
Abstract
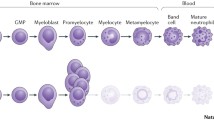
Hereditary neutropenia includes many disorders of distinct origin and variable prognosis, characterized by a reduction of the absolute neutrophil count (ANC) below 0.5 × 109/l, that predisposes patients to bacterial infections of variable severity, in particular pyogenic infections, such as cutaneous cellulitis, deep abscesses, pneumonia, and sepsis. The diagnosis of severe congenital neutropenia (SCN) is usually made on the basis of patient history and physical examination, and of a severe neutropenia with normal or near normal hemoglobin level and platelet count, while bone marrow examination reveals in most cases the typical defect of neutrophils, with myeloid cell differentiation arrest at the promyelocyte stage, and very few myelocytes and metamyelocytes.
The most common monogenic congenital neutropenia are usually classified according to the presence or absence of association with innate or adaptive immunodeficiency, or with extrahematopoietic manifestations, like involvement of the pancreas, central nervous system, heart, muscle, and skin.
Most of the forms of congenital neutropenia are extremely rare, about six cases per one million; some genes have been only found in a few families. The inheritance is monogenic, which may be autosomal (dominant or recessive), or X-linked. During the last few years, several genetic causes of neutropenia have been elucidated.
Similar content being viewed by others


1 Salient Points
-
Hereditary neutropenia is characterized by a reduction of the absolute neutrophil count (ANC) below 0.5 × 109/l.
-
Neutropenia can lead to bacterial infections of variable severity, in particular pyogenic infections, such as cutaneous cellulitis, deep abscesses, pneumonia, and sepsis.
-
The diagnosis of severe congenital neutropenia (SCN) is usually made on the basis of patient history and physical examination, and of a severe neutropenia with normal or near normal hemoglobin level and platelet count.
-
Bone marrow examination reveals in most cases the typical defect of neutrophils with myeloid cell differentiation arrest at the promyelocyte stage, and very few myelocytes and metamyelocytes.
-
The most common monogenic congenital neutropenia are usually classified according to the presence or absence of association with innate or adaptive immunodeficiency, or with extrahematopoietic manifestations, like involvement of the pancreas, central nervous system, heart, muscle, and skin.
-
Most of the forms of congenital neutropenia are extremely rare, the estimated frequency is about six per one million; some genes have been only found in a few families.
-
The inheritance is monogenic, which may be autosomal (dominant or recessive) or X-linked. During the last few years, several genetic causes of neutropenia have been elucidated.
2 Introduction
Hereditary neutropenia includes many disorders of distinct origin and variable prognosis, characterized by a reduction of the absolute neutrophil count (ANC) below 0.5 × 109/l, that predisposes patients to bacterial infections, in particular pyogenic infections, such as cutaneous cellulitis, deep abscesses, pneumonia, and sepsis (Welte et al. 2006). Susceptibility to bacterial infections, even in patients with severe neutropenia, can be quite variable, depending on the underlying etiology (Boztug et al. 2008). The diagnosis of severe congenital neutropenia (SCN) is usually made on the basis of the observation at birth, or in the following months of life, of a severe reduction of ANC (<0.5 × 109/l) with normal or near normal levels of hemoglobin and platelets, while bone marrow examination reveals in most cases the typical defect of neutrophils with myeloid cell differentiation arrest at the promyelocyte stage and very few myelocytes and metamyelocytes (Badolato et al. 2004). Congenital neutropenia may be associated with innate or adaptive immunodeficiency or with extrahematopoietic manifestations, like involvement of the pancreas, central nervous system, heart, muscle, and skin.
All the forms of congenital neutropenia are extremely rare, except for ethnic neutropenia, which is a frequent mild congenital form, with probably polygenic inheritance. The minimal prevalence of congenital neutropenia is six cases per million inhabitants (Dale et al. 2003); the inheritance is monogenic which may be autosomal (dominant or recessive) or X- linked.
During the last few years, several genetic causes of neutropenia have been elucidated. In this respect, scientific research is continuously evolving; some genes are extremely rare and have been only found in a few families. The classification of the most common causes of monogenic congenital neutropenia is presented below (Table 1).
3 Congenital Neutropenia Without Extrahematopoietic Manifestations and with Normal Adaptive Immunity
3.1 Severe Congenital Neutropenia (OMIM Code 202700)
Severe congenital neutropenia (SCN) is a rare disease characterized by a chronic neutropenia due to a genetic defect. The absolute neutrophil count (ANC) is short of 200 cells/mm3 from birth. This predisposes to severe life-threatening infections (Zeidler et al. 2003; Lekstrom-Himes and Gallin 2000).
The incidence is estimated to be approximately two cases per million of the population (Xia et al. 2009). Affected patients regularly have episodes of fever, skin infections, stomatitis, pneumonia, and perirectal abscesses that usually begin in the first months of life and lead to death during infancy and childhood. Eosinophylia, monocytosis, and splenomegaly may be present. The disorder remains a paradigm in the field of congenital neutropenia. The bone marrow examination reveals the typical arrest of the myeloid cell differentiation at the promyelocytic stage with a marked depletion of mature neutrophils (Xia et al. 2009; Dale et al. 2000). Recent studies on the genetic basis of SCN have detected mutation in the neutrophil elastase gene, ELA2 (ELANE) ; this gene defect is inherited as an autosomal dominant trait in about 60–80% of patients (Horwitz et al. 2007). More than 50 separate ELANE mutations have been hitherto found in SCN patients (Germeshausen et al. 2013).
ELA2 encodes a serine protease synthesized during the promyelocyte/myelocyte stage, which is stored in the primary granules (Horwitz et al. 2007). Correct localization of the neutrophil elastase (NE) in the primary granules requires interaction of NE with the adaptor protein complex AP3 that shuttles transmembrane cargo proteins from the Golgi network to the lysosomes. Most mutations in ELA2 remove the tyrosine-based recognition sequence for the AP3μ subunit, thereby favoring the misallocation of the enzyme to the membrane instead of inside the granules. It was proposed that mislocalized NE must drastically reduce the production of neutrophils from the myeloid progenitor cells in favor of monocytes.
The introduction of hematopoietic growth factors has greatly improved both life span and quality of life (Freedman et al. 2000). However, patients with SCN have a heterogeneous response to G-CSF therapy and often require a gradual increase of the dosage that generally ranges from 11 to 13 μg/kg/die. In addition, 3–5% of SCN patients are refractory to G-CSF treatment.
Finally, a significant number of children who are receiving G-CSF therapy are developing myelodysplastic syndrome (MDS) or acute myeloid leukemia (AML). The exact relationship between G-CSF treatment and the risk of leukemic transformation is still unclear (Rosenberg et al. 2010). A subset of SCN patients, namely those with more severe disease, are more prone to develop MDS, AML, or both, probably because of exposure to other leukemogenic factors such as monosomy of chromosome 7, alterations of chromosome 21, activating mutations of the oncogene ras, and mutation in the G-CSF receptor (CSF3R) (Donadieu et al. 2005). At risk are patients with mutations in ELANE, HAX1, or WAS (Rosenberg et al. 2008, 2010; Devriendt et al. 2001) (see below).
The cumulative incidence of malignant transformations in SCN after 10 years of treatment with G-CSF amounts to 21% (Rosenberg et al. 2006) according to the Severe Chronic Neutropenia International Registry data. It does not appear to be a direct consequence of the treatment; indeed, cases of malignant transformation were reported in SCN patients who were not treated with G-CSF, while patients with CyN or idiopathic neutropenia showed no signs of malignant conversion after prolonged use of recombinant G-CSF (Rosenberg et al. 2006; Germeshausen et al. 2005).
Patients with SCN showed mutations in an area of the CSF3R gene that codes for the intracytoplasmic domain of the G-CSFR which were initially thought to be the cause of SCN (Dong et al. 1995). Later, those mutations turned out to be acquired somatic mutations which are associated with the transformation of leukemia in SCN (Germeshausen et al. 2001).
In the light of this finding, G-CSF treatment seams a safe option for the vast majority of patients with neutropenia, while patients at risk for leukemic transformation would benefit from hematopoietic stem cell transplantation (Fioredda et al. 2015).
3.2 Cyclic Neutropenia (OMIM Code 162800)
Cyclic neutropenia is an autosomal dominant or sporadic condition, characterized by periodic episodes of severe neutropenia with a nadir of less than 0.2 × 109 cells/L that occur usually every 21 days (Dale and Hammond 1988).
The neutropenic periods persist for 3–5 days and are accompanied by malaise, fever, gingivitis, oral ulcers, and lymphoadenopathy. There are also regular oscillations of the other leukocyte subsets, reticulocytes, and platelets. Symptoms begin during the first year of life, are often milder after puberty, and are usually less severe than in SCN (Horwitz et al. 2007; Germeshausen et al. 2013; Freedman et al. 2000; Rosenberg et al. 2006, 2008, 2010; Donadieu et al. 2005; Devriendt et al. 2001; Germeshausen et al. 2005; Dong et al. 1995; Germeshausen et al. 2001; Fioredda et al. 2015; Dale and Hammond 1988), even though fatal Clostridium bacteremia has been reported in untreated patients (Dale et al. 2003). A variety of studies suggest that cyclic neutropenia occurs because of a periodic disruption in cell production in the bone marrow (Dale et al. 2003). It has been shown that a heterozygous mutation of ELA2 is the genetic cause of the disease. The mutations reported in cyclic neutropenia are located in the same gene associated with SCN, but in different exons resulting in preferential accumulation of NE in granules.
In the management of these patients the use of prophylactic G-CSF treatment is recommended; it has been very effective in improving peripheral blood neutrophil counts and avoiding symptoms and infections (Freedman et al. 2000). The required dose of G-CSF is usually lower than in SNC: 2–3 μg/kg/day or on alternate days; the occurrence of MDS or leukemia has not been reported in patients with cyclic neutropenia (Freedman et al. 2000).
4 Congenital Neutropenia with Innate and Adaptive Deficiency but Without Extrahematopoietic Manifestations
The same genetic mutations that lead to myeloid cell defects may also be associated with aberrations in cells involved in innate and adaptive immunity.
To identify the affected babies, a complete white blood cell (WBC) count and differential, with lymphocyte subpopulations and immunoglobulin levels, should be obtained.
4.1 Neutropenia Associated with GFI1 Mutations (OMIM Code 202700)
This cause of congenital neutropenia is extremely rare, having hitherto been found in just four patients (Karsunky et al. 2002; Person et al. 2003). The clinical phenotype is not particularly homogeneous: severe neutropenia – and marked monocytosis – was diagnosed in one patient at 4 months of age, while the father, who showed the same mutation, had a moderate form of neutropenia. On the other hand, another patient, who was diagnosed at 56 years of age with idiopathic neutropenia, was not clearly liable to infections. All those patients had moderate lymphopenia (CD3 cells were 1–1.4 G/l), normal memory cells, and humoral immunity. Heterozygous GFI1 mutations – i.e., dominant ones – cause an increased expression of ELANE, just as ELANE mutations do.
GFI1, a transcriptional repressor and splicing control factor, plays an outstanding role in the control of the normal differentiation of hematopoietic cells (van der Meer et al. 2010). In some patients, monoallelic mutations have been observed which may lead to SCN (Armistead et al. 2010; Zhuang et al. 2006). Furthermore, GFI1 mutations are linked to aberrations in lymphoid and myeloid cells. Unsurprisingly, a defect in a significant transcriptional master switch factor provokes the impaired regulation of a number of pathways that may contribute to atypically differentiated neutrophil granulocytes.
4.2 Myelocathexis and WHIM Syndrome (OMIM Code193670)
Myelocathexis is a rare autosomal dominant disorder characterized by moderate to severe chronic neutropenia resulting from the retention of mature neutrophils in the bone marrow. Myelocathexis is often associated with hypogammaglobulinemia, leucopenia, and warts, a clinical picture recognized as WHIM syndrome (warts, hypogammaglobulinemia, infections, myelokathexis) (Aprikyan et al. 2000), an autosomal dominant disease. In WHIM syndrome ANCs are usually <0.5 × 109/L, but during infectious episodes their number rises suddenly, allowing a more benign course with recurrent mild respiratory infections. Early death related to infections has not been reported. WHIM syndrome is caused by heterozygous mutations in the gene coding for the chemokine receptor CXCR4. Cells expressing the mutated CXCR4 have an increased responsiveness to chemokines and thus are not released from the bone marrow to the circulating blood pool (Aprikyan et al. 2000). The neutropenia associated with myelocathexis and WHIM is only partially corrected by administration of G-CSF (Gorlin et al. 2000).
4.3 SNC Due to WAS Mutation (OMIM code 300299)
Pernicious mutations in WASp are the usual causes of the Wiskott–Aldrich syndrome, which is an X-linked syndrome showing immunodeficiency, eczema, and hemorrhage (Notarangelo et al. 2008). The Wiskott–Aldrich syndrome protein (WASp) is an adaptor protein steering the polymerization of actin filaments (Thrasher and Burns 2010). Cells lacking WASp show flawed locomotion and delocalization of receptors. Unlike WASp deficiency, rare WAS mutations cause a constitutive activation of WASp, thus leading to increased actin polymers and, as a result, to abnormal cell divisions (Moulding et al. 2007; Westerberg et al. 2010). Those patients fail to show the classical signs of the Wiskott–Aldrich syndrome, but rather present congenital neutropenia (Devriendt et al. 2001; Beel et al. 2009), with myelodysplasia (Devriendt et al. 2001; Beel et al. 2009; Ancliff et al. 2006), increased myeloid cell apoptosis (Devriendt et al. 2001; Moulding et al. 2007; Westerberg et al. 2010; Beel et al. 2009; Ancliff et al. 2006) and abnormalities in lymphoid cells (Devriendt et al. 2001; Westerberg et al. 2010).
5 Congenital Neutropenia with Extrahematopoietic Manifestations
5.1 Kostmann Syndrome (OMIM code 610738)
Rolf Kostmann described, in 1956, an autosomal recessive hematological disease characterized by a chronic neutropenia with an absolute neutrophil count (ANC) below of 500 cells/mm3 and an early eruption of severe bacterial infections in a large Swedish family with frequent intermarriages (Kostmann 1956), which was later referred to as the “Kostmann syndrome.” The underlying genetic defect in this syndrome was found to be a mutation of the HAX1 gene (Klein et al. 2007; Germeshausen et al. 2008; Kostman 1975). Its precise frequency is not known, but, apparently, it is much lower than ELANE neutropenia, except in some areas, such as Sweden and Kurdistan. A following study published by Kostmann in 1975, which focused on the same pedigree in northern Sweden, described an improvement in survival rates linked to the use of antibiotics. On the other hand, neurological disorders began to be observed in the second decade, in particular neuropsychological deficits and epilepsy (Carlsson and Fasth 2001). The syndrome is described more accurately in a recent study of the same pedigree, where five out of six patients presented with neurological issues (Germeshausen et al. 2008; Kostman 1975; Carlsson and Fasth 2001). A HAX1gene mutation with pleiotropic functions has been found in this syndrome, while the detailed working of the HAX1 deficiency are not fully understood. HAX1 regulates the potential of the inner mitochondrial membrane (Klein et al. 2007; Han et al. 2010), while also being a cytoplasmic protein with numerous interaction partners. For instance, HAX1 binds to multiple virus proteins. Patients showing mutations only affecting isoform A have SCN without any further neuron impairments, while mutations of both isoforms (A and B) are found to be linked to neurological abnormalities, such as development delays and seizures (Germeshausen et al. 2008). Sometimes, neutropenia with ELANE mutations is improperly referred as Kostmann syndrome.
5.2 Hermansky-Pudlak Syndrome2 (OMIM Code 608233)
This autosomal recessive disease is characterized by neutropenia, oculocutaneous albinism, and moderate bleeding disorders (Dell’Angelica et al. 1999). It is caused by mutations of the gene encoding for the beta3 component of the AP3 complex, again preventing the transport of NE (and other proteins) from the Golgi network to the lysosomes in hematopoietic cells and in melanocytes (Dell’Angelica et al. 1999).
The severe neutropenia is responsive to G-CSF treatment.
5.3 Shwachman-Diamond Syndrome (OMIM Code 260400)
The Shwachman-Diamond syndrome (SDS) is a rare multiorgan disease inherited as an autosomal recessive trait that combines neutropenia, exocrine pancreatic insufficiency, skeletal abnormalities, and short stature (Dror and Freedman 2002).
The symptoms begin early in infancy with bacterial infections (pneumonia, otitis media, osteomyelitis, skin infections, sepsis) and failure to thrive because of intestinal malabsorption.
Chronic neutropenia is constantly observed in all patients and two thirds of them have a neutrophil count less than 1 × 109 cells/L; so while the neutropenia can be intermittent, it is never cyclic. Mild anemia and thrombocytopenia are described commonly (Dror and Freedman 2002).
Cytopenia reflects hematopoietic dysplasia, which, in association with cytogenetic abnormalities, may increase the risk of transformation in MDS/AML mainly in older children. For this reason, annual bone marrow aspirates are recommended in patients with SDS. Indeed, follow-up studies of SDS patients demonstrate that some cytogenetic changes my spontaneously regress.
The causative gene of the disease has been identified and was named SDBS. It is expressed in both hematopoietic and nonhematopoietic tissues. Treatment of patients suffering from SDS includes pancreatic enzyme replacement and administration of G-CSF, which increases ANC to normal levels and should be started in case of severe infections (Paley et al. 1991).
5.4 Neutropenia Associated with Glucose-6-Phosphatase Complex Disorders
Patients with glycogen-storage disease of type Ib, which is caused by mutations in the gene coding for the glucose-6-phosphate-transporter (G6PT) protein (Gerin et al. 1997), have long been known to suffer not only from hypoglycemia and glycogen storage symptoms but also from congenital neutropenia. Glucose 6 phosphatase is a complex of three proteins bound to the endoplasmic reticulum responsible for glycogenolysis. Two of these three proteins are associated with congenital neutropenia: the translocase (SLC37A4), and G6PC3 that is a catalytic protein. Neutrophil granulocytes are particularly vulnerable to defects in glucose metabolism. The association between these molecular changes and neutropenia is not clear, because the glycogenolysis pathway is not the source of energy normally used by neutrophils, which mainly use the pentose pathway; this raises the hypothesis that the involved proteins perform a different function in neutrophils.
5.4.1 Glycogen Storage Disease Type Ib
It is a metabolic disorder characterized by hepatic glycogen accumulation, intolerance of fasting, hypoglycemic events, and hyperlactacidemia, as well as susceptibility to infections (Ambruso et al. 2003) and colitis resembling Crohn’s. This susceptibility to infections is due to neutropenia and, sometimes, to neutrophil dysfunction (defective chemotactism). The origin of the neutropenia and neutrophil dysfunction is unknown.
5.4.2 G6PC3 Mutations (OMIM Code 612541)
Patients with G6PC3 mutations, which are prevalent in Turkey, are affected by a variant of congenital neutropenia disease with neutropenia from severe to moderate, typical arrested myeloid maturation, and several other congenital abnormalities. Apart from heart and urogenital deficiencies, face dysmorphic disorders, increasingly visible superficial veins, hearing loss from inner ear dysfunction, aberrant endocrine function, or myopathy may be observed (Notarangelo et al. 2014).
G6PC3 deficiency’s molecular physiopathology includes an activation of the unfolded protein response and an increased stress of the endoplasmic reticulum, in addition to abnormal intracellular glucose homeostasis.
Mutations of the G6PC3 gene are generally homozygous, but a double heterozygote has been described (Boztug et al. 2009).
5.5 Neutropenia Associated with Poikilodermia, Clericuzio Type (OMIM Code 604173)
Clericuzio type is a neutropenia with genodermatosis that onset in the first year of life. Recurrent infections occur, especially pneumonia. The neutropenia is often severe. The poikilodermia includes skin atrophy and popular erythematous rash. Composite mutations of the C16ORF57 gene are responsible of pathology (Volpi et al. 2010). The Clericuzio type was first described among Navajos.
5.6 Barth’s Disease (OMIM Code 302060)
This X-linked disease causes dilated cardiomyopathy as well as endomyocardial fibrosis (resulting in an early death in a few cases), myopathy, and moderate or severe neutropenia, which occasionally cause serious infections. An acidopathy involving a number of organic acids, including the 3-methylglutaconic acid, may also be observed. This syndrome is linked to G4-5 gene mutations coding for the tafazzin protein, which contributes to the phospholipid membrane homeostasis (Donadieu et al. 2011).
References
Ambruso DR, McCabe ER, Anderson DC et al (2003) Infectious and bleeding complications in patients with glycogen Ib. Am J Dis Child 139:691–697
Ancliff PJ, Blundell MP, Cory GO et al (2006) Two novel activating mutations in the Wiskott-Aldrich syndrome protein result in congenital neutropenia. Blood 108:2182–2189
Aprikyan A, Liles W, Park J et al (2000) Myelocatexis, a congenital disorder of severe neutropenia characterized by accelerated apoptosis and defective expression of bcl-x in neutrophil precursors. Blood 95:320–327
Armistead PM, Wieder E, Akande O et al (2010) Cyclic neutropenia associated with T cell immunity to granulocyte proteases and a double de novo mutation in GFI1, a transcriptional regulator of ELANE. Br J Haematol 150:716–719
Badolato R, Fontana S, Notarangelo LD, Savoldi G (2004) Congenital neutropenia: advances in diagnosis and treatment. Curr Opin Allergy Clin Immunol 4:513–521
Beel K, Cotter MM, Blatny J et al (2009) A large kindred with X-linked neutropenia with an I294T mutation of the Wiskott-Aldrich syndrome gene. Br J Haematol 144:120–126
Boztug K, Welte K, Zeidler C, Klein C (2008) Congenital neutropenia syndromes. Immunol Allergy Clin North Am 28:259–275
Boztug K, Appaswamy G, Ashikov A et al (2009) A syndrome with congenital neutropenia and mutations in G6PC3. N Engl J Med 360:32–43
Carlsson G, Fasth A (2001) Infantile genetic agranulocytosis, morbus Kostmann: presentation of six cases from the original “Kostmann family” and a review. Acta Paediatr 90:757–764
Dale DC, Hammond WP (1988) Cyclic neutropenia: a clinical review. Blood Rev 2:178–185
Dale DC, Person RE, Bolyard AA et al (2000) Mutation in the gene encoding neutrophil elastase in congenital and cyclic neutropenia. Blood 96:2317–2322
Dale DC, Cottle TE, Fier CJ, Bolyard AA, Bonilla MA, Boxer LA, Cham B, Freedman MH, Kannourakis G, Kinsey SE et al (2003) Severe chronic neutropenia: treatment and follow-up of patients in the Severe Chronic Neutropenia International Registry. Am J Hematol 72:82–93
Dell’Angelica EC, Shotelersuk V, Aguilar RC et al (1999) Altered trafficking of lysosomal proteins in Hermansky-Pudlak syndrome due to mutations in the beta 3A subunit of the AP-3 adaptor. Mol Cell 3:11–21
Devriendt K, Kim AS, Mathijs G et al (2001) Constitutively activating mutation in WASP causes X-linked severe congenital neutropenia. Nat Genet 27:313–317
Donadieu J, Leblanc T, Bader Meunier B, French Severe Chronic Neutropenia Study Group et al (2005) Analysis of risk factors for myelodysplasias, leukemias and death from infection among patients with congenital neutropenia. Experience of the French Severe Chronic Neutropenia Study Group. Haematologica 90:45–53
Donadieu J, Fenneteau O, Beaupain B, Mahlaoui N, Chantelot CB (2011) Congenital neutropenia: diagnosis, molecular bases and patient management. Orphanet J Rare Dis 6:26
Dong F, Brynes RK, Tidow N, Welte K, Lowenberg B, Touw IP (1995) Mutations in the gene for the granulocyte colony-stimulating-factor receptor in patients with acute myeloid leukemia preceded by severe congenital neutropenia. N Engl J Med 333:487–493
Dror Y, Freedman MH (2002) Shwachman-Diamond syndrome. Br J Haematol 118:701–713
Fioredda F, Iacobelli S, van Biezen A, Gaspar B, Ancliff P, Donadieu J et al (2015) Stem cell transplantation in severe congenital neutropenia: an analysis from the European Society for Blood and Marrow Transplantation. Blood 126:1885–1892
Freedman MH, Bonilla MA, Fier C et al (2000) Myelodisplasia syndrome and acute myeloid leukemia in patients with congenital neutropenia receiving G-CSF therapy. Blood 96:429–436
Gerin I, Veiga-da-Cunha M, Achouri Y et al (1997) Sequence of a putative glucose 6- phosphate translocase, mutated in glycogen storage disease type Ib. FEBS Lett 419:235–238
Germeshausen M, Ballmaier M, Welte K (2001) Implications of mutations in hematopoietic growth factor receptor genes in congenital cytopenias. Ann N Y Acad Sci 938:305–320
Germeshausen M, Schulze H, Kratz C et al (2005) An acquired G-CSF receptor mutation results in increased proliferation of CMML cells from a patient with severe congenital neutropenia. Leukemia 19:611–617
Germeshausen M, Grudzien M, Zeidler C, Abdollahpour H, Yetgin S, Rezaei N et al (2008) Novel HAX1 mutations in patients with severe congenital neutropenia reveal isoform-dependent genotype-phenotype associations. Blood 111:4954–4957
Germeshausen M, Deerberg S, Peter Y, Reimer C, Kratz CP, Ballmaier M (2013) The spectrum of ELANE mutations and their implications in severe congenital and cyclic neutropenia. Hum Mutat 34:905–914
Gorlin RJ, Gelb B, Diaz GA et al (2000) WHIM syndrome, an autosomal dominant disorder: clinical, hematological, and molecular studies. Am J Med Genet 91:368–376
Han J, Goldstein LA, Hou W et al (2010) Deregulation of mitochondrial membrane potential by mitochondrial insertion of granzyme B and direct Hax-1 cleavage. J Biol Chem 285:22461–22472
Horwitz MS, Duan Z, Korkmaz B et al (2007) Neutrophil elastase in cyclic and severe congenital neutropenia. Blood 109:1817–1824
Karsunky H, Zeng H, Schmidt T, Zevnik B, Kluge R, Schmid KW, Dührsen U, Möröy T (2002) Inflammatory reactions and severe neutropenia in mice lacking the transcriptional repressor Gfi1. Nat Genet 30:295–300
Klein C, Grudzien M, Appaswamy G, Germeshausen M, Sandrock I, Schäffer AA et al (2007) HAX1 deficiency causes autosomal recessive severe congenital neutropenia (Kostmann disease). Nat Genet 39:86–92
Kostman R (1975) Infantile genetic agranulocytosis. A review with presentation of ten new cases. Acta Paediatr Scand 64:362–368
Kostmann R (1956) Infantile genetic agranulocytosis (agranulocytosis infantilis hereditaria): a new recessive lethal disease in man. Almqvist and Wiksells Boktryckeri, Uppsala
Lekstrom-Himes JA, Gallin JI (2000) Immunodeficiency diseases caused by defects in phagocytes. N Engl J Med 343:1703–1714
Moulding DA, Blundell MP, Spiller DG et al (2007) Unregulated actin polymerization by WASp causes defects of mitosis and cytokinesis in X-linked neutropenia. J Exp Med 204:2213–2224
Notarangelo LD, Miao CH, Ochs HD (2008) Wiskott-Aldrich syndrome. Curr Opin Hematol 15:30–36
Notarangelo LD, Savoldi G, Cavagnini S, Bennato V, Vasile S, Pilotta A, Plebani A, Porta F (2014) Severe congenital neutropenia due to G6PC3 deficiency: early and delayed phenotype in two patients with two novel mutations. Ital J Pediatr 40:80
Paley C, Murphy S, Karayalcin G et al (1991) Treatmemt of neutropenia in Shwachman-Diamond syndrome (SDS) with recombinant human granulocyte colony-stimulating factor (RH-GCSF). Blood 78:3a
Person RE, Li FQ, Duan Z, Benson KF, Wechsler J, Papadaki HA, Eliopoulos G, Kaufman C, Bertolone SJ, Nakamoto B et al (2003) Mutations in proto-oncogene GFI1 cause human neutropenia and target ELA2. Nat Genet 34:308–312
Rosenberg PS, Alter BP, Bolyard AA et al (2006) The incidence of leukemia and mortality from sepsis in patients with severe congenital neutropenia receiving long-term G-CSF therapy. Blood 107:4628–4635
Rosenberg PS, Alter BP, Link DC et al (2008) Neutrophil elastase mutations and risk of leukaemia in severe congenital neutropenia. Br J Haematol 140:210–213
Rosenberg PS, Zeidler C, Bolyard AA et al (2010) Stable long-term risk of leukaemia in patients with severe congenital neutropenia maintained on G-CSF therapy. Br J Haematol 150:196–199
Thrasher AJ, Burns SO (2010) WASP: a key immunological multitasker. Nat Rev Immunol 10:182–192
van der Meer LT, Jansen JH, van der Reijden BA (2010) Gfi1 and Gfi1b: key regulators of hematopoiesis. Leukemia 24:1834–1843
Volpi L, Roversi G, Colombo EA et al (2010) Targeted next-generation sequencing appoints c16orf57 as clericuziotype poikiloderma with neutropenia gene. Am J Hum Genet 86:72–76
Welte K, Zeidler C, Dale DC (2006) Severe congenital neutropenia. Semin Hematol 43:189–195
Westerberg LS, Meelu P, Baptista M et al (2010) Activating WASP mutations associated with X-linked neutropenia result in enhanced actin polymerization, altered cytoskeletal responses, and genomic instability in lymphocytes. J Exp Med 207:1145–1152
Xia J, Bolyard AA, Rodger E et al (2009) Prevalence of mutations in ELANE, GFI1, HAX1, SBDS, WAS and G6PC3 in patients with severe congenital neutropenia. Br J Haematol 147:535–542
Zeidler C, Schwinzer B, Welte K (2003) Congenital neutropenias. Rev Clin Exp Hematol 7:72–83
Zhuang D, Qiu Y, Kogan SC, Dong F (2006) Increased CCAAT enhancer-binding protein epsilon (C/EBPepsilon) expression and premature apoptosis in myeloid cells expressing Gfi-1 N382S mutant associated with severe congenital neutropenia. J Biol Chem 281:10745–10751
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this entry

Cite this entry
Chirico, G., D’Ippolito, C. (2018). Neonatal Hereditary Neutropenia. In: Buonocore, G., Bracci, R., Weindling, M. (eds) Neonatology. Springer, Cham. https://doi.org/10.1007/978-3-319-29489-6_244
Download citation
DOI: https://doi.org/10.1007/978-3-319-29489-6_244
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-29487-2
Online ISBN: 978-3-319-29489-6
eBook Packages: MedicineReference Module Medicine