
Abstract
An important goal in the development of highly functional organic materials is to design self-assembling molecules that can reproducibly display chemical signals across length scales. Within the biomedical field, biomolecules are highly attractive candidates to serve as bioactive building blocks for the next generation of biomaterials. The peptide amphiphiles (PAs) developed by the Stupp Laboratory at Northwestern University generated a highly versatile self-assembly code to create well-defined bioactive nanofibers that have been proven to be very effective at signaling cells in vitro and in vivo. Here, we describe the basic steps necessary for synthesis and assembly of PA molecules into functional nanostructures.
Similar content being viewed by others


Key words
1 Introduction
Regenerative therapies are of enormous interest given their potential to increase quality of life in the world’s population (1–4). Therefore, bioactive materials that can trigger or control biological processes could become important components of regenerative therapies. One promising approach is the development of bioactive molecules to form nanostructures that can interact specifically and reproducibly with cell receptors and proteins to control processes such as cell survival, cell proliferation, cell differentiation, and de-differentiation in the context of tissue and organ regeneration. These biomolecules can be designed not only to signal cells but also to self-assemble into well-defined supramolecular structures and materials. Furthermore, an attractive feature of these self-assembling systems is the possibility to deliver them through minimally invasive procedures, for example, by injecting them as dissolved molecules in aqueous media that rapidly form at the targeted tissues the signaling structures through programmed self-assembly. A number of research groups are currently taking advantage of the increased understanding of molecular self-assembly and nanoscience to develop bioactive, biomimetic, and multifunctional materials for regenerative medicine (5–13). The Stupp Laboratory at Northwestern developed a very broad class of peptide amphiphiles (PAs) capable of forming bioactive nanoscale filaments that mimic those in extracellular matrices. These filaments can display in tuneable densities peptide signals that promote regenerative processes (5, 6, 14–31). These molecules are composed of a peptide segment and a hydrophobic segment, typically an alkyl tail that provides a key driving force for self-assembly through hydrophobic collapse in water. Since an alkyl tail is always more hydrophobic than any peptide segment, it promotes the formation of nanostructures in water that display on their surfaces the peptide segments. The peptide segment contains at least two domains, one designed to create β-sheets and a terminal domain that is displayed on the nanostructure’s surface containing the bioactive signal. The switch for self-assembly is electrolyte screening of charges that are introduced in the sequence if they are not present in the bioactive domain. The self-assembly process involves the formation of hydrogen bonding among PA molecules in the shape of long fibers and not spherical micelles as a result of β-sheet formation. The fibrous nanostructures are much more bioactive than nanospheres displaying a similar density of biological signals (32). In addition to bioactivity, the PA nanoscale fibers designed in the Stupp Laboratory are both biocompatible and biodegradable (33), and have been shown to have in vivo efficacy in neuronal regeneration (21), blood vessel regeneration (16), cartilage regeneration (29), bone regeneration (28), and bone marrow derived cell delivery (27). This chapter summarizes the key steps that are used to fabricate these PA molecules using Fmoc solid-phase peptide synthesis.
2 Materials
2.1 Method 1: PAs with the Alkyl Tail Located at the N Terminus
-
1.
Preloaded Wang resin (Novabiochem, EMD Chemicals, Darmstadt, Germany).
-
2.
N,N-Dimethylformamide (DMF).
-
3.
Dichloromethane.
-
4.
N,N-Diisopropylethylamine (DIEA), redistilled.
-
5.
Fmoc-protected amino acids (Novabiochem, EMD Chemicals, Darmstadt, Germany).
-
6.
2-(1H-benzotriazole-1-yl) -1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU).
-
7.
Piperidine, redistilled.
-
8.
Fluorenylmethoxycarbonyl-O-(benzylphospho)-serine (Fmoc-Ser(PO3Bnzl)).
-
9.
1-(Mesitylene-2-sulfonyl)-3-nitro-1H-1,2,4-triazole.
-
10.
Fatty acid (e.g., palmitic acid).
-
11.
Ninhydrin (2,2-dihydroxyindane-1,3-dione).
-
12.
Diethyl ether.
-
13.
Dithiothreitol (DTT).
-
14.
Triflouroacetic acid (TFA).
-
15.
Triisopropylsilane (TIPS).
-
16.
Ethanedithiol.
-
17.
Water (deionized with a Millipore Milli-Q water purifier, resistance of 18 MW).
-
18.
Automated peptide synthesizer (CS Bio Peptide Synthesizer, Menlo Park, CA).
-
19.
Ultrasonicator (Branson 2510 bath sonicator, Danbury, CT).
-
20.
Nuclear magnetic resonance (NMR) spectrometer (Varian Mercury 400 MHz, Palo Alto, CA).
-
21.
Reverse-phase high-performance liquid chromatography (RP-HPLC) equipped with a Waters Atlantis C18 column (Agilent HP 1050 system, Santa Clara, CA).
-
22.
Electrospray ionization (ESI) mass spectrometer (MSD, Agilent, Santa Clara, CA).
-
23.
Matrix-assisted laser desorption/ionization (MALDI-TOF) (Apex III, Bruker, Bremen, Germany).
-
24.
Rotary evaporator (RE-11 Rotovapor with a Welch 2025 dry vacuum pump, Büchi Laboretechnik, Postfach, Switzerland).
-
25.
Lyophilizer (Labconco Freezone 6, Kansas City, MO).
2.2 Method 2: PAs with the Alkyl Tail Located at the C Terminus
-
1.
N-Carbobenzyloxy-l-aspartic anhydride.
-
2.
Dichloromethane.
-
3.
Dodecylamine.
-
4.
Triethylamine.
-
5.
Ethanol.
-
6.
Dioxane.
-
7.
Hydrochloric acid.
-
8.
Chloroform.
-
9.
Magnesium sulfate.
-
10.
Celite.
-
11.
Fmoc-O-succinimide (Fmoc-OSu).
-
12.
Rink resin.
-
13.
Piperidine.
-
14.
2-(1H-benzotriazole-1-yl) -1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU).
3 Methods
3.1 Method 1: PAs with the Alkyl Tail Located at the N Terminus
Figure 1 illustrates a PA molecule synthesized with the following protocol (24).

Chemical structure of a PA molecule containing the alkyl tail on the N terminus. Reprinted with permission from ref. 34. Copyright 2005, American Chemical Society.
-
1.
PAs are prepared on a 0.25-mmol scale by using standard fluorenylmethoxycarbonyl (Fmoc) chemistry using Wang resin pre-loaded with the first (C terminal) amino acid (see Note 1). The Wang resin should be swelled by soaking in dichloromethane for 30 min prior to use. The synthesis proceeds either by manual synthesis in a peptide synthesis vessel (e.g., ChemGlass Part #CG-1860) or on an automated peptide synthesizer.
-
2.
The Fmoc group is cleaved with a 30% solution of piperidine in DMF for 10 min and the beads washed thoroughly with DMF. If using manual synthesis, several beads can be removed and the Kaiser (ninhydrin) test, which is used to detect ammonia or primary and secondary amines. The deprotection reaction is repeated until the ninhydrin test is positive.
-
3.
The next amino acid is then activated by dissolving 4 equivalents of the amino acid, 3.95 equivalents 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU), and 6 equivalents of DIEA in DMF (see Note 2). The activated ester solution is added to the reaction vessel and the reaction proceeds for 2 h. A Kaiser test should give a negative result when the coupling is complete.
-
4.
The deprotect–coupling procedure is repeated to form the full-length peptide sequences.
-
5.
After the peptide portion of the molecule is prepared, the Wang resin is removed from the automated synthesizer and transferred to a peptide synthesis vessel.
-
6.
The N terminus is capped with a fatty acid containing 6, 10, 16, or 22 carbon atoms. The alkylation reaction is accomplished by using 8 equivalents of the fatty acid, 8 equivalents HBTU, and 12 equivalents of DIEA in enough dichloromethane and DMF to fully dissolve the acid.
-
7.
The acylation reaction is allowed to proceed for 6 h. Complete reaction is indicated by a negative Kaiser test.
-
8.
Cleavage and deprotection of the PAs is achieved with a mixture of triflouroacetic acid (TFA), water, and triisopropylsilane in a ratio of 91:2.5:2.5 for 3 h at room temperature. For PAs containing cysteine, a mixture of TFA, water, triisopropylsilane, and ethanedithiol in a ratio of 91:3:3:3 is used.
-
9.
The cleavage mixture and two subsequent dichloromethane washings are filtered into a round-bottom flask.
-
10.
The solution is concentrated in vacuo by rotary evaporation to afford a viscous solution.
-
11.
This solution is precipitated with cold diethyl ether.
-
12.
The white precipitate is collected by filtration, washed with copious cold ether, and dried under vacuum.
-
13.
The purity of the crude material is determined by analytical reverse-phase high-performance liquid chromatography (RP-HPLC) equipped with a Waters Atlantis C18 column (5 μm particle size, 150 × 4.6 mm or 250 × 4.6 mm) (see Note 3). The crude residue is then dissolved and purified by preparative reverse-phase HPLC with a Waters Atlantis C18 preparative column (5 μm particle size, 250 × 30.0 mm) (see Note 4).
-
14.
The appropriate fractions are then collected, concentrated in vacuo by rotary evaporation to remove the organic solvent, followed by freezing with liquid nitrogen and lyophilization to give a fluffy white solid (see Notes 5 and 6). The identity of the purified material is confirmed by ESI and MALDI-TOF mass spectrometers (see Note 7).
3.2 Method 2: PAs with the Alkyl Tail Located at the C Terminus
Figure 2 illustrates a PA molecule synthesized with the following protocol (34).

Chemical structure of a PA molecule containing the alkyl tail on the C terminus. Reprinted with permission from ref. 34. Copyright 2005, American Chemical Society.
-
1.
The first step consists of synthesizing the molecule illustrated in Fig. 3
Fig. 3. 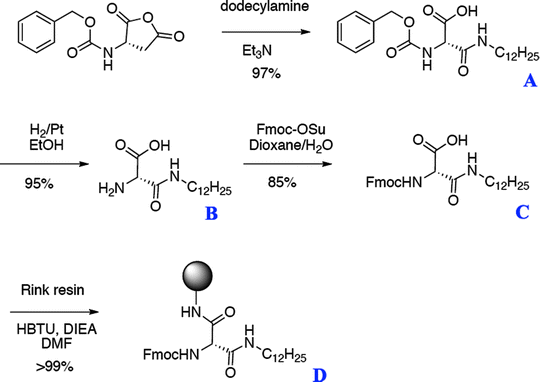
Steps to synthesize the fatty acid amino acid D, which serves as the first amino acid in the synthesis of PAs with the alkyl tail at the N terminus. Reprinted with permission from ref. 34. Copyright 2005, American Chemical Society.
Product A (N-Dodecyl-2-carbobenzyloxyaminosuccinamic acid):
-
(a)
N-Carbobenzyloxy-l-aspartic anhydride (1 mmol) is dissolved in 50 mL of dichloromethane.
-
(b)
Addition of 1.05 equivalents of dodecylamine and 1.1 equivalents of triethylamine.
-
(c)
The reaction is sealed with a tight cap to prevent evaporation, and stirred for 12 h until no trace of starting material is detected by thin-layer chromatography (TLC) (5% MeOH in CH2Cl2).
-
(d)
The reaction is quenched with 20 mL of 1 M hydrochloric acid followed by extraction with chloroform (5×).
-
(e)
The organic layer is dried over magnesium sulfate, and product A is obtained as a white solid (yield 97%).
Product B (2-Amino-N-dodecylsuccinamic acid):
-
(a)
In 100 mL of ethanol, 100 mmol of product A are dissolved and transferred to a reaction vessel containing palladium on carbon (10 wt%).
-
(b)
The vessel is then placed under hydrogen (35 Torr) for 3 h. The reaction mixture is filtered over Celite, and the product is obtained as a white solid after evaporation to dryness under reduced pressure (yield 95%).
Product C (N-Dodecyl-2-Fmoc-aminosuccinamic acid):
-
(a)
In 200 mL of a water/dioxane (1:1 v/v) mixture, 6.6 mmol of product B are dissolved with 1.3 mL (1.5 equivalents) of triethylamine and 1 equivalent of Fmoc-O-succinimide (Fmoc-OSu).
-
(b)
The reaction is monitored by TLC (10% MeOH in CH2Cl2) and after 2–3 h all of the Fmoc-OSu is consumed.
-
(c)
The reaction is quenched with acid, resulting in a white precipitate (product C) that is collected by filtration (yield 85%).
Product D:
-
(a)
The standard Rink resin is placed in a reaction vessel, deprotected three times with 30% piperidine in NMP.
-
(b)
The resin is then coupled with 2 equivalents of product C overnight using HBTU as a coupling reagent.
-
(c)
The coupling is repeated until a ninhyndrin test is negative.
-
(a)
-
2.
The amino acids are added to product D using standard Fmoc solid-phase techniques on the automated synthesizer to give the full-length peptide.

3.3 Method 3: Self-Assembly of Peptide Amphiphiles
These molecules self-assemble into nanofibers with no detectable critical micelle concentration (cmc). Electrostatic screening of the charged residues with salt or by changing the pH results in greater bundling of the nanofibers and is observed macroscopically as gelation (Fig. 4) (see Note 8).

Electrostatic screening of the PA molecules leads to self-assembly into well-defined nanofibers and subsequent gel formation (6).
-
1.
Gelation of peptide amphiphiles with basic residues
-
(a)
The solid PA is dissolved in water to the desired concentration (typically about 1 wt%). Ultrasonication (Branson 2510 bath sonicator, Danbury, CT) or gentle heating may be required for complete dissolution (see Note 9).
-
(b)
Gelation is induced by lowering the pH by introducing aqueous HCl or by exposing the solution to HCl vapors in a sealed container for approximately 10 min (see Note 10). Gelation is indicated in samples that are self-supporting upon inversion of the container.
-
(a)
-
2.
Gelation of peptide amphiphiles with acidic residues
-
(a)
The solid PA is dissolved in water to the desired concentration (typically on the order of 1 wt%). Sonication or gentle heating may be required for complete dissolution.
-
(b)
Gelation is induced by raising the pH by introducing aqueous NaOH or by exposing the solution to ammonia vapors in a sealed container for approximately 10 min (see Notes 10 and 11).
-
(a)
4 Notes
-
1.
All reactions described in Subheadings 3.1– 3.3 are conducted at room temperature.
-
2.
Using an excess of HBTU can result in capping of the N terminus (34).
-
3.
These peptide amphiphile molecules can be purified by HPLC using either water–acetonitrile or water–methanol solvent gradients. However, the self-assembly behavior after purification is most consistent using water–acetonitrile.
-
4.
In some cases, the peptide amphiphile can be difficult to solubilize, adding a small amount of a co-solvent like methanol or hexafluoroisopropanol (HFIP) can be used to aid solubility. Furthermore, using a horn sonicator can also be quite useful.
-
5.
The solid material after lyophilization is very low density. It is recommended that static electricity be minimized to avoid loss of material or weighing errors. An antistatic gun is very useful for this purpose.
-
6.
The lyophilized material can be stored in a freezer at −20°C for many months without any significant degradation.
-
7.
The preferred matrix for MALDI of these compounds is α-cyano-4-hydroxycinnamic acid.
-
8.
If the samples will be used for cell studies, then the trifluoracetate counterions should be exchanged for chloride. Typically, 600 mg of PA is dissolved in 600 mL of 0.01 M HCl and the sample is immediately frozen immediately and then lyophilized. Cold packs placed beside the samples can help to minimize melting. The concentration of residual TFA can be determined by fluorine-19 NMR (Varian Mercury 400 MHz, Palo Alto, CA) in methanol-d4 with trifluoroethanol, hexafluorobenzene, or 4-trifluoromethylacetanilide as an external standard.
-
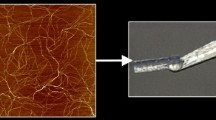
9.
Lower PA concentrations also result in nanofiber formation, but gelation may not be observed macroscopically. The nanofiber formation can be observed by transmission electron microscopy or atomic force microscopy.
-
10.
Gelation can be induced by adding aqueous NaOH 1 μL at a time (for positively charged PAs) or adding aqueous HCl 1 μL at a time (for negatively charged PAs) until a self-supporting material is observed.
-
11.
For sequences with acidic residues, gelation can also be induced by addition of CaCl2 (0.5 molar equivalents of Ca2+ per carboxylic acid) (35).
References
Huebsch, N., Mooney, D. (2009) Inspiration and application in the evolution of biomaterials Nature 462, 426–432.
Stevens, M.M., George, J.H. (2005) Exploring and engineering the cell surface interface Science 310, 1135–1138.
Furth, M. E., Atala, A. (2008) Current and future perspectives of regenerative medicine. Principles of Regenerative Medicine. Burlington MA: Elsevier.
Stupp, S. I. (2005) Biomaterials for regenerative medicine MRS Bulletin 30, 546–553.
Hartgerink, J.D., Beniash, E., Stupp, S. I. (2001) Self-assembly and mineralization of peptide-amphiphile nanofibers Science 294, 1684–1688.
Hartgerink, J.D., Beniash, E., Stupp, S.I. (2002) Peptide-amphiphile nanofibers: A versatile scaffold for the preparation of self-assembling materials Proceedings of the National Academy of Sciences of the United States of America 99, 5133–5138.
Paramanov, S.E., Gauba, V., Hartgerink, J.D. (2005) Synthesis of collagen-like peptide polymers by native chemical ligation Macromolecules 38, 7555–7561.
Betre, H., Setton, L.A., Meyer, D.E., Chilkoti, A. (2002) Characterization of a genetically engineered elastin-like polypeptide for cartilaginous tissue repair Biomacromolecules 3, 910–916.
Zhao, X., Zhang, S. (2007) Designer self-assembling peptide materials Macromolecular Biosciences 7, 13–22.
Aggeli, A., Bell, M., Carrick, L.M., Fishwick, C.W.G., Harding, R., Mawer, P.J., Radford, S.E., Strong, A.E., Boden, N. (2003) pH as a trigger of peptide b-sheet self-assembly and reversible switching between nematic and isotropic phases Journal of the American Chemical Society 125, 9619–9628.
Williams, R.J., Smith, A.M., Collins, R., Hodson, N., Das, A.K., Ulijn, R.V. (2009) Enzyme-assisted self-assembly under thermodynamic control Nature Nanotechnology 4, 19–24.
Schneider, J.P., Pochan, D.J., Ozbas, B., Rajagopal, K., Pakstis, L., Kretsinger, J. (2002) Responsive hydrogels from the intramolecular folding and self-assembly of a designed peptide Journal of the American Chemical Society 124, 15030–15037.
Zhang, S., Holmes, T., Lockshin, C., Rich, A. (1993) Spontaneous assembly of a self-complementary oligopeptide to form a stable macroscopic membrane Proceeding of the National Academy of Sciences of the United States of America 90, 3334–3338.
Silva, G.A., Czeisler, C., Niece, K.L., Beniash, E., Harrington, D.A., Kessler, J. A., Stupp, S. I. (2004) Selective differentiation of neural progenitor cells by high-epitope density nanofibers Science 303(5662), 1352–1355.
Guler, M.O., Soukasene, S., Hulvat, J.F., Stupp, S.I. (2005) Presentation and recognition of biotin on nanofibers formed by branched peptide amphiphiles. Nano Letters 5(2), 249–252.
Rajangam, K., Behanna, H.A., Hui, M.J., Han, X., Hulvat, J.F., Lomasney, J.W., Stupp, S.I. (2006) Heparin binding nanostructures to promote growth of blood vessels Nano Letters 6, 2086–2090.
Storrie, H., Guler, M.O., Abu-Amara, S.N., Volberg, T., Rao, M., Geiger, B., Stupp, S.I. (2007) Supramolecular crafting of cell adhesion Biomaterials 28, 4608–4618.
Stendahl, J.C., Want, L.J., Chow, L.W., Kaufman, D.B., Stupp, S.I. (2008) Growth factor delivery from self-assembling nanofibers to facilitate islet transplantation Transplantation 86(3), 478–481.
Capito, R., Azevedo, H., Gelichko, Y., Mata, A., Stupp, S.I. (2008) Self-assembly of large and small molecules into hierarchically ordered sacs and membranes Science 319, 1812–1816.
Kapadia, M. R., Chow, L.W., Tsihlis, N.D., Ahanchi, S.S., Eng, J.W., Murar, J., Martinez, J., Popowich, D.A., Jiang, Q., Hrabie, J.A., Saavedra, J.E., Keefer, L.K., Hulvat, J.F., Stupp, S.I., Kibbe, M.R. (2008) Nitric oxide and nanotechnology: A novel approach to inhibit neointimal hyperplasia Journal of Vascular Surgery 47(1), 173–182.
Tysseling-Mattiace, V.M., Sahni, V., Niece, K.L., Birch, D., Czeisler, C., Fehlings, M.G., Stupp, S.I., Kessler, J.A. (2008) Self-assembling nanofibers inhibit glial scar formation and promote axon elongation after spinal cord injury The Journal of Neuroscience 28(14), 3814–3823.
Rajangam, K., Arnold, M.S., Rocco, M.A., Stupp, S.I. (2008) Peptide amphiphile nanostructure-heparin interactions and their relationship to bioactivity Biomaterials 29, 293298–293305.
Sargeant, T.D., Guler, M.O., Oppenheimer, S.M., Mata, A., Satcher, R.L., Dunand, D.C., Stupp, S.I. (2008) Hybrid bone implants: Self-assembly of peptide amphiphile nanofibers within porous titanium Biomaterials 29(2), 161–171.
Sargeant, T. D., Rao, M.S., Koh, C.Y., Stupp, S.I. (2008) Covalent functionalization of NiTi surfaces with bioactive peptide amphiphile nanofibers Biomaterials 29(8), 1085–1098.
Spoerke, E.D., Anthony, S.G., Stupp, S.I. (2009) Enzyme Directed Templating of Artificial Bone Mineral Advanced Materials 21(4), 425–430.
Mata, A., Hsu, L., Capito, R., Aparicio, C., Henrikson, K., Stupp, S.I. (2009) Micropatterning of bioactive self-assembling gels Soft Matter 5, 1228–1236.
Webber, M.J., Tongers, J., Renault, M.A., Roncalli, J.G., Losordo, D.W., Stupp, S.I. (2009) Development of bioactive peptide amphiphiles for therapeutic cell delivery Acta Biomateriala 6, 3–11.
Mata, A., Geng, Y., Henrikson, K., Aparicio, C., Stock, S., Satcher, R., Stupp, S.I. (2010) Bone regeneration mediated by biomimetic mineralization of a nanofiber matrix Biomaterials 31, 6004–6012.
Shah, R.N., Shah, N.A., Del Rosario Lim, M.M., Hsieh, C., Nuber, G., Stupp, S.I. (2010) Supramolecular design of self-assembling nanofibers for cartilage regeneration Proceedings of the National Academy of Sciences of the United States of America 107(8), 3293–3298.
Chow, L.W., Wang, L.J., Kaufman, D.B., Stupp, S.I. (2010) Self-assembling nanostructures to deliver angiogenic factors to pancreatic islets Biomaterials 31(24), 6154–6161.
Standley, S.M., Toft, D.T., Cheng, H., Soukasene, S., Chen, J., Raja, S.M., Band, V., Band, H., Cryns, V.L, Stupp, S.I. Induction of Cancer Cell Death by Self-assembling Nanostructures Incorporating a Cytotoxic Peptide Cancer Research. (available online Res. 0: 0008–5472.CAN-09-3267v1).
Muraoka, T., Koh, C.Y., Cui, H., Stupp, S.I. (2009) Light-triggered bioactivity in three-dimensions Angewante Chemie International Edition 48, 5946–5949.
Ghanaati, S., Webber, M.J., Unger, R.E., Orth, C., Hulvat, J.F., Kiehna, S.E., Barbeck, M., Rasic, A., Stupp, S.I. (2009) Dynamic in vivo biocompatibility of angiogenic peptide amphiphile nanofibers Biomaterials 30, 6202–6212.
Behanna, H.A., Donners, J.J.J.M, Gordon, A.C., Stupp, S.I. (2005) Coassembly of amphiphiles with opposite polarities into nanofibers Journal of the American Chemical Society 127, 1193–1200.
Greenfield, M.A., Hoffman, J.R., de la Cruz, M.O., Stupp, S.I. (2010) Tunable Mechanics of Peptide Nanofiber Gels Langmuir 26(5), 3641–3647.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2012 Springer Science+Business Media, LLC
About this protocol
Cite this protocol
Mata, A., Palmer, L., Tejeda-Montes, E., Stupp, S.I. (2012). Design of Biomolecules for Nanoengineered Biomaterials for Regenerative Medicine. In: Navarro, M., Planell, J. (eds) Nanotechnology in Regenerative Medicine. Methods in Molecular Biology, vol 811. Humana Press. https://doi.org/10.1007/978-1-61779-388-2_3
Download citation
DOI: https://doi.org/10.1007/978-1-61779-388-2_3
Published:
Publisher Name: Humana Press
Print ISBN: 978-1-61779-387-5
Online ISBN: 978-1-61779-388-2
eBook Packages: Springer Protocols