
Abstract
RNA interference (RNAi) is an evolutionarily conserved mechanism of gene silencing induced by double-stranded RNAs (dsRNAs). Among the widely used dsRNAs, small interfering RNAs (siRNAs) and short hairpin RNAs have evolved as extremely powerful and the most popular gene silencing reagents. The key challenge to achieving efficient gene silencing especially for the purpose of therapeutics is mainly dependent on the effectiveness and specificity of the selected RNAi-targeted sequences. Practically, only a small number of dsRNAs are capable of inducing highly effective and sequence-specific gene silencing via RNAi mechanism. In addition, the efficiency of gene silencing induced by dsRNAs can only be experimentally examined based on inhibition of the target gene expression. Therefore, it is essential to develop a fully robust and comparative validation system for measuring the efficacy of designed dsRNAs. In this chapter, we focus our discussion on a reliable and quantitative reporter-based siRNA validation system that has been previously established in our laboratory. The system consists of a short synthetic DNA fragment containing an RNAi-targeted sequence of interest and two expression vectors for targeting reporter and triggering siRNA expressions. The efficiency of siRNAs is determined by their abilities to inhibit expression of the targeting reporters with easily quantified readouts including enhanced green fluorescence protein and firefly luciferase. Since only a readily available short synthetic DNA fragment is needed for constructing this reliable and efficient reporter-based siRNA validation system, this system not only provides a powerful strategy for screening highly effective RNAi-targeted sequences from mammalian genes but also implicates the use of RNAi-based dsRNA reagents for reverse functional genomics and molecular therapeutics.
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others


Key words
- Gene silencing
- RNA interference (RNAi)
- Small interfering RNA (siRNA)
- Short hairpin RNA (shRNA)
- pDual
- Enhanced green fluorescence protein (EGFP)
- Firefly luciferase (Fluc)
- siRNA validation system
Introduction
RNA interference (RNAi) is an evolutionarily conserved mechanism of posttranscriptional gene silencing induced by double-stranded RNAs (dsRNAs) that mediate sequence-specific cleavage of the cognate RNA transcripts and in turn trigger complete degradation of the disruptive RNA fragments, resulting in reduction or loss of gene activities [1, 2]. During the processes of RNAi-mediated gene silencing, the dsRNAs are first recognized and cleaved into 21- to 23-nucleotide (nt) small interfering RNA (siRNA) duplexes with symmetrical 2-nt 3′ overhangs by dsRNA-specific RNase III-related endonuclease, Dicer [3, 4]. The produced siRNAs are efficiently incorporated into RNA-induced silencing complex (RISC) to form a ribonucleoprotein complex that first unwinds the siRNA duplexes and selectively degrades the sense strand of siRNA. The single antisense siRNA-coupled RISC is afterwards guided to catalyze the endonucleolytic cleavage of homologous RNA transcripts at the site where the antisense siRNAs is complementarily bound [5, 6]. Subsequently, the resulting cleaved RNA fragments are immediately subjected to exonucleolytic destruction by the action of exoribonuclease.
Originally, RNAi was identified as involved in the response to exogenous pathogenic and endogenous parasitic nucleic acids [7, 8] and also as participating in the basic cellular functions, including gene regulation and heterochromatin formation [9–11]. Recently, it has become an extremely powerful study tool for reverse functional genomics [12–15] and a remarkably potent strategy for gene silencing-based therapeutics [16, 17]. Compared to other gene silencing reagents, such as antisense oligonucleotides (ODNs), ribozymes, and DNAzymes, dsRNAs including siRNAs and short hairpin RNAs (shRNAs) have apparently become the most powerful and widely used gene silencing reagents for manipulating gene activity in mammalians [18]. There are mainly two approaches in generating active dsRNAs in mammalian cells by exogenous delivery of synthetic siRNAs [19, 20] or shRNAs [21] and endogenous vector-expressed shRNAs [22–25] or siRNAs formed by annealing two complementary sense and antisense RNAs [26–28].
A large number of studies have shown that not all of the RNAi-targeted sequences selected from a target gene exhibit the same efficiency in inhibiting gene expression. Practically, only a small number of siRNAs are capable of inducing highly effective gene silencing in a sequence-specific manner [29]. Moreover, the silencing efficiency of siRNAs is dependent on the specificity of the target sequences within a gene and can only be measured experimentally based on the inhibition of the target gene expression. In order to select functional and effective siRNAs, it is necessary to design, synthesize, and screen many distinct siRNAs, which is expensive due to the cost of chemical synthesis of RNA oligonucleotides. Several previous studies have suggested that the secondary structures of mRNA and mRNA-binding proteins might interfere with the target site accessibility for RISC; therefore, the rational design strategy for selecting effective siRNAs is not fully programmable [30–33]. In addition, extensive large-scale and systematic analyses of the siRNA-specific features revealed that siRNA might have sequence-specific characteristics associated with its functionality [29, 34–37]. Thus, these studies indicate that the efficacy of siRNA is not totally secondary structure dependent and strongly suggest that the sequence properties of siRNA may play the major and most important role in determining inhibition efficiency.
The key challenge in achieving highly effective gene silencing, particularly for the purpose of the therapeutics, is mainly dependent on the effectiveness and specificity of the RNAi-targeted sequence. Previously, the effective siRNAs are identified on the basis of their abilities to inhibit the expression of cognate sequences in an ectopically expressed target gene-reporter fusion chimeric mRNA [38]. The described target gene-reporter-based siRNA validation system totally depends on the availability of cDNA clones, and this may limit the high-throughput application of the method. Besides, the chimeric mRNA of target gene-reporter fusion construct may encode an impaired fusion protein that exhibits a low reporter activity, which interferes with the screening and identification of effective siRNAs. Recently, a reporter-based siRNA validation system has been published in which the validating system is constructed by fusing a short synthetic DNA fragment containing an RNAi-targeted sequence, instead of cDNA, with a reporter gene [39, 40]. However, to generate the corresponding triggering siRNA, it is necessary to have either a synthetic siRNA (or shRNA) or another synthetic DNA fragment for constructing siRNA (or shRNA) expression vector. Thus, this system is inefficient and cost intensive, especially for large-scale studies.
To facilitate large-scale functional genomics in mammals, especially for novel genes, it is important to have a rational design strategy for selecting potentially effective siRNAs and measure the efficacy of the designed siRNAs by a simple and fully robust validating system. In this chapter, we focus our discussion on a reliable and quantitative reporter-based siRNA validation system that has been developed in our laboratory for functional screening and identification of effective RNAi-targeted sequences in mammalian genes [41]. In this system, only a short synthetic DNA fragment is needed to construct both the targeting reporter and triggering siRNA expression vectors. Because only a readily available short synthetic DNA fragment is needed to construct both the targeting reporter and triggering siRNA expression vectors, the protocols described in this chapter provide a novel system that not only greatly facilitates large-scale loss-of-function genetic screens in mammalian cells but also provides the basis for an improved approach to screen and identify the most potent siRNA for therapeutic purposes.
Materials
Cell Culture
-
1.
Mammalian cell lines of interest obtained from the American Type Culture Collection (Manassas, VA, USA) and stored in liquid nitrogen or at −80 °C freezer.
-
2.
Cell line-specific growth media supplemented with or without the heat-inactivated various percentages of fetal calf serum (FCS) and 1% antibiotic/antimycotic solution and stored at 4 °C refrigerator.
-
3.
Phosphate-buffered saline (PBS) stored at room temperature.
-
4.
Trypsin solution (0.25%) and ethylenediamine tetraacetic acid (EDTA, 1 mM) stored in aliquots at −30 °C freezer.
-
5.
Cell scrapers and spatulas (Techno Plastic Products AG, Trasadingen, Switzerland).
Plasmid Vectors
-
1.
pEGFP-3′UTR, pEGFP-3′UTR-PGK, pFluc-3′UTR, pFluc-3′UTR-PGK, pDual, pDual-PGK, pDual-siEGFP, pDual-siFluc, pHsH1, pHsH1-PGK, pHsH1-shEGFP, and pHsH1-shFluc expression vectors (see Fig. 3; Note 1) stored in aliquots at −30 °C freezer.
Fig. 1 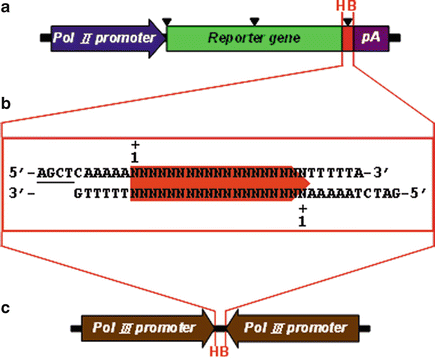
Strategy and experimental design for screening effective RNAi-targeted sequences using reporter-based siRNA validation system. This reporter-based siRNA validation system is composed of a short synthetic DNA fragment (b) and two expression vectors for targeting reporter (a) and triggering siRNA (c) expression. The short synthetic DNA fragment contains a unique RNAi-targeted sequence with 19 nt in length and two specific restriction enzyme-compatible ends HindIII and BglII for cloning into the HindIII/BglII-digested targeting reporter and triggering siRNA expression vectors simultaneously. The restriction enzyme-compatible ends of HindIII (5′-AGCT) and BglII (5′-GATC) are underlined. The targeting reporter vector not only contains the reporter gene expression cassette driven by RNA Pol II promoter but also includes two unique restriction enzyme sites, HindIII (H) and BglII (B), for cloning of the RNAi-targeted sequence at the 5′- or 3′-UTR or insertion within the reporter gene without disrupting its activity. The positions for inserting the RNAi-targeted sequence are marked with black triangles. The triggering siRNA expression vector contains two convergent RNA Pol III promoters to drive the expression of both the sense and antisense strands of siRNA, respectively. The short synthetic DNA fragment is also cloned into the HindIII (H) and BglII (B) sites in triggering siRNA expression vector
Fig. 2 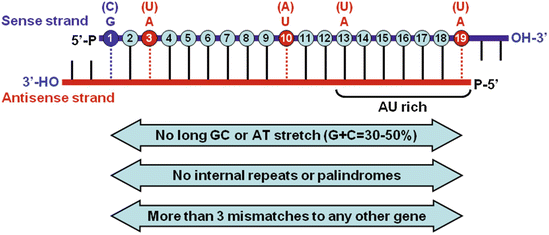
Sequence-specific features for the selection and design of effective siRNAs. The mature siRNA is a 21- to 23-nt dsRNA that contains a 19-nt duplexed region, symmetrical 2-nt 3′ overhangs, and 5′-phosphate (P) and 3′-hydroxyl (OH) groups. The positions of each nucleotide in the 19-nt duplexed region of the sense strand are numbered. On the basis of currently established selection and design rules, an effective siRNA has high stability at the 5′ terminus of the sense strand and lower stability at the 5′ antisense terminus and at the cleavage site. In addition, the sequence-specific preferences at the following positions on the sense strand are important including the presence of a G (or C) at position 1 and an A (or U) at positions 3, 10, 13, and 19
Fig. 3 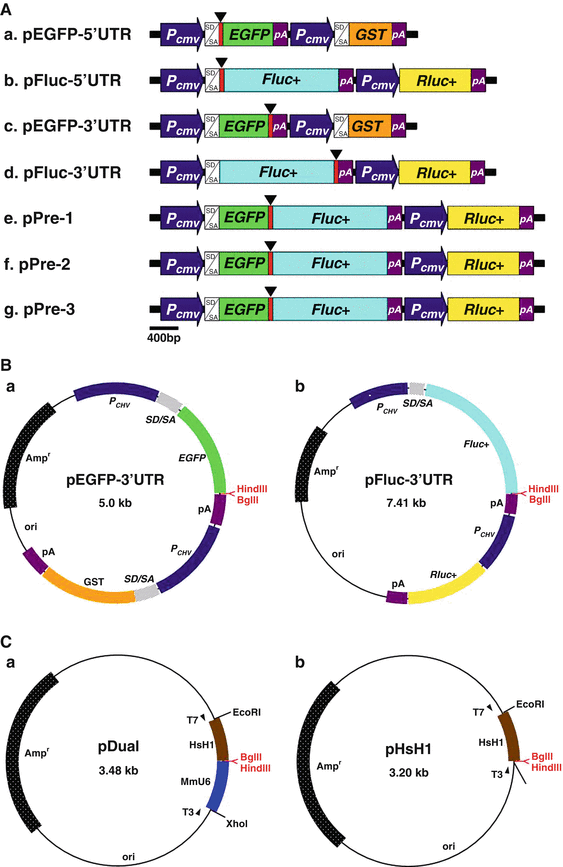
Design and construction of targeting reporter and triggering siRNA expression vectors. (a) Constructs of targeting reporter expression vectors. The vectors contain two independent expression cassettes for expression of the targeting reporter and reference protein as well as two unique restriction enzyme sites, HindIII and BglII, located either at the 5′-UTR (a and b) or the 3′-UTR (c and d) of the reporter gene or within a chimeric fusion reporter gene (e, f, and g) for cloning the RNAi-targeted sequence. The unique restriction enzyme sites, HindIII and BglII, for inserting the RNAi-targeted sequence are in red. pEGFP-5′UTR (a) and pEGFP-3′UTR (c) contain the EGFP as targeting reporter and GST as reference protein. pFluc-5′UTR (b) and pFluc-3′UTR (d) contain the firefly luciferase (Fluc+) as targeting reporter and Renilla luciferase (Rluc+) as reference protein. Fig. 3 (continued) pPre-1 (e), -2(f), and -3 (g) vectors contain the chimeric EGFP–Fluc fusion protein as targeting reporter and Rluc as reference protein, as well as they possess three different reading frame sequences between EGFP and Fluc+ fused sites for inserting RNAi-targeted sequences with different reading frames. (b) Selective constructs of targeting reporter expression vectors used in this siRNA validation system. (c) Constructs of triggering siRNA and shRNA expression vectors. The siRNA expression plasmid pDual (a) contains two convergent RNA Pol III promoters, mouse U6 (MmU6) and human H1 (HsH1), to drive the expression of both the sense and antisense strands of the siRNA, respectively. The shRNA expression plasmid pHsH1 (b) contains only the human H1 (HsH1) promoter to drive the transcription of the consecutive sequence of the sense, a loop, and the antisense regions
Fig. 4 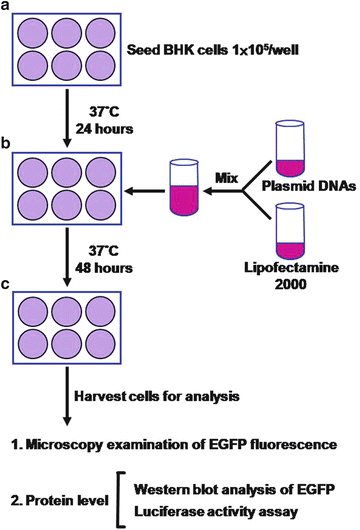
Experimental procedures for assessing the inhibition efficiency of shRNA expression constructs. (a) Seeding of targeting BHK cell line. 24 h before transfection, the targeting cell line is subcultured and plated into 6-well culture plate at 1 × 105 cells per well. (b) Co-transfection of targeting reporter and triggering siRNA expression constructs. The cultured cells are co-transfected with 0.5 μg of targeting reporter expression construct and 1.5 μg of trigger siRNA expression construct by using Lipofectamine 2000 according to the manufacturer’s instructions. (c) Assessment of inhibition efficiency of triggering siRNA against targeting reporter. After 48-h incubation, the EGFP-expressed cells are first examined under an inverted fluorescence microscopy and then harvested and lysed for protein-level analysis by using Western blot. In addition, the luciferase-expressed cells are harvested and lysed for protein-level analysis by using functional reporter assay (luciferase activity)
-
2.
Competent cells of Escherichia coli strain XL 1-blue (Stratagene, La Jolla, CA, USA) stored in aliquots at −80 °C freezer.
-
3.
Luria-Bertani (LB) broth stored at room temperature.
-
4.
Ampicillin stock solution (100 mg/mL) stored in aliquots at −30 °C freezer.
-
5.
BglII and HindIII restriction enzymes (Promega, Madison, WI, USA) as well as T4 DNA ligase (Promega) stored at −30 °C freezer.
-
6.
Agarose gel stored at room temperature.
-
7.
Tris–acetate (TAE) stock solution (50×) stored at room temperature.
-
8.
Gel-loading buffer (6×): 0.25% bromophenol blue, 0.25% xylene cyanol FF, 15% Ficoll type 400; stored at room temperature.
-
9.
Plasmid Mini and Maxi Purification Kits (Viogene, Sunnyvale, CA, USA) as well as Gel extraction (Viogene) stored at room temperature.
-
10.
Phenol/chloroform/isoamyl alcohol (25/24/1) and chloroform/isoamyl alcohol (24/1) stored at 4 °C and room temperature, respectively.
-
11.
Sodium acetate (3 M, pH 4.8) stored at room temperature.
-
12.
Ethanol (100 and 70%, vol/vol) stored at −30 °C freezer.
-
13.
TE: 10 mM Tris–HCl, pH 8.0 and 1 mM EDTA; stored at room temperature.




Transfection and Functional Assessments
-
1.
Lipofectamine 2000™ (Invitrogen, Carlsbad, CA, USA) stored at 4 °C refrigerator.
-
2.
Protein lysis buffer: 50 mM NaCl, 50 mM Tris–HCl, pH 7.4, 2 mM EDTA, 0.5% sodium deoxycholate, 1% NP-40, and 0.1% SDS; stored at room temperature.
-
3.
Protease inhibitors (Roche Molecular Biochemicals, Mannheim, Germany) stored in aliquots at −80 °C freezer.
-
4.
Micro bicinchoninic acid (Micro BCA) assay (Pierce, Rockford, IL, USA) stored at room temperature.
-
5.
Bovine serum albumin (BSA; Sigma, St Louis, MO, USA) stored at room temperature.
-
6.
Dual-luciferase Reporter Assay System (Promega) stored in aliquots at −80 °C freezer.
-
7.
Enhanced chemiluminescence (ECL) Western blotting detection reagents (Amersham Biosciences, Arlington Heights, IL, USA) stored at 4 °C refrigerator.
Instruments
-
1.
Microcentrifuges.
-
2.
Dri-block heater (Techne DRI-BLOCK DB 20; Techne, Cambridge, UK).
-
3.
Handheld UV lamp (VL-4.L; Vilber Lourmat, Marne-la-Vallée, France).
-
4.
UV image system (UV illuminator; Vilber Lourmat).
-
5.
Spectrophotometer.
-
6.
Microplate reader (Dynatech MR5000; Dynatech Laboratories, Chantilly, VA, USA).
-
7.
Luminometer (MiniLumat LB 9506; EG&G Berthold, Wildbach, Germany).
-
8.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) apparatus (Mighty Small II 8 × 7 cm; Hoefer Scientific Instruments, San Francisco, CA, USA).
-
9.
Electrophoresis power supply.
-
10.
Semidry transfer apparatus (Semiphor Transphor Unit; Amersham Pharmacia Biotech).
-
11.
Inverted fluorescence microscopy (Olympus IX71; Olympus Co., Tokyo, Japan).
Methods
The methods described in this section outline (1) the strategy and experimental design for screening and identifying effective RNAi-targeted sequences, (2) the molecular characteristics of designed and selected RNAi-targeted sequences, (3) the construction of reporter-based siRNA validation system including targeting reporter and triggering siRNA expression vectors, (4) the functional assessment of designed and selected RNAi-targeted sequences in this reporter-based siRNA validation system, and (5) the application of this reporter-based siRNA validation system to identify highly effective novel siRNAs.
Strategy and Experimental Design for Screening and Identifying Effective RNAi-Targeted Sequences
To screen and identify the effective RNAi-targeted sequences more robustly and cost effectively, we have established a reliable and quantitative reporter-based siRNA validation system requiring only a short synthetic DNA fragment [41]. This system is composed of a short synthetic DNA fragment and two expression vectors for targeting reporter and triggering siRNA expressions (see Fig. 1). The short synthetic DNA fragment is generated by annealing two complementary sense- and antisense-oligonucleotides that contain a unique RNAi-targeted sequence with 19 nt in length flanked by five consecutive adenosine and thymidine residues (As and Ts) at the 5′ and 3′ ends as an efficient termination signal for transcription of the antisense and sense RNA, respectively (see Fig. 1b).
The unique RNAi-targeted sequence is selected and designed from protein-coding region or 3′-untranslated region (3′-UTR) of the target gene according to the sequence-specific characteristics of the effective siRNAs as described briefly in Subheading 1 and thoroughly in Subheading 3.2. To construct the targeting reporter vector, a short synthetic DNA fragment containing a unique RNAi-targeted sequence of interest is fused with a reporter gene at the 5′- and 3′-UTR or is inserted within the reporter gene without interfering in its activity (see Fig. 1a). To make the greatest value and utility of this short synthetic DNA fragment, simultaneously, it is also cloned into a specific triggering siRNA expression vector that contains two functional convergent RNA Pol III promoters (see Fig. 1c; Note 2).
Efficacy of the siRNAs is measured by their abilities to inhibit expression of the targeted reporter gene, which contains the corresponding short synthetic DNA fragment, with easily quantified readouts including EGFP or Fluc. In addition, to make this system complete, it is convenient to include an excellent in vitro cell model, which not only provides an easy cell culture system but also has high transfection efficiency. Because this system is used for screening the highly effective RNAi-targeted sequences directly against genes with a wide range of biological functions, it is better to perform the screening in a nonhuman and non-mouse cell line. To fit these criteria, the baby hamster kidney fibroblast BHK is chosen as an in vitro experimental model. In addition, the experimental procedures including cell culture conditions and transfection protocols have been optimized and standardized.
The Molecular Characteristics of Designed and Selected RNAi-Targeted Sequences
The efficiency of RNAi-induced gene silencing is mainly dependent on the effectiveness and specificity of the RNAi-targeted sequences. To gain the functionally effective siRNAs, it is necessary to design, synthesize, and screen a number of different RNAi-targeted sequences from a target gene. Large-scale and systematic analyses of the specific features from the effective siRNAs reveal that siRNA might have sequence-specific features associated with its functionality. These molecular characteristics generally include low-to-medium G/C content (30–50%), high internal stability at the sense strand 5′-terminus, low internal stability at the sense strand 3′-terminus, absence of internal repeats or palindromes, and base preferences at the sense strand positions 1 (G/C), 3, 10, 13, and 19 (A/U) (see Fig. 2) [29, 34–37].
-
1.
Retrieve the nucleotide sequence of target gene from the National Center for Biotechnology Information (NCBI) nucleotide database (GenBank) at
-
2.
Screen any 19-nt sequence (see Note 3) within the amino acid-coding region and 3′-UTR that fulfill the above sequence-specific characteristics and in particular do not contain stretches of four or more consecutive adenosines (As) or thymidines (Ts).
-
3.
Select any 19-nt sequence containing more than three mismatches to any other gene in the same species and also avoiding any known single-nucleotide polymorphism (SNP) by searching the non-redundant NCBI database at
http://www.ncbi.nlm.nih.gov/BLAST/
with the selected sequence.
-
4.
Choose particularly two to four 19-nt sequences with a G/C at the sense strand position 1 and an A/T at the sense strand positions 3, 10, 13, and 19.
-
5.
Design the sense and antisense oligonucleotides for siRNA: siGene-S:
5′-AGCTCAAAAANNNNNNNNNNNNNNNNNNNTTTTTA-3′
and siGene-AS:
5′-GATCTAAAAANNNNNNNNNNNNNNNNNNNTTTTTG-3′
(see Note 4).
-
6.
Design the sense and antisense oligonucleotides for shRNA: shGene-S:
5′-CGNNNNNNNNNNNNNNNNNNttcaagagaNNNNNNNNNNNNNNNNNNCTTTTTGGAAA-3′
and shGene-AS:
5′-AGCTTTTCCAAAAAGNNNNNNNNNNNNNNNNNNtctcttgaaNNNNNNNNNNNNNNNNNN-3′.
Construction of the Reporter-Based siRNA Validation System Including Targeting Reporter and Triggering siRNA Expression Vectors
This reporter-based siRNA validation system includes two expression vectors for targeting reporter and triggering siRNA expressions (see Fig. 1a, c). To fully normalize the transfection variation and accurately evaluate the efficacy of the RNAi-targeted sequences, the targeting reporter vectors all contain two independent expression cassettes for transcription of the targeting reporter (enhanced green fluorescence protein, EGFP, or firefly luciferase, Fluc) and reference protein (glutathione S-transferase, GST, or Renilla luciferase, Rluc) genes (see Fig. 3a). The easy and sensitive EGFP fluorescence detection or Fluc activity assay combined with well-documented and easily analyzed reference protein, GST or Rluc, provides a simple and reliable readout for the system. To simply and efficiently perform this reporter-based siRNA validation system, this method has only focused on the targeting reporter expression vectors, pEGFP-3′UTR and pFluc-3′UTR (see Fig. 3b), and the triggering siRNA and shRNA expression vectors, pDual and pHsH1 (see Fig. 3c). To enhance the convenience of constructing this reporter-based siRNA validation system and facilitate the screening of recombinant clones, all the vectors are further improved by inserting a stuffer of phosphoglycerate kinase (PGK) gene between the unique cloning sites, BglII and HindIII, which makes the preparation of the DNA vectors simple and easy by only removing the stuffer of PGK DNA fragment with BglII and HindIII double digestion (see Note 1).
To fully utilize the short synthetic DNA fragment for producing the triggering siRNA, we have constructed a particular siRNA expression vector, pDual (see Fig. 3c), which contains two convergent RNA Pol III promoters, mouse U6 and human H1, to drive the expression of both sense and antisense strands of siRNA using the short synthetic DNA fragment as template, respectively. In addition, to simply and efficiently clone the short synthetic DNA fragment containing the RNAi-targeted sequence of interest into this vector, the pDual vector also contains the same HindIII and BglII restriction enzyme sites located between mouse U6 and human H1 promoters in which the sense and antisense strands of siRNA are transcribed by U6 and H1 promoters, respectively. It has been shown that shRNA exhibits slightly better effect on the inhibition of gene expression as compared with that of the siRNA [21, 42]. To make this system complete, we have particularly used a highly effective shRNA expression vector, pHsH1 (see Fig. 3c) [41], which contains only the human H1 promoter to drive transcription of the consecutive sequence of the sense, a loop, and the antisense regions.
In addition, to objectively and accurately examine the efficiency of selected RNAi-targeted sequence-mediated inhibition of the targeting reporter gene expression in both the EGFP- and Fluc-based siRNA validation systems, it is important to have highly effective RNAi-targeted sequences directly against EGFP and Fluc expressions. For this purpose, we have particularly constructed four effective siRNA and shRNA expression vectors for silencing EGFP (pDual-siEGFP and pHsH1-shEGFP) and Fluc (pDual-siFluc and pHsH1-shFluc) expressions that could serve as references for positive controls [41]. These effective RNAi-targeted sequences directly target on the coding regions of EGFP and Fluc mRNA transcripts and exhibit strong inhibition effects with a silencing efficiency of more than 90% (see Table 1).
Preparation of the Targeting Reporter and Triggering siRNA Expression Vectors
-
1.
Digest 10 μg of pEGFP-3′UTR-PGK, pFluc-3′UTR-PGK, or pDual-PGK in a 1.5 mL Eppendorf tube in a reaction with 5 μL of 10× restriction enzyme buffer, 10 units of BglII and HindIII, and distilled H2O to total 50 μL in 37 °C water bath for 2 h (see Note 1).
-
2.
Analyze 1 μL of digested DNA mixtures on a 0.8% (wt/vol) agarose gel with an appropriate molecular weight marker.
-
3.
Inactivate the restriction enzymes by incubation on a 70 °C heat block for 10 min.
-
4.
Isolate the digested vector by using electrophoresis on a 0.8% (wt/vol) agarose gel.
-
5.
Recover the DNA fragment from the agarose gel by using the gel extraction kit, and elute the DNA fragment with 50 μL of TE (pH 8.0).
Preparation of the RNAi-Targeted Sequences (and siRNA Expression Templates)
-
1.
Mix 5 μL of the complementary oligonucleotides (100 μM) in a 1.5 mL Eppendorf tube in a reaction with 2 μL of 10× annealing buffer (T4 DNA ligase ligation buffer) and distilled H2O to total 20 μL (see Note 5).
-
2.
Place the Eppendorf tube in a 95 °C heat block for 10 min.
-
3.
Remove the Eppendorf tube from the heat block and allow to cool to room temperature on the bench.
-
4.
Spin down briefly the Eppendorf tube to recover the reaction solution and store on ice at 4 °C until ready to use (see Note 6).
Cloning of the Gene-Specific Targeting Reporter and Triggering siRNA Expression Vectors
-
1.
Mix 2 μL of BglII/HindIII-digested vectors and 8 μL of annealed RNAi-targeted sequences (or siRNA expression templates) in a 1.5 mL Eppendorf tube in a reaction with 2 μL of 10× ligation buffer and distilled H2O to total 19 μL.
-
2.
Add 1 μL of T4 DNA ligase.
-
3.
Incubate in 16 °C water bath overnight.
-
4.
Transform 200 μL of XL 1-blue competent cells with 20 μL of ligated mixtures.
-
5.
Plate on LB agar plates containing 100 μg/mL of ampicillin.
-
6.
Incubate in 37 °C incubator overnight.
Screening of the RNAi-Targeted Sequence and siRNA Expression Template-Positive Clones
-
1.
Inoculate four selected colonies into 3 mL LB broth containing 100 μg/mL of ampicillin (see Note 7).
-
2.
Incubate in 37 °C incubator overnight.
-
3.
Purify plasmid DNAs from 1.5 mL overnight culture by using plasmid mini purification kit, and elute the plasmid DNAs with 50 μL of TE (pH 8.0).
-
4.
Check isolated plasmid DNAs by single digestion with restriction enzyme BglII or HindIII. Digest 2 μL of purified plasmid DNA in a 1.5 mL Eppendorf tube in a reaction with 2 μL of 10× restriction enzyme buffer, 2 units of BglII or HindIII, and distilled H2O to total 20 μL in 37 °C water bath for 1 h.
-
5.
Analyze 10 μL of digested DNAs on a 0.8% (wt/vol) agarose gel with an appropriate molecular weight marker. The positive RNAi-targeted sequence and siRNA expression template clones containing restriction enzyme BglII site but usually losing restriction enzyme HindIII site are digested only with BglII and not with HindIII. Plasmids showing this restriction enzyme-digestion pattern are presumably correct and should be confirmed by direct sequencing.
-
6.
Sequence selected plasmid DNAs by using an automated DNA sequencer that uses the dideoxy sequencing method with fluorescent dyes. The oligonucleotides used for sequencing RNAi-targeted sequences in both targeting reporters pEGFP-3′UTR and pFluc-3′UTR and triggering siRNA and shRNA in pDual and pHsH1 are EGFP-primer:
5′-GCATCAAGGTGAACTTCAAGATC-3′,
Fluc-primer:
5′-GGCGAACTGTGTGTGAGAGGTCC-3′,
and HsH1-primer:
5′-GGGCCCAGTGTCACTAGGCGG-3′.
This sequencing can be carried out by an institutional core sequencing facility or a professional sequencing service.
Functional Assessment of Designed and Selected RNAi-Targeted Sequences in This Reporter-Based siRNA Validation System
Much evidence has already shown that not all of the RNAi-targeted sequences selected from a target gene display the same potencies on inducing gene silencing. Only a small number of siRNAs are capable of inducing highly efficient target gene silencing in a sequence-specific manner. The silencing efficacy of siRNAs is dependent on the specificity of the target sites within the gene and can only be determined experimentally based on the inhibition of the target gene expression. Several widely used approaches can be utilized to analyze the efficiency of gene silencing induced by DNA vector-based siRNA or shRNA expression, including (1) Northern blot, (2) quantitative reverse transcription (RT)-PCR or real-time RT-PCR, (3) Western blot, (4) immuno-staining, and (5) functional activity assay. In general, the effect of gene silencing can be detected 24–48 h after transfection, dependent on the abundance and the stability of the proteins encoded by the target genes (see Fig. 4).
Transfection of Targeting Reporter and Triggering siRNA Expression Vectors
-
1.
Subculture and plate 1 × 105 cells per well in 2 mL growth medium onto a 6-well culture plate at 24 h before transfection.
-
2.
Co-transfect 0.5 μg of targeting reporter and 1.5 μg of triggering siRNA expression vectors by using Lipofectamine 2000 following the manufacturer’s protocol.
-
3.
Incubate the transfected cells at 37 °C in a CO2 incubator for 48 h.
Examination of Transfected Cells Under an Inverted Fluorescence Microscopy
-
1.
Remove growth medium, and wash the transfected cells three times with PBS.
-
2.
Examine the expressed EGFP in cells by using inverted fluorescence microscopy.
-
3.
Confirm the expression levels of EGFP and GST in the total protein extracts by using Western blot analysis.
Preparation of Total Cell Lysates for Western Blot Analysis
-
1.
Remove growth medium, and wash the transfected cells three times with PBS.
-
2.
Harvest the transfected cells from the plate by using cell scrapers or spatulas into a 50 mL culture tube.
-
3.
Prepare total cell lysates from the transfected cells by using protein lysis buffer containing protease inhibitors.
-
4.
Perform Western blot analysis with specific antibodies for EGFP and GST according to standard protocols.
Preparation of Total Cell Lysates for Luciferase Activity Assay
-
1.
Remove growth medium, and wash the transfected cells three times with PBS.
-
2.
Harvest the transfected cells from the plate by using cell scrapers or spatulas into a 50 mL culture tube.
-
3.
Prepare total cell lysates from the transfected cells by using cell lysis buffer.
-
4.
Perform the dual-luciferase reporter assay system as described by the manufacturer.
Application of This Reporter-Based siRNA Validation System to Identify Highly Effective siRNAs
This system is composed of two expression vectors for targeting reporter and triggering siRNA expressions as well as two highly effective siRNAs, siEGFP and siFluc, which serve as references for positive controls [41]. As compared with the inhibition levels of these positive references, one could easily evaluate the efficacy of selected and designed RNAi-targeted sequences of interest.
There are many strategies to build the profile of a gene and its function. One of the best and simple ways to elucidate gene function is to disrupt or inhibit the gene and characterize the phenotype of resulting mutant. To efficiently apply RNAi technology for reverse functional genomics [12–15], in particular the novel or the putative genes with only available nucleotide sequences in databases but without cDNA clones in hand, one could simply identify the effective RNAi-targeted sequences directly against the novel or the putative genes by using this system. Once the highly effective RNAi-targeted sequences are identified, one could easily establish the specific gene knockdown in the in vitro cell lines or in vivo animal models that provide a loss-of-function mutation of the novel or the putative genes. Subsequently, many different molecular, cellular, biochemical, and other analyses could be performed to examine the inhibition effects on the in vitro or in vivo models.
siRNAs could be used clinically to inhibit gene expression as a therapeutic agent in many diseases characterized by elevated gene function. Inhibition of virus-specific genes by siRNAs has proven to be a potential therapeutic strategy against virus-induced diseases. A number of extremely virulent viruses including Ebola, Lassa, severe acute respiratory syndrome (SARS), avian influenza (H5N1), West Nile, and smallpox viruses are highly infectious and cause extraordinarily deadly diseases [43, 44]. Furthermore, there are currently no vaccines or effective therapies available, and in particular these viruses require special containment for safe research. To develop an extremely potent RNAi-based therapeutics for these virulent viruses with safety, this reporter-based siRNA validation system could provide a simple and powerful approach for screening and identification of highly effective siRNAs directly against viral gene expression without the need of direct virus culture.
Notes
-
1.
The main advantage of cloning procedures presented in this method is that preparation of the inserting vectors is simple and efficient by only double digestion with restriction enzymes BglII and HindIII to remove the stuffer of PGK gene from pEGFP-3′UTR-PGK, pFluc-3′UTR-PGK, pDual-PGK, and pHsH1-PGK vectors. This will largely increase the cloning efficiency to more than 75%.
-
2.
The siRNA validation systems described in this method are cost effective and convenient in that any annealed oligonucleotide duplexes can be simultaneously cloned into both targeting reporter and triggering siRNA expression vectors.
-
3.
The length of duplex region for a siRNA (shRNA) is relatively flexible from 19 to 29 nt. Although increasing the length of duplex region for a relatively ineffective 19-nt siRNA (shRNA) can increase its effectiveness, increasing the length of an effective 19-nt siRNA (shRNA) may not further improve the inhibition effect.
-
4.
The oligonucleotides used for constructing the systems can be purchased from any local commercial suppliers without any further modification or treatment.
-
5.
The annealing of two complementary oligonucleotides can be efficiently carried out in 1× T4 DNA ligase buffer, which can be obtained from any T4 DNA ligase commercial suppliers.
-
6.
The annealed oligonucleotide duplexes should not be phosphorylated before the ligation step, because it might result in multiple copies of insertion.
-
7.
Using this protocol for cloning the targeting reporter and triggering siRNA or shRNA expression cassettes is efficient and cost effective in that only four colonies are selected and screened for the positive clones containing the RNAi-targeted sequence, siRNA or shRNA expression sequence.
References
Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC (1998) Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391:806–811
Meister G, Tuschl T (2004) Mechanisms of gene silencing by double-stranded RNA. Nature 431:343–349
Bernstein E, Caudy AA, Hammond SM, Hannon GJ (2001) Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 409:363–366
Carmell MA, Hannon GJ (2004) RNase III enzymes and the initiation of gene silencing. Nat Struct Mol Biol 11:214–218
Martinez J, Patkaniowska A, Urlaub H, Luhrmann R, Tuschl T (2002) Single-stranded antisense siRNAs guide target RNA cleavage in RNAi. Cell 110:563–574
Schwarz DS, Hutvagner G, Haley B, Zamore PD (2002) Evidence that siRNAs function as guides, not primers, in the Drosophila and human RNAi pathways. Mol Cell 10:537–548
Schramke V, Allshire R (2003) Hairpin RNAs and retrotransposon LTRs effect RNAi and chromatin-based gene silencing. Science 301:1069–1074
Soifer HS, Zaragoza A, Peyvan M, Behlke MA, Rossi JJ (2005) A potential role for RNA interference in controlling the activity of the human LINE-1 retrotransposon. Nucleic Acids Res 33:846–856
Reinhart BJ, Slack FJ, Basson M et al (2000) The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 403:901–906
Volpe TA, Kidner C, Hall IM, Teng G, Grewal SI, Martienssen RA (2002) Regulation of heterochromatic silencing and histone H3 lysine-9 methylation by RNAi. Science 297:1833–1837
Matzke MA, Birchler JA (2005) RNAi-mediated pathways in the nucleus. Nat Rev Genet 6:24–35
Tuschl T (2003) Functional genomics: RNA sets the standard. Nature 421:220–221
Berns K, Hijmans EM, Mullenders J et al (2004) A large-scale RNAi screen in human cells identifies new components of the p53 pathway. Nature 428:431–437
Paddison PJ, Silva JM, Conklin DS et al (2004) A resource for large-scale RNA-interference-based screens in mammals. Nature 428:427–431
Silva J, Chang K, Hannon GJ, Rivas FV (2004) RNA-interference-based functional genomics in mammalian cells: reverse genetics coming of age. Oncogene 23:8401–8409
Shuey DJ, McCallus DE, Giordano T (2002) RNAi: gene-silencing in therapeutic intervention. Drug Discov Today 7:1040–1046
Hannon GJ, Rossi JJ (2004) Unlocking the potential of the human genome with RNA interference. Nature 431:371–378
Scherer LJ, Rossi JJ (2003) Approaches for the sequence-specific knockdown of mRNA. Nat Biotechnol 21:1457–1465
Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T (2001) Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411:494–498
Dorsett Y, Tuschl T (2004) siRNAs: applications in functional genomics and potential as therapeutics. Nat Rev Drug Discov 3:318–329
Siolas D, Lerner C, Burchard J et al (2005) Synthetic shRNA as potent RNAi triggers. Nat Biotechnol 23:227–231
Brummelkamp TR, Bernards R, Agami R (2002) A system for stable expression of short interfering RNAs in mammalian cells. Science 296:550–553
Sui G, Soohoo C, Affar EB, Gay F, Shi Y, Forrester WC, Shi Y (2002) A DNA vector-based RNAi technology to suppress gene expression in mammalian cells. Proc Natl Acad Sci USA 99:5515–5520
Paddison PJ, Caudy AA, Bernstein E, Hannon GJ, Conklin DS (2002) Short hairpin RNAs (shRNAs) induce sequence-specific silencing in mammalian cells. Genes Dev 16:948–958
Paul CP, Good PD, Winer I, Engelke DR (2002) Effective expression of small interfering RNA in human cells. Nat Biotechnol 20:505–508
Lee NS, Dohjima T, Bauer G et al (2002) Expression of small interfering RNAs targeted against HIV-1 rev transcripts in human cells. Nat Biotechnol 20:500–505
Miyagishi M, Taira K (2002) U6 promoter-driven siRNAs with four uridine 3′ overhangs efficiently suppress targeted gene expression in mammalian cells. Nat Biotechnol 20:497–500
Zheng L, Liu J, Batalov S, Zhou D, Orth A, Ding S, Schultz PG (2004) An approach to genomewide screens of expressed small interfering RNAs in mammalian cells. Proc Natl Acad Sci USA 101:135–140
Reynolds A, Leake D, Boese Q, Scaringe S, Marshall WS, Khvorova A (2004) Rational siRNA design for RNA interference. Nat Biotechnol 22:326–330
Holen T, Amarzguioui M, Wiiger MT, Babaie E, Prydz H (2002) Positional effects of short interfering RNAs targeting the human coagulation trigger tissue factor. Nucleic Acids Res 30:1757–1766
Harborth J, Elbashir SM, Vandenburgh K, Manninga H, Scaringe SA, Weber K, Tuschl T (2003) Sequence, chemical, and structural variation of small interfering RNAs and short hairpin RNAs and the effect on mammalian gene silencing. Antisense Nucleic Acid Drug Dev 13:83–105
Kretschmer-Kazemi Far R, Sczakiel G (2003) The activity of siRNA in mammalian cells is related to structural target accessibility: a comparison with antisense oligonucleotides. Nucleic Acids Res 31:4417–4424
Heale BSE, Soifer HS, Bowers C, Rossi JJ (2005) siRNA target site secondary structure predictions using local stable substructures. Nucleic Acids Res 33:e30
Khvorova A, Reynolds A, Jayasena SD (2003) Functional siRNAs and miRNAs exhibit strand bias. Cell 115:209–216
Schwarz DS, Hutvagner G, Du T, Xu Z, Aronin N, Zamore PD (2003) Asymmetry in the assembly of the RNAi enzyme complex. Cell 115:199–208
Mittal V (2004) Improving the efficiency of RNA interference in mammals. Nat Rev Genet 5:355–365
Ui-Tei K, Naito Y, Takahashi F et al (2004) Guidelines for the selection of highly effective siRNA sequences for mammalian and chick RNA interference. Nucleic Acids Res 32:936–948
Kumar R, Conklin DS, Mittal V (2003) High-throughput selection of effective RNAi probes for gene silencing. Genome Res 13:2333–2340
Du Q, Thonberg H, Zhang HY, Wahlestedt C, Liang Z (2004) Validating siRNA using a reporter made from synthetic DNA oligonucleotides. Biochem Biophys Res Commun 325:243–249
Smart N, Scambler PJ, Riley PR (2005) A rapid and sensitive assay for quantification of siRNA efficiency and specificity. Biol Proced Online 7:1–7
Hung C-F, Lu K-C, Cheng T-L, Wu R-H, Huang L-Y, Teng C-F, Chang W-T (2006) A novel siRNA validation system for functional screening and identification of effective RNAi probes in mammalian cells. Biochem Biophys Res Commun 346:707–720
Wu M-T, Wu R-H, Hung C-F, Cheng T-L, Tsai W-H, Chang W-T (2005) Simple and efficient DNA vector-based RNAi systems in mammalian cells. Biochem Biophys Res Commun 330:53–59
Kuiken T, Fouchier R, Rimmelzwaan G, Osterhaus A (2003) Emerging viral infections in a rapidly changing world. Curr Opin Biotechnol 14:641–646
Morens DM, Folkers GK, Fauci AS (2004) The challenge of emerging and re-emerging infectious diseases. Nature 430:242–249
Acknowledgments
This work was supported by grants from the National Science Council of Taiwan, ROC (to Wen-Tsan Chang).
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media, LLC
About this protocol
Cite this protocol
Tsai, WH., Chang, WT. (2014). Construction of Simple and Efficient siRNA Validation Systems for Screening and Identification of Effective RNAi-Targeted Sequences from Mammalian Genes. In: Ochs, M. (eds) Gene Function Analysis. Methods in Molecular Biology, vol 1101. Humana Press, Totowa, NJ. https://doi.org/10.1007/978-1-62703-721-1_15
Download citation
DOI: https://doi.org/10.1007/978-1-62703-721-1_15
Published:
Publisher Name: Humana Press, Totowa, NJ
Print ISBN: 978-1-62703-720-4
Online ISBN: 978-1-62703-721-1
eBook Packages: Springer Protocols