
Abstract
Cell enumeration using the hemocytometer is applicable when determining the number of cells in a suspension, and when the number of samples to be analyzed is relatively small. Hemocytometry is also useful for determining the proportion of singly dispersed cells in a suspension, and for estimating the frequency of viable cells.
1 Hemocytometry and Determination of Number of Viable Cells by Dye Exclusion
Cell enumeration using the hemocytometer is applicable when determining the number of cells in a suspension, and when the number of samples to be analyzed is relatively small. Hemocytometry is also useful for determining the proportion of singly dispersed cells in a suspension, and for estimating the frequency of viable cells.
To prepare a cell suspension, it is important to base dilution on the number of viable cells, rather than on the total number of cells. The number of viable cells in any given cell suspension varies, depending on the age and species of the animal from which the cells are isolated, the type of tissue, and the procedure for cell disaggregation. In suspensions prepared from cell cultures, the number of viable cells present depends on whether the cells in the culture were in the logarithmic or stationary phase of growth.
The average error in counting cells, using the hemocytometer, approaches 15–20% but it may be kept as low as 5–8%. Errors inherent in cell enumeration by this method are caused by inadequate suspension of cells, inaccurate dilution, overfilling the hemocytomter chambers, too few or too many cells in the sample to be counted, and inaccurate counting. The optimal number of cells to be counted is 1 × 105 cells/mL.
1.1 Determination of Number of Viable Cells by Dye Exclusion
-
1.
Materials:
-
Trypan blue (0.4%) or nigrosin (0.3%).
Note: Caution should be used with trypan blue, because it is a cancer suspect agent and teratogen.
-
Test tube (1).
-
Capillary or Pasteur pipet.
-
Serological pipets, sterile, 1-mL.
-
Improved Neubauer hemocytometer and coverslip.
-
Ethanol, 95%.
-
Soft lint-free gauze or tissue.
Preparation for counting cells:
-
a.
Cleaning the hemocytometer:
-
i.
Using a lint-free gauze or tissue, clean the surfaces of the counting chamber of the hemocytometer and the coverglass with water. Do not scratch the counting surfaces.
-
ii.
Repeat the cleaning, using 95% ethanol. Completely dry surfaces and coverglass.
-
i.
-
b.
Mount the coverslip over the two chambers (Fig. 1).
Note: To ensure accuracy in the depth of the chamber, it is advisable to moisten the edges of the coverslip, then gently slip the coverslip over the chamber. If the cover-slip is properly placed, an interference pattern (rainbow color rings) should appear where the coverslip touches the hemocytometer.
-
c.
Prepare an aliquot of the cell sample for counting cells:
-
i.
Add 0.2 mL nigrosin to a test tube.
-
ii.
Mix the cell suspension thoroughly by triturating, and immediately add 0.8 mL cell suspension to a test tube with nigrosine.
Note: Smaller amounts can be used, but the proportion of nigrosine to cell suspension must be maintained.
-
iii.
Mix the contents of the tube by gentle agitation by hand.
Note: If the cells remain in the dye solution too long, the cells may settle out, or may be injured. Such suspensions will give wrong estimates of viable cell numbers.
-
iv.
Gently mix the cell suspension with the pipet, and immediately fill a capillary pipet or tip of a Pasteur pipet.
-
i.
-
d.
Fill both halves of the counting chamber: Place the tip of the pipet on the edge of the hemocytometer chamber, being careful not to move the coverglass, and allow the cell suspension to fill the chamber by capillary action. The rate of flow can be regulated by placing a finger over the top of the pipet. A micropipet can also be used to fill the chamber (approx 20 µL cell suspension is necessary to fill one chamber). Fill the other half of the chamber.
Note: This step should be done quickly to avoid settling of the cells in the pipet, which would cause uneven distribution of cells in the hemocytometer.
Note: Be careful not to overfill the chambers, because this will cause counting errors. If the chamber is filled improperly, clean the hemocytometer and coverglass and repeat the procedure.
-
-
3.
Counting viable and dead cells: Using a 10× microscope objective, count all unstained cells (viable cells) and all stained cells (nonviable cells) in the four large corner squares in both counting chambers (Fig. 1).
Note: Trypan blue and nigrosin are vital dyes, and they do not penetrate the cell membrane. However, the cell membrane of injured or dead cells is permeable, and the dyes enter the cells and stain them. Therefore, in the dye exclusion test, the viable cells are unstained and nonviable cells are stained.
-
4.
Determining the cell number:
-
a.
The hemocytometer is divided into nine 1-mm2 (large) squares (Fig. 1). The height of the chamber is 0.1 mm, and the volume over one large corner square is 0.1 mm3 (0.0001 mL). Using the 10× microscope objective, count all cells in the four large (1-mm2) corner squares in both halves of the counting chamber (total of eight large corner squares). Count cells that touch the outer left and upper lines (if the chamber has triple outside lines, count the cells that touch the middle of the three outside lines on these two sides), and disregard those touching the right and lower outer lines.
-
b.
If the cell counts in the two chambers differ by more than 20% of the mean, clean and refill the chambers, and repeat the counts.
-
c.
Remove the coverslip, and place both the coverslip and the hemocytometer into a container of distilled water. Clean and dry the hemocytometer and coverslip.
-
a.

Calculation of cell number by using the hemocytometer and the dye exclusion method. One large corner square of the hemocytometer is enlarged, to illustrate the counting of cells. Count all cells within the area bounded by the triple lines, and, in addition, count the cells that touch the middle of the outer triple boundary lines on the top and on the left side (open circles). Do not count cells that touch the middle outer boundary lines on the bottom and right side of the square, nor any on the other sides that do not touch the middle outer boundary line (filled circles).
1.2 Calculation of Cell Number
-
1.
Add up the number of viable and nonviable cells in the eight large squares, and divide by eight to obtain the mean number of the cells in one large corner square.
-
2.
Multiply the mean number of cells by 10 to obtain the number of cells in 1 mm3 (0.001 mL).
-
3.
Multiply by 1000 to convert from 1 mm3 (0.001 mL) to 1 mL.
-
4.
Finally, to determine the number of cells in 1 mL of the original cell suspension, multiply by the factor of cell dilution used to prepare the cell-dye suspension. In the procedure described, the dilution factor is 10:8.
-
5.
Equation for calculating total number of cells/mL:
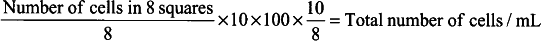
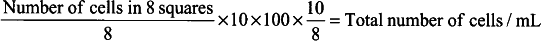
1.3 Calculation of Frequency of Viable Cells
-
1.
The percentage of viable cells in suspension is a good indicator of the effect of treatment on the viability of cells and variability in the procedures used, and is useful in standardizing procedures.
-
2.
Equation for calculating frequency of viable cells:
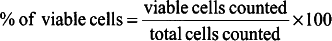
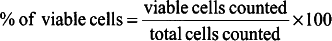
2 Electronic Cell Counter
Counters operating on the gating principle, introduced by Coulter in 1956, have an aperture through which electric current flows. Cells suspended in an electrolyte solution are drawn in a measured amount by vacuum through the aperture. When a cell passes through the aperture, the flow of current through the aperture changes because of increased resistance, causing a voltage pulse. This voltage pulse is directly proportional to the volume (size) of the cell. Therefore, when cells pass through the aperture, there is a voltage pulse for each cell. The pulses are recorded on a counter, and the relationship between the number and the size of the cells can be visualized on the oscilloscope.
Interpretation of oscilloscope patterns can be of valuable assistance in setting up the counter to determine aperture current and amplification settings, to detect blockage of the aperture, and to get an overview of distribution of the cells in the sample. An upper and lower threshold can be set in such a way that no particles smaller than the lower threshold settings, and no particles larger than the upper threshold settings, are counted. This excludes counting cell debris or cell clumps.
The electronic cell counter is particularly useful when a large number of samples must be counted. The procedure is fast, the counts are reproducible, and, when operated properly, yield single counts with an error of no more than 10% and multiple counts with an error as low as 1–2%. As mentioned, in addition, counters are capable of providing information about the size of cells. For accurate cell enumeration, optimum instrument settings should be determined for each cell type, the aperture should be examined for blockage, the cell suspension should be examined for cell debris and cell aggregates that may be indiscriminately counted, and sampling errors, resulting from improper mixing, should be eliminated. Most electronic cell counters cannot distinguish between viable and nonviable cells, and therefore the percentage of viable cells is best estimated using the dye exclusion technique and hemocytometry.
3 Counting Cell Nuclei in Cultures
It is often of great advantage to be able to estimate the number of cells in a culture, especially to determine how many cells are present in a colony or a clone in colony cultures. The Hoechst 33258 fluorescent stain (Sigma, cat. no. B 1155) is a benzimidazole derivative that intercalates in the A–T region of DNA, and makes cell nuclei visible. Staining cells with Hoechst 33258 is rapid, easy, and gives sensitivity of 10 ng/mL of intact double-stranded DNA. An additional advantage is that the Hoechst 33258 fluorochrome is readily distinguishable from fluorescein and rhodamine. Therefore, the cells can be double- or triple-immunostained, in addition to the Hoechst staining, thereby allowing determination of the frequency of different cell types in the culture. Hoechst 33258 exhibits fluorescent excitation at 350–365 nm, and emission over 450 nm. If the cultures are densely populated, it is possible to count only predetermined areas in the culture, then to extrapolate the number obtained to the whole culture.
Hoechst 33258 can also be used to detect contamination in cultures. In noncontaminated cultures, only cell nuclei are stained. Cells infected with mycoplasma have punctate or filamentous staining in the cytoplasm or adsorbed to the cell membrane. Bacteria and fungi also stain brightly, and are easy to recognize. Care should be taken that cell nuclear debris are not confused with the contaminating organisms.
3.1 Procedure
The dye is nontoxic to cells, except in the presence of light, when the cells have incorporated 5-bromodeoxyuridine (see Chapter 25).
-
1.
Materials:
-
Hoechst 33258 working solution of 1 µg/mL.
-
Dulbecco’s phosphate-buffered saline (DPBS) (Gibco-BRL, cat. No. 21600-010).
Procedure:
-
a.
Staining with Hoechst 33258 dye can be done alone or in conjunction with other immunocytochemical procedures that use probes labeled with fluorescein, rhodamine, or Texas red. When used in conjunction with other immunostains, use Hoechst staining as the last step.
-
b.
Stain cells with Hoechst 33258 dye for 5 min at room temperature.
-
c.
Wash cells twice with DPBS for 5 min.
-
d.
Mount and observe cultures with the fluorescent microscope, equipped with excitation filter 350–365 nm and emission filter 450 nm.
-
-
3.
Solutions:
-
a.
Hoechst 33258 stock solution.
Note: Hoechst 33258 is toxic. Wear rubber gloves, and make up solution under a fume hood.
-
i.
Dissolve 1 mg Hoechst 33258 in 10 mL DPBS. This stock solution of 100 µg/mL is 100× more concentrated than the working solution.
-
ii.
The bottle of stock solution should be wrapped in aluminum foil and stored in the dark at 0–5°C. The solution is stable for approx 2 wk under these conditions. The stock solution may also be frozen in aliquots at −20°C. At this temperature, the stock may be stored indefinitely.
-
i.
-
b.
Hoechst 33258 working solution: Dilute 0.1 mL Hoechst 33258 stock with 9.9 mL DPBS. This gives a working solution of 1 µg/mL.
-
a.
4 Growth Assays
The following growth assay is rapid, sensitive, reproducible, avoids the use of enzymes, and makes possible the processing of large numbers of cultures. A known number of cells (as a single-cell suspension) are plated in multiwell culture plates, and the agent to be tested is added in serial dilutions. The effects on growth or cytotoxicity effects can be evaluated by direct determinations in cell cultures, and values obtained are read from standard curves. Precautions should be taken to avoid increased evaporation in edge wells in multiwell plates during incubation (see Chapter 20).
4.1 [3H]-Thymidine Incorporation Assay
Tritiated thymidine is incorporated into cells that are synthesizing DNA during the S phase of the cell cycle. This assay therefore measures the degree of DNA synthesis that has occurred during the time the cells have been incubated with tritiated thymidine. The assay does not determine the number of cells present in the culture at the end of the experiment.
-
1.
Materials:
-
96-well plate containing cells at a suitable stage of growth.
-
[3H]-thymidine solution.
-
Scintillation solution.
-
Scintillation vials.
-
Multichannel pipet.
-
Absorbent glass-filter paper.
-
Cell harvester.
Procedure:
-
a.
Add 20 µL [3H]-thymidine solution to each well of the multiwell plate, using a multichannel pipet.
-
b.
Incubate the multiwell plate at 37°C for 4 h.
-
c.
Harvesting of cells:
-
i.
Nonadherent cells: Nonadherent cells may be harvested directly onto absorbent glass-filter paper, using a cell harvester.
-
ii.
Adherent cells: Add 100 µL 0.1% Triton X-100 to each well of the culture plate, gently agitate the multiwell plate for 2 min, and harvest onto absorbent glass-filter paper, using a cell harvester.
-
i.
-
d.
Dry the absorbent glass-filter paper containing the cells for 30 min in a drying oven (or air-dry overnight).
-
e.
Cut circles containing the cells from the glass-filter paper. Place each circle of glass-filter paper into a scintillation vial.
-
f.
Add 1 mL liquid scintillation solution.
-
g.
Measure the radioactivity of each sample by using a scintillation counter.
-
3.
[3H]-thymidine solution:
-
a.
Stock solution: 1 µCi [3H]-thymidine (New England Nuclear, Boston, MA) with a specific activity of 20 Ci/mmol.
-
b.
Dilute stock solution 1:19 in culture medium for use.
Note: It may be necessary to increase the amount of [3H]-thymidine when using medium that contains thymidine as a nutrient.
-
a.
-
5 Other Assays
This chapter describes four ways to enumerate cell numbers in culture: hemocytometry, electron cell counting, fluorescent staining of cells in cultures, and [3H]-thymidine incorporation. Additional assays are described in this volume. Chapter 15 describes the use of aggregate neural cell cultures in biochemical and molecular biological assays; Chapter 20 deals with the use of multiple well plates in biological assays; Chapter 21 deals with cell number determination in microplate culture, and Chapter 23, the use of colony cultures for determination of plating efficiency.
Further Reading
Bainbridge, D. R. and Macey, M. M. (1983), Hoechst 33258: a fluorescent nuclear counterstain suitable for double-labelling immunofluorescence. J. Immunol. Methods 62, 193–195.
Baserga, R. (1989), Measuring parameters of growth, in Cell Growth and Division, Baserga, R., ed., IRL, New York.
Branch, D. R. and Guilbert, L. J. (1986), Practical in vitro assay systems for the measurement of hematopoietic growth factors J. Tissue Culture Methods 10, 101–108.
Hamilton, L. H. (1956), Errors in blood cell counting. 1. Technical errors. 2. Statistical errors. Can. J. Med. Tech. 18, 8–14.
Hanks, J. H. and Wallace, R. E. (1958), Determination of cell viability. Proc. Soc. Exp. Biol Med. 98, 188–192.
Harris, M. (1959), Growth measurements on monolayer cultures with an electronic cell counter. Can. Res. 19, 1020–1024.
Jones, K. H. and Senft, J. A. (1985), An improved method to determine cell viability by simultaneous staining with fluorescein diacetate propidium iodide. J. Hist. Cytol. 33, 77–79.
Kaltenbach, J. P., Kaltenbach, M. H., and Lyons, W. B. (1958), Nigrosin as a dye for differentiating live and dead ascites cells. Exp. Cell Res. 15, 112–117.
Latt, S. A. and Stetten, G. (1976), Spectral studies on 33258 Hoechst and related bisbenzimidazole dyes useful for fluorescent detection of deoxyribonucleic acid synthesis. J. Histochem. Cytochem. 24, 24–33.
Phillips, H. J. and Terryberry, J. E. (1957), Counting actively metabolizing tissue cultured cells. Exp. Cell Res. 13, 341–347.
Schrek, R. (1944), Studies in vitro on the physiology of normal and of cancerous cells. II. The survival and the glycolysis of cells under anaerobic conditions. Arch. Pathol. 37, 319–327.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2001 Humana Press Inc., Totowa, NJ
About this protocol
Cite this protocol
Richardson, A., Fedoroff, S. (2001). Quantification of Cells in Culture. In: Fedoroff, S., Richardson, A. (eds) Protocols for Neural Cell Culture. Springer Protocols Handbooks. Humana Press. https://doi.org/10.1385/1-59259-207-4:333
Download citation
DOI: https://doi.org/10.1385/1-59259-207-4:333
Publisher Name: Humana Press
Print ISBN: 978-0-89603-902-5
Online ISBN: 978-1-59259-207-4
eBook Packages: Springer Book Archive