142014
Ogongo P, Porterfield JZ, Leslie A (2019) Lung tissue resident memory T-cells in the immune response to Mycobacterium tuberculosis. Front Immunol 10:992
Lockhart E, Green AM, Flynn JL (2006) IL-17 production is dominated by γδ T cells rather than CD4 T cells during <em>Mycobacterium tuberculosis</em> infection. J Immunol 177:4662–4669
Okamoto Yoshida Y, Umemura M, Yahagi A, O’Brien RL, Ikuta K, Kishihara K, Hara H, Nakae S, Iwakura Y, Matsuzaki G (2010) Essential role of IL-17A in the formation of a mycobacterial infection-induced granuloma in the lung. J Immunol 184:4414–4422
Wenzel SE, Schwartz LB, Langmack EL, Halliday JL, Trudeau JB, Gibbs RL, Chu HW (1999) Evidence that severe asthma can be divided pathologically into two inflammatory subtypes with distinct physiologic and clinical characteristics. Am J Respir Crit Care Med 160:1001–1008
Kuruvilla ME, Lee FE, Lee GB (2019) Understanding asthma phenotypes, endotypes, and mechanisms of disease. Clin Rev Allergy Immunol 56:219–233
Assaf SM, Hanania NA (2020) Eosinophilic vs. neutrophilic asthma. Curr Pulmonol Rep 9:28–35
Zasłona Z, Przybranowski S, Wilke C, van Rooijen N, Teitz-Tennenbaum S, Osterholzer JJ, Wilkinson JE, Moore BB, Peters-Golden M (2014) Resident alveolar macrophages suppress, whereas recruited monocytes promote, allergic lung inflammation in murine models of asthma. J Immunol 193:4245–4253
Staples KJ, Hinks TS, Ward JA, Gunn V, Smith C, Djukanović R (2012) Phenotypic characterization of lung macrophages in asthmatic patients: overexpression of CCL17. J Allergy Clin Immunol 130:1404–12.e7
Fricker M, Gibson PG (2017) Macrophage dysfunction in the pathogenesis and treatment of asthma. Eur Respir J 50:1700196
Fitzpatrick AM, Holguin F, Teague WG, Brown LA (2008) Alveolar macrophage phagocytosis is impaired in children with poorly controlled asthma. J Allergy Clin Immunol 121(1372–8):8.e1–3
Simpson JL, Gibson PG, Yang IA, Upham J, James A, Reynolds PN, Hodge S (2013) Impaired macrophage phagocytosis in non-eosinophilic asthma. Clin Exp Allergy 43:29–35
Huynh ML, Malcolm KC, Kotaru C, Tilstra JA, Westcott JY, Fadok VA, Wenzel SE (2005) Defective apoptotic cell phagocytosis attenuates prostaglandin E2 and 15-hydroxyeicosatetraenoic acid in severe asthma alveolar macrophages. Am J Respir Crit Care Med 172:972–979
Lay JC, Alexis NE, Zeman KL, Peden DB, Bennett WD (2009) In vivo uptake of inhaled particles by airway phagocytes is enhanced in patients with mild asthma compared with normal volunteers. Thorax 64:313–320
Zizzo G, Hilliard BA, Monestier M, Cohen PL (2012) Efficient clearance of early apoptotic cells by human macrophages requires M2c polarization and MerTK induction. J Immunol 189:3508–3520
Erriah M, Pabreja K, Fricker M, Baines KJ, Donnelly LE, Bylund J, Karlsson A, Simpson JL (2019) Galectin-3 enhances monocyte-derived macrophage efferocytosis of apoptotic granulocytes in asthma. Respir Res 20:1
Draijer C, Boorsma CE, Robbe P, Timens W, Hylkema MN, Ten Hacken NH, van den Berge M, Postma DS, Melgert BN (2017) Human asthma is characterized by more IRF5+ M1 and CD206+ M2 macrophages and less IL-10+ M2-like macrophages around airways compared with healthy airways. J Allergy Clin Immunol 140:280–3.e3
Kawano H, Kayama H, Nakama T, Hashimoto T, Umemoto E, Takeda K (2016) IL-10-producing lung interstitial macrophages prevent neutrophilic asthma. Int Immunol 28:489–501
Eisenbarth SC (2019) Dendritic cell subsets in T cell programming: location dictates function. Nat Rev Immunol 19:89–103
Nakano H, Free ME, Whitehead GS, Maruoka S, Wilson RH, Nakano K, Cook DN (2012) Pulmonary CD103+ dendritic cells prime Th2 responses to inhaled allergens. Mucosal Immunol 5:53–65
Mishra A, Brown AL, Yao X, Yang S, Park S-J, Liu C, Dagur PK, McCoy JP, Keeran KJ, Nugent GZ, Jeffries KR, Qu X, Yu Z-X, Levine SJ, Chung JH (2015) Dendritic cells induce Th2-mediated airway inflammatory responses to house dust mite via DNA-dependent protein kinase. Nat Commun 6:6224
Medoff BD, Seung E, Hong S, Thomas SY, Sandall BP, Duffield JS, Kuperman DA, Erle DJ, Luster AD (2009) CD11b<sup>+</sup> myeloid cells are the key mediators of Th2 cell homing into the airway in allergic inflammation. J Immunol 182:623–635
Fear VS, Lai SP, Zosky GR, Perks KL, Gorman S, Blank F, von Garnier C, Stumbles PA, Strickland DH (2016) A pathogenic role for the integrin CD103 in experimental allergic airways disease. Physiol Rep 4:e13021
Lombardi V, Speak AO, Kerzerho J, Szely N, Akbari O (2012) CD8α+β− and CD8α+β+ plasmacytoid dendritic cells induce Foxp3+ regulatory T cells and prevent the induction of airway hyper-reactivity. Mucosal Immunol 5:432–443
Brown AS, Bourges D, Ang DK, Hartland EL, van Driel IR (2014) CD8 subunit expression by plasmacytoid dendritic cells is variable, and does not define stable subsets. Mucosal Immunol 7:200–201
Khare A, Krishnamoorthy N, Oriss TB, Fei M, Ray P, Ray A (2013) Cutting Edge: Inhaled Antigen Upregulates Retinaldehyde Dehydrogenase in Lung CD103<sup>+</sup> but Not Plasmacytoid Dendritic Cells To Induce Foxp3 De Novo in CD4<sup>+</sup> T Cells and Promote Airway Tolerance. J Immunol 191:25–29
Bernatchez E, Gold MJ, Langlois A, Lemay A-M, Brassard J, Flamand N, Marsolais D, McNagny KM, Blanchet M-R (2015) Pulmonary CD103 expression regulates airway inflammation in asthma. Am J Physiol-Lung Cell Mol Physiol 308:L816–L826
Conejero L, Khouili SC, Martínez-Cano S, Izquierdo HM, Brandi P, Sancho D (2017) Lung CD103+ dendritic cells restrain allergic airway inflammation through IL-12 production. JCI Insight 2:e90420
Engler DB, Reuter S, Yv W, Urban S, Kyburz A, Maxeiner J, Martin H, Yogev N, Waisman A, Gerhard M, Cover TL, Taube C, Müller A (2014) Effective treatment of allergic airway inflammation with <i>Helicobacter pylori</i> immunomodulators requires BATF3-dependent dendritic cells and IL-10. Proc Natl Acad Sci 111:11810–11815
de Heer HJ, Hammad H, Soullié T, Dl H, Vos N, Willart MAM, Hoogsteden HC, Lambrecht BN (2004) Essential role of lung plasmacytoid dendritic cells in preventing asthmatic reactions to harmless inhaled antigen. J Exp Med 200:89–98
Oriss TB, Ostroukhova M, Seguin-Devaux C, Dixon-McCarthy B, Stolz DB, Watkins SC, Pillemer B, Ray P, Ray A (2005) Dynamics of dendritic cell phenotype and interactions with CD4<sup>+</sup> T cells in airway inflammation and tolerance. J Immunol 174:854–863
Hosseini B, Berthon BS, Starkey MR, Collison A, McLoughlin RF, Williams EJ, Nichol K, Wark PA, Jensen ME, Da Silva Sena CR, Baines KJ, Mattes J, Wood LG (2021) Children With Asthma Have Impaired Innate Immunity and Increased Numbers of Type 2 Innate Lymphoid Cells Compared With Healthy Controls. Front Immunol 12:664668
Kim J, Chang Y, Bae B, Sohn KH, Cho SH, Chung DH, Kang HR, Kim HY (2019) Innate immune crosstalk in asthmatic airways: Innate lymphoid cells coordinate polarization of lung macrophages. J Allergy Clin Immunol 143:1769–82.e11
Nussbaum JC, Van Dyken SJ, von Moltke J, Cheng LE, Mohapatra A, Molofsky AB, Thornton EE, Krummel MF, Chawla A, Liang H-E, Locksley RM (2013) Type 2 innate lymphoid cells control eosinophil homeostasis. Nature 502:245–248
Han M, Ishikawa T, Bermick JR, Rajput C, Lei J, Goldsmith AM, Jarman CR, Lee J, Bentley JK, Hershenson MB (2020) IL-1β prevents ILC2 expansion, type 2 cytokine secretion, and mucus metaplasia in response to early-life rhinovirus infection in mice. Allergy 75:2005–2019
Sugita K, Steer CA, Martinez-Gonzalez I, Altunbulakli C, Morita H, Castro-Giner F, Kubo T, Wawrzyniak P, Ruckert B, Sudo K, Nakae S, Matsumoto K, O’Mahony L, Akdis M, Takei F, Akdis CA (2018) Type 2 innate lymphoid cells disrupt bronchial epithelial barrier integrity by targeting tight junctions through IL-13 in asthmatic patients. J Allergy Clin Immunol 141(300–10):e11
Wallrapp A, Riesenfeld SJ, Burkett PR, Abdulnour R-EE, Nyman J, Dionne D, Hofree M, Cuoco MS, Rodman C, Farouq D, Haas BJ, Tickle TL, Trombetta JJ, Baral P, Klose CSN, Mahlakõiv T, Artis D, Rozenblatt-Rosen O, Chiu IM, Levy BD, Kowalczyk MS, Regev A, Kuchroo VK (2017) The neuropeptide NMU amplifies ILC2-driven allergic lung inflammation. Nature 549:351–356
Nagashima R, Kosai H, Masuo M, Izumiyama K, Noshikawaji T, Morimoto M, Kumaki S, Miyazaki Y, Motohashi H, Yamamoto M, Tanaka N (2019) Nrf2 suppresses allergic lung inflammation by attenuating the type 2 innate lymphoid cell response. J Immunol 202:1331–1339
Han M, Hong JY, Jaipalli S, Rajput C, Lei J, Hinde JL, Chen Q, Hershenson NM, Bentley JK, Hershenson MB (2017) IFN-γ blocks development of an asthma phenotype in rhinovirus-infected baby mice by inhibiting type 2 innate lymphoid cells. Am J Respir Cell Mol Biol 56:242–251
Haworth O, Cernadas M, Levy BD (2011) NK cells are effectors for resolvin E1 in the timely resolution of allergic airway inflammation. J Immunol 186:6129–6135
Barnig C, Cernadas M, Dutile S, Liu X, Perrella Mark A, Kazani S, Wechsler Michael E, Israel E, Levy Bruce D (2013) Lipoxin A4 regulates natural killer cell and type 2 innate lymphoid cell activation in asthma. Sci Transl Med 5:174ra26-ra26
Molofsky AB, Van Gool F, Liang HE, Van Dyken SJ, Nussbaum JC, Lee J, Bluestone JA, Locksley RM (2015) Interleukin-33 and interferon-γ counter-regulate group 2 innate lymphoid cell activation during immune perturbation. Immunity 43:161–174
Bi J, Cui L, Yu G, Yang X, Chen Y, Wan X (2017) NK cells alleviate lung inflammation by negatively regulating group 2 innate lymphoid cells. J Immunol 198:3336–3344
Farhadi N, Lambert L, Triulzi C, Openshaw PJM, Guerra N, Culley FJ (2014) Natural killer cell NKG2D and granzyme B are critical for allergic pulmonary inflammation. J Allergy Clin Immunol 133:827–35.e3
Gorska MM (2017) Natural killer cells in asthma. Curr Opin Allergy Clin Immunol 17:50–54
Molet S, Hamid Q, Davoine F, Nutku E, Taha R, Pagé N, Olivenstein R, Elias J, Chakir J (2001) IL-17 is increased in asthmatic airways and induces human bronchial fibroblasts to produce cytokines. J Allergy Clin Immunol 108:430–438
Qian Y, Zhu Y, Li Y, Li B (2020) Legend of the sentinels: development of lung resident memory T cells and their roles in diseases. Front Immunol 11:624411
Turner DL, Verter J, Turner R, Cao M (2017) Tissue resident memory B cells established in lungs in allergic asthma. J Immunol 198(71):73
Schramm CM, Puddington L, Yiamouyiannis CA, Lingenheld EG, Whiteley HE, Wolyniec WW, Noonan TC, Thrall RS (2000) Proinflammatory roles of T-cell receptor (TCR) γδ and TCR αβ lymphocytes in a murine model of asthma. Am J Respir Cell Mol Biol 22:218–225
Svensson L, Lilliehöök B, Larsson R, Bucht A (2003) γδ T cells contribute to the systemic immunoglobulin E response and local B-cell reactivity in allergic eosinophilic airway inflammation. Immunology 108:98–108
Hahn YS, Ji XY, Woo SI, Choi YK, Song MS, Shin KS, Jin N, O’Brien RL, Born WK (2008) Vγ1+ γδ T cells reduce IL-10-producing CD4+CD25+ T cells in the lung of ovalbumin-sensitized and challenged mice. Immunol Lett 121:87–92
Henriques MDG, Penido C (2012) γδ T lymphocytes coordinate eosinophil influx during allergic responses. Front Pharmacol 3:200
Nakada EM, Shan J, Kinyanjui MW, Fixman ED (2014) Adjuvant-dependent regulation of interleukin-17 expressing γδ T cells and inhibition of Th2 responses in allergic airways disease. Respir Res 15:90
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F (2021) Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: Cancer J Clin 71:209–49
Giannou AD, Marazioti A, Kanellakis NI, Giopanou I, Lilis I, Zazara DE, Ntaliarda G, Kati D, Armenis V, Giotopoulou GA, Krontira AC, Lianou M, Agalioti T, Vreka M, Papageorgopoulou M, Fouzas S, Kardamakis D, Psallidas I, Spella M, Stathopoulos GT (2017) NRAS destines tumor cells to the lungs. EMBO Mol Med 9:672–686
Chen H, Stoltzfus KC, Lehrer EJ, Horn SR, Siva S, Trifiletti DM, Meng M-B, Verma V, Louie AV, Zaorsky NG (2021) The epidemiology of lung metastases. Front Med 8:723396
Altorki NK, Markowitz GJ, Gao D, Port JL, Saxena A, Stiles B, McGraw T, Mittal V (2019) The lung microenvironment: an important regulator of tumour growth and metastasis. Nat Rev Cancer 19:9–31
Balkwill FR, Capasso M, Hagemann T (2012) The tumor microenvironment at a glance. J Cell Sci 125:5591–5596
Jenkins SJ, Ruckerl D, Cook PC, Jones LH, Finkelman FD, van Rooijen N, MacDonald AS, Allen JE (2011) Local macrophage proliferation, rather than recruitment from the blood, is a signature of TH2 inflammation. Science 332:1284–1288
Loyher P-L, Hamon P, Laviron M, Meghraoui-Kheddar A, Goncalves E, Deng Z, Torstensson S, Bercovici N, Baudesson de Chanville C, Combadière B, Geissmann F, Savina A, Combadière C, Boissonnas A (2018) Macrophages of distinct origins contribute to tumor development in the lung. J Exp Med 215:2536–2553
Fu Y, Pajulas A, Wang J, Zhou B, Cannon A, Cheung CCL, Zhang J, Zhou H, Fisher AJ, Omstead DT, Khan S, Han L, Renauld J-C, Paczesny S, Gao H, Liu Y, Yang L, Tighe RM, Licona-Limón P, Flavell RA, Takatsuka S, Kitamura D, Sun J, Bilgicer B, Sears CR, Yang K, Kaplan MH (2022) Mouse pulmonary interstitial macrophages mediate the pro-tumorigenic effects of IL-9. Nat Commun 13:3811
Qian B, Deng Y, Im JH, Muschel RJ, Zou Y, Li J, Lang RA, Pollard JW (2009) A distinct macrophage population mediates metastatic breast cancer cell extravasation Establishment and Growth. PLoS ONE 4:e6562
Sharma SK, Chintala NK, Vadrevu SK, Patel J, Karbowniczek M, Markiewski MM (2015) Pulmonary alveolar macrophages contribute to the premetastatic niche by suppressing antitumor T cell responses in the lungs. J Immunol 194:5529–5538
Duhen T, Duhen R, Montler R, Moses J, Moudgil T, de Miranda NF, Goodall CP, Blair TC, Fox BA, McDermott JE, Chang SC, Grunkemeier G, Leidner R, Bell RB, Weinberg AD (2018) Co-expression of CD39 and CD103 identifies tumor-reactive CD8 T cells in human solid tumors. Nat Commun 9:2724
Ganesan A-P, Clarke J, Wood O, Garrido-Martin EM, Chee SJ, Mellows T, Samaniego-Castruita D, Singh D, Seumois G, Alzetani A, Woo E, Friedmann PS, King EV, Thomas GJ, Sanchez-Elsner T, Vijayanand P, Ottensmeier CH (2017) Tissue-resident memory features are linked to the magnitude of cytotoxic T cell responses in human lung cancer. Nat Immunol 18:940–950
O’Brien SM, Klampatsa A, Thompson JC, Martinez MC, Hwang W-T, Rao AS, Standalick JE, Kim S, Cantu E, Litzky LA, Singhal S, Eruslanov EB, Moon EK, Albelda SM (2019) Function of human tumor-infiltrating lymphocytes in early-stage non-small cell lung cancer. Cancer Immunol Res 7:896–909
Heim L, Friedrich J, Engelhardt M, Trufa DI, Geppert CI, Rieker RJ, Sirbu H, Finotto S (2018) NFATc1 promotes antitumoral effector functions and memory CD8+ T-cell differentiation during non–small cell lung cancer development. Can Res 78:3619–3633
Corgnac S, Boutet M, Kfoury M, Naltet C, Mami-Chouaib F (2018) The emerging role of CD8(+) tissue resident memory T (T(RM)) cells in antitumor immunity: a unique functional contribution of the CD103 integrin. Front Immunol 9:1904
Karaki S, Blanc C, Tran T, Galy-Fauroux I, Mougel A, Dransart E, Anson M, Tanchot C, Paolini L, Gruel N, Gibault L, Lepimpec-Barhes F, Fabre E, Benhamouda N, Badoual C, Damotte D, Donnadieu E, Kobold S, Mami-Chouaib F, Golub R, Johannes L, Tartour E (2021) CXCR6 deficiency impairs cancer vaccine efficacy and CD8<sup>+</sup> resident memory T-cell recruitment in head and neck and lung tumors. J Immunother Cancer 9:e001948
Amsen D, van Gisbergen KPJM, Hombrink P, van Lier RAW (2018) Tissue-resident memory T cells at the center of immunity to solid tumors. Nat Immunol 19:538–546
Amsen D, Hombrink P, van Lier RAW (2017) Tumor immunity requires border patrol to fight the enemy within. Nat Immunol 18:870–872
Oja AE, Piet B, van der Zwan D, Blaauwgeers H, Mensink M, de Kivit S, Borst J, Nolte MA, van Lier RAW, Stark R, Hombrink P (2018) Functional heterogeneity of CD4+ tumor-infiltrating lymphocytes with a resident memory phenotype in NSCLC. Front Immunol 9:2654
Christian LS, Wang L, Lim B, Deng D, Wu H, Wang XF, Li QJ (2021) Resident memory T cells in tumor-distant tissues fortify against metastasis formation. Cell Rep 35:109118
Enamorado M, Iborra S, Priego E, Cueto FJ, Quintana JA, Martínez-Cano S, Mejías-Pérez E, Esteban M, Melero I, Hidalgo A, Sancho D (2017) Enhanced anti-tumour immunity requires the interplay between resident and circulating memory CD8+ T cells. Nat Commun 8:16073
Edwards J, Wilmott JS, Madore J, Gide TN, Quek C, Tasker A, Ferguson A, Chen J, Hewavisenti R, Hersey P, Gebhardt T, Weninger W, Britton WJ, Saw RPM, Thompson JF, Menzies AM, Long GV, Scolyer RA, Palendira U (2018) CD103(+) tumor-resident CD8(+) T cells are associated with improved survival in immunotherapy-naïve melanoma patients and expand significantly during anti-PD-1 treatment. Clin Cancer Res 24:3036–3045
Anderson KG, Sung H, Skon CN, Lefrancois L, Deisinger A, Vezys V, Masopust D (2012) Cutting edge: intravascular staining redefines lung CD8 T cell responses. J Immunol 189:2702–2706
Tomura M, Yoshida N, Tanaka J, Karasawa S, Miwa Y, Miyawaki A, Kanagawa O (2008) Monitoring cellular movement <i>in vivo</i> with photoconvertible fluorescence protein & #x201c;Kaede” transgenic mice. Proc Natl Acad Sci 105:10871–10876
Finerty JC (1952) Parabiosis in physiological studies. Physiol Rev 32:277–302
Masopust D, Choo D, Vezys V, Wherry EJ, Duraiswamy J, Akondy R, Wang J, Casey KA, Barber DL, Kawamura KS, Fraser KA, Webby RJ, Brinkmann V, Butcher EC, Newell KA, Ahmed R (2010) Dynamic T cell migration program provides resident memory within intestinal epithelium. J Exp Med 207:553–564
Klein SL, Flanagan KL (2016) Sex differences in immune responses. Nat Rev Immunol 16:626–638
Scotland RS, Stables MJ, Madalli S, Watson P, Gilroy DW (2011) Sex differences in resident immune cell phenotype underlie more efficient acute inflammatory responses in female mice. Blood 118:5918–5927
Kadel S, Kovats S (2018) Sex hormones regulate innate immune cells and promote sex differences in respiratory virus infection. Front Immunol 9:1653
Loering S, Cameron GJM, Bhatt NP, Belz GT, Foster PS, Hansbro PM, Starkey MR (2021) Differences in pulmonary group 2 innate lymphoid cells are dependent on mouse age, sex and strain. Immunol Cell Biol 99:542–551
Zazara DE, Arck PC (2019) Developmental origin and sex-specific risk for infections and immune diseases later in life. Seminars in Immunopathology 41:137–151
Keselman A, Fang X, White PB, Heller NM (2017) Estrogen signaling contributes to sex differences in macrophage polarization during asthma. J Immunol 199:1573–1583
Laffont S, Blanquart E, Guéry J-C (2017) Sex differences in asthma: a key role of androgen-signaling in group 2 innate lymphoid cells. Front Immunol 8:1069
Funding
Open Access funding enabled and organized by Projekt DEAL. This work was supported by the German Research Foundation to DEZ (SO1413/1–2). DEZ and ADG are supported by the Clinician Scientist program of the RU5068 and the Medical Faculty of the University of Hamburg. ADG is also supported by an Else Kröner Memorial Fellowship, Werner Otto and Erich und Gertrud Roggenbuck Foundation, Hamburger Krebsgesellschaft Foundation, the Jung Foundation for Science and Research (Ernst Jung Career Development Award 2022) and the Deutsche Krebshilfe Foundation.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is a contribution to the special issue on: Heterogeneity of tissue-resident immunity across organs and in health and disease - Guest Editors: Federica Sallusto & Petra Arck
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zazara, D.E., Belios, I., Lücke, J. et al. Tissue-resident immunity in the lung: a first-line defense at the environmental interface. Semin Immunopathol 44, 827–854 (2022). https://doi.org/10.1007/s00281-022-00964-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-022-00964-2