
Abstract
Infectious bronchitis virus (IBV), a group 3 coronavirus, produces three proteins (IBV E, IBV 3a, and IBV 3b) from subgenomic mRNA 3 during infection. IBV E, a viral envelope protein, plays a role in virus budding, possibly by altering membrane morphology at the virus assembly site. In addition to this role, IBV E may also function as a viroporin, although no data from infected cells have confirmed this possibility definitively. Conversely, the IBV 3a and IBV 3b proteins are nonstructural proteins. These proteins are dispensable for replication in cell culture, but are thought to be important for infection of the natural host. This chapter details methods for generating and screening antibodies to these gene 3 proteins. Antibodies were raised in rabbits following inoculation with IBV-specific peptides and GST fusion proteins, and were screened by immunofluorescence, radioimmunoprecipitation, and immunoblotting.
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others


Key Words
1 Introduction
Coronaviruses have large, positive-sense, single-stranded RNA genomes (1). During infection, four structural proteins are expressed: spike (S), membrane (M), nucleocapsid (N), and envelope (E). The viral envelope E protein is thought to alter membrane morphology at the virus assembly site, thus promoting virus budding (2–(10). Additionally, more infectious bronchitis virus (IBV) E protein is produced in infected cells than is incorporated into virions, suggesting that IBV E may have a function apart from its role in virus budding (3). Consistent with this idea, studies have indicated that the E protein from several coronaviruses may act as a viroporin (1 (1–(1 (4). However, no conclusive experiments in infected cells have confirmed this possibility. In addition to the coronavirus structural proteins, several other open reading frames (ORFs) located between the structural protein ORFs produce proteins of undetermined function during infection (1 (5). These proteins are thought to be nonstructural, accessory proteins that are essential for productive virus infection in the natural host, but dispensable for virus growth in cell culture (1 (6–(2 (3). Therefore, study of these accessory proteins, in addition to study of the coronavirus structural proteins, may more fully elucidate pathogenesis mechanisms and lead to the development of more effective antiviral drugs.
IBV, a group 3 coronavirus that infects chickens, produces two nonstructural proteins, 3a and 3b, from ORFs located between the S and E genes (2 (4). Both of these proteins are translated from subgenomic mRNA 3, with IBV 3a translation initiated via the first AUG, and the downstream IBV 3b translation initiated via leaky ribosomal scanning (2 (5). Subgenomic mRNA 3 also produces the IBV E protein through an internal ribosomal entry site. We have developed antibodies specific to each of these IBV gene 3 proteins for use in characterization and functional studies. IBV E was found to localize to Golgi membranes via its C-terminal tail (3,(2 (6). IBV E also promoted the release of virus-like particles through the interaction of its C-terminal tail with the IBV M protein (2 (7). Studies on the IBV 3a protein demonstrated that one pool of IBV 3a localized cytoplasmically, while another pool associated with membranes at the smooth endoplasmic reticulum (ER) (2 (8). The short length of IBV 3a appeared to preclude efficient association with signal recognition particle (SRP), causing inefficient co-translational insertion of IBV 3a into membranes. Thus, IBV may limit 3a levels at smooth ER membranes by a novel mechanism.
Conversely, IBV 3b localized to the nucleus in mammalian cells when using a vaccinia virus expression system (1 (8,(2 (9). However, IBV 3b was undetectable via microscopy of IBV-infected or transiently transfected mammalian cells (2 (9). Surprisingly, IBV 3b, which was readily detected in chicken cells, localized to the cytoplasm with apparent nuclear exclusion. The half-life of IBV 3b was greatly reduced in mammalian cells as compared to chicken cells. This rapid turnover in mammalian cells was proteasome-dependent, whereas turnover in avian cells was proteasome-independent. Thus, the importance of using cells derived from the natural host when studying coronavirus nonstructural proteins is highlighted. This chapter details methods for generating polyclonal antibodies to the IBV gene 3 proteins, including protocols for generating peptides or fusion proteins for immunizing rabbits, prescreening of nonimmunized rabbit sera, general rabbit immunization procedures, and screening sera from immunized rabbits.
2 Materials
2.1 Preparing Peptides for Injection into Rabbits
-
1.
Protein sequence
2.2 Preparing Fusion Proteins for Injection into Rabbits
-
1.
Plasmid DNA: pGEX-2T (Amersham) (encodes the GST gene) and pGEX-2T with the gene of interest cloned in frame with the GST gene (see Note 1).
-
2.
Competent BL21 Escherichia coli bacteria from the GST gene fusion system (Amersham). Store in 250-μl aliquots at –80°C. Thaw only once after aliquoting.
-
3.
YT-amp broth: 0.8% (w/v) BactoTryptone (BD Biosciences, San Diego, CA), 1% (w/v) Bacto yeast extract (BD Biosciences), and 0.5% (w/v) NaCl in dH2O. Autoclave and add 80 μg/ml ampicillin when the solution has cooled to below 65°C. Store at 4°C (see Note 2).
-
4.
LB-amp plates: 1% (w/v) BactorTryptone, 1% (w/v) Bacto yeast extract, 0.5% (w/v) NaCl, 0.1 M NaOH, and 1.3% BactoAgar (BD Biosciences) in dH2O. Autoclave and add 80 μg/ml ampicillin when the solution has cooled to below 65°C. Pour into Petri dishes and store at 4°C when the agar has solidified.
-
5.
Inducing solution (1000X): 100 mM isopropyl-β-D-thiogalactopyranoside (IPTG) (Amersham) in dH2O. Make fresh before each use.
-
6.
Phosphate buffered saline (PBS). Store at room temperature.
-
7.
PBSP: Protease inhibitors (Sigma, catalog number P8340) are added to PBS right before use.
-
8.
Glutathione sepharose beads: Glutathione sepharose 4B (Amersham). Store at 4°C.
-
9.
Elution solution: 10 mM reduced glutathione (Sigma), 50 mM Tris-HCl (pH 8) in dH2O. Make fresh when needed.
-
10.
Loading sample buffer (LSB) (4X): 200 mM Tris-HCl (pH 6.8), 8% (w/v) SDS, 60% (v/v) glycerol, and 0.02% (w/v) bromophenol blue in dH2O. Store at 4°C. Since the buffer solidifies at 4°C, it has to be heated briefly at 65°C before use.
-
11.
2-mercaptoethanol. Store at room temperature.
-
12.
Destain solution: 40% (v/v) methanol and 10% (v/v) acetic acid in dH2O. Store at room temperature.
-
13.
Coomassie solution: 0.25% (w/v) Coomassie brilliant blue R (Sigma) in destain solution. Store at room temperature.
2.3 Prescreening Rabbit Sera before Injection of Peptides or Proteins
-
1.
Sera from four nonimmunized rabbits.
-
2.
Phosphate buffered saline (PBS). Store at room temperature.
-
3.
All materials listed in Sections 2.13 and 2.14 if prescreening sera using SDS-PAGE and immunoblotting.
-
4.
All materials listed in Section 2.11 if prescreening sera using indirect immunofluorescence microscopy.
2.4 Rabbit Immunization
-
1.
A minimum of 1.75 mg of peptide or a minimum of 875 μg of GST fusion protein with a concentration of no less than 0.25 mg/ml.
2.5 Screening Sera from Immunized Rabbits
2.5.1 Screening Sera from Immunized Rabbits Using in vitro Transcribed and Translated Protein
-
1.
All of the materials listed in Section 2.10.
-
2.
All of the materials listed in Section 2.12, except for the [35S]-methionine-cysteine and the methionine and cysteine-free DMEM.
2.5.2 Screening Sera from Immunized Rabbits Using Other Methods
-
1.
All of the materials listed in Sections 2.7 (IBV infection), Section 2.8 (vTF7-3 infection/transfection), or Section 2.9 (transient transfection), depending on the preferred methods of screening.
-
2.
All of the materials listed in Sections 2.13 and 2.14 if screening sera using immunoblotting.
-
3.
All of the materials listed in Section 2.12 if screening by immunoprecipitation.
-
4.
All of the materials listed in Section 2.11 if screening by indirect immunofluorescence microscopy.
2.6 Affinity Purification of Sera
-
1.
Kit equilibration buffer #1 from the Reduce-Imm Immobilized Reductant Kit (Pierce).
-
2.
Kit equilibration buffer #2 from the Reduce-Imm Immobilized Reductant Kit (Pierce).
-
3.
DTT solution: 10 mM dithiothreitol in kit equilibration buffer #1. Make this solution fresh before using.
-
4.
Diluted peptide: Dissolve 5–10 mg of peptide in kit equilibration buffer #1. Make this solution fresh before using.
-
5.
Diluted protein: Dissolve 5–6 mg of GST fusion protein in kit equilibration buffer #2. Make this solution fresh before using.
-
6.
Ellman’s Reagent (Pierce)
-
7.
Reduce-Imm kit reductant column (Pierce)
-
8.
SulfoLink coupling column (Pierce)
-
9.
SulfoLink equilibration buffer (Pierce): 0.1 M sodium phosphate and 5 mM EDTA [pH 6.0] in dH2O.
-
10.
SulfoLink coupling buffer (Pierce): 5 mM EDTA and 50 mM Tris-HCl [pH 8.5] in dH2O.
-
11.
Cysteine buffer: 15.8 mg of L-cysteine • HCl in 2 ml of SulfoLink coupling buffer.
-
12.
Wash buffer (Pierce): 1.0 M NaCl and 0.5% (w/v) NaN3 in dH2O.
-
13.
0.05% NaN3:0.05% (w/v) NaN3 in dH2O. Store at 4°C.
-
14.
0.05% degassed NaN3: aspirate 0.05% NaN3 for 5 min.
-
15.
Storage buffer: 10 mM EDTA, 0.05% NaN3, 50% (v/v) glycerol in dH2O.
-
16.
Rabbit sera.
-
17.
WB 1: 0.2% deoxycholic acid and 10 mM Tris-HCl [pH 7.4] in dH2O. Store at 4°C.
-
18.
WB 2: 0.5 M NaCl and 10 mM Tris-HCl [pH 7.4] in dH2O. Store at 4°C.
-
19.
Elution solution: 4 M MgCl2 in dH2O. Store at 4°C.
-
20.
BSA: 30% bovine serum albumin (Sigma). Store at 4°C.
-
21.
Dialysis bags (Pierce–10 K MWCO): Make sure that bags are knotted and clipped before beginning the elution in step 5 of Section 3.6.
-
22.
Phosphate buffered saline (PBS) (pH 7.4). Store at room temperature.
2.7 IBV Infection
-
1.
The Vero-adapted Beaudette strain of IBV is used to infect Vero cells (30). The egg-adapted strain of IBV (American Type Tissue Culture VR-22) is used to infect DF1 cells. Work with IBV should be done using Biosafety Level 2 (BL-2) precautions, including the use of biological safety cabinets, autoclaving of all disposable items that have been in contact with virus, and inactivation of all virus-containing solutions with bleach. Store virus in 1-ml aliquots at –80°C. Do not subject to freeze/thaw more than twice.
-
2.
Serum-free DMEM: High-glucose Dulbecco’s modified Eagle’s medium (Invitrogen Life Technologies, Carlsbad, CA). Store at 4°C.
-
3.
Cells: Vero (African green monkey kidney epithelial cells) and DF1 (UMNSAH/DF1, immortalized chicken embryo fibroblast cells).
-
4.
Normal growth medium: 10% (v/v) fetal calf serum (FBS) (Atlanta Biologicals, Norcross, GA) and 0.1 mg/ml Normocin (Invivogen, San Diego, CA) in serum-free DMEM. Store at 4°C.
2.8 vTF7-3 Infection/Transfection
-
1.
vTF7-3: vaccinia virus encoding T7 RNA polymerase (31). Store in 200-μl aliquots at –80°C. Do not subject to freeze/thaw more than five times. Work with vaccinia virus should be done using BL-2 precautions. These precautions include the use of biological safety cabinets, autoclaving of all disposable items that have been in contact with virus, and inactivation of all virus-containing solutions with bleach. Additionally, a virus-only laminar flow hood can be used to prevent the accidental contamination of cell lines with virus for routine tissue culture work. Finally, institutional or governmental regulations on researcher vaccination and virus containment should be consulted before beginning any projects involving vaccinia virus.
-
2.
Plasmid DNA: Gene of interest must be cloned behind the T7 RNA polymerase promoter.
-
3.
Serum-free DMEM: High glucose Dulbecco’s modified Eagle’s medium (Invitrogen). Store at 4°C.
-
4.
Cells: Vero (African green monkey kidney epithelial cells) and DF1 (UMNSAH/DF1, immortalized chicken embryo fibroblast cells).
-
5.
Opti-MEM® I (Invitrogen). Store at 4°C.
-
6.
Fugene 6 (Roche Molecular Biochemicals, Indianapolis, IN). Store at 4°C.
-
7.
Normal growth medium: 10% (v/v) fetal calf serum (FBS) (Atlanta Biologicals) and 0.1 mg/ml Normocin (Invivogen) in serum-free DMEM. Store at 4°C.
2.9 Transient Transfection of Cells
-
1.
Cells: Vero (African green monkey kidney epithelial cells) or DF1 (UMNSAH/DF1, immortalized chicken embryo fibroblast cells) (see Note 3).
-
2.
Plasmid DNA
-
3.
Fugene 6 (Roche): Store at 4°C.
-
4.
Opti-MEM® I (Invitrogen): Store at 4°C.
2.10 in vitro Transcription and Translation
-
1.
TNT master mix from the quick-coupled transcription/translation T7 system (Promega Corporation, Madison, WI): Store in 40-μl aliquots at –80°C. Only use the aliquots once after thawing them.
-
2.
Plasmid DNA: Gene of interest must be cloned behind the T7 RNA polymerase promoter.
-
3.
[35S]-Redivue-methionine (Amersham): Store at 4°C.
2.11 Indirect Immunofluorescence Microscopy
-
1.
Phosphate buffered saline (PBS) [pH 7.4] (10X): 0.1 M NaH2PO4, 0.1 M Na2HPO4, and 1.5 M NaCl in dH2O: Store at room temperature.
-
2.
3% paraformaldehyde solution: Add 3 g of paraformaldehyde to 80 ml of dH2O and 50 μl of 5 M NaOH. Heat the solution to 50°C until the paraformaldehyde is dissolved. Add 10 ml of 10X PBS and bring the total volume up to 100 ml with dH2O. Check with pH paper (see Note 4) that the pH is 7.4. Store in 15-ml aliquots at –20°C.
-
3.
PBS/Gly: 10 mM glycine and 0.02% (w/v) NaN3 in 1X PBS. Store at room temperature.
-
4.
Permeabilization solution: 0.5% (v/v) TritonX-100 in PBS/Gly. Make fresh before each use.
-
5.
Primary and secondary antibody dilution buffers: 1% (v/v) bovine serum albumin (BSA) (Sigma-Aldrich Co., St. Louis, MO) in PBS/Gly. Make fresh before each use.
-
6.
Mounting solution: 0.1 M N-propyl gallate in glycerol. Store at room temperature protected from light (see Note 5).
2.12 Immunoprecipitation
-
1.
[35S]-methionine-cysteine: pro-mix (Amersham). Store in 35-μl aliquots at –80°C.
-
2.
Methionine and cysteine-free DMEM (Invitrogen)
-
3.
Phosphate buffered saline (PBS). Store at room temperature.
-
4.
Detergent solution: 62.5 mM EDTA, 50 mM Tris-HCl [pH 8], 0.4 % deoxycholic acid (CalBiochem, La Jolla, CA), and 1% nonidet P40 substitute (Fluka Chemie AG, Buchs, Switzerland) in dH2O. Store at 4°C. Add protease inhibitors (Sigma, cat. no. P8340) immediately before each use.
-
5.
10% SDS: 10% (w/v) sodium dodecylsulfate (SDS) in dH2O. Store at room temperature.
-
6.
2-mercaptoethanol (BioRad). Store at room temperature.
-
7.
WP buffer 1: 2% (w/v) SDS and 1% (v/v) 2-mercaptoethanol in dH2O. Make fresh before use.
-
8.
WP buffer 2: 20 mM Tris-HCl [pH 8], 1 M NaCl, 1% (v/v) TritonX-100, and 0.02% (w/v) NaN3. Make fresh before use.
-
9.
Washed pansorbin (to reduce background binding): 25 ml of standardized pansorbin cells (Calbiochem) are mixed with 5 ml of 10% SDS and 270 μl of 2-mercaptoethanol. Cells are then taken through a heating procedure [heating to 100°C for 15 min, centrifugation at 7840 × g for 7 min at 4°C, and resuspension in 30 ml of WP buffer 1]. This heating procedure is then repeated once. Cells are washed five times in 30 ml of a WP buffer 2 before being resuspended in 25 ml of WP buffer 2 and stored in 1-ml aliquots at –20°C (see Note 6).
-
10.
Primary antibodies: Undiluted rabbit serum
-
11.
RIPA buffer: 10 mM Tris-HCl [pH 7.4], 0.1% (w/v) SDS, 1% (w/v) deoxycholine, 1% (v/v) nonidet P 40 substitute, and 150 mM NaCl in dH2O. Store at 4°C.
-
12.
Loading sample buffer (LSB) (4X): 200 mM Tris-HCl [pH 6.8], 8% (w/v) SDS, 60% (v/v) glycerol, and 0.02% (w/v) bromophenol blue in dH2O. Store at 4°C. Since the buffer solidifies at 4°C, it has to be heated briefly at 65°C before use.
2.13 SDS Polyacrylamide Gel Electrophoresis (SDS-PAGE)
-
1.
Ethanol (the Warner-Graham Company, Cockeysville, MD). Store at room temperature in a flammables cabinet.
-
2.
Acrylamide: 40% acrylamide (BioRad Laboratories, Hercules, CA), 2% bis-acrylamide (BioRad), 30% polyacrylamide:bis (37.5:1 with 2.6% C, BioRad). Store at 4°C. Note that acrylamide is a neurotoxin before it polymerizes, so gloves should be worn when handling these chemicals.
-
3.
TEMED (BioRad): Store at room temperature.
-
4.
Separating gel buffer (4X): 1.5 M Tris-HCl [pH 8.8] in dH2O. Store at room temperature.
-
5.
10% SDS: 10% (w/v) sodium dodecylsulfate (SDS) in dH2O. Store at room temperature.
-
6.
30% APS: 30% (w/v) ammonium persulfate (APS) in dH2O. Store at 4°C. Make no more than 2 ml of this solution at a time, as it will generally last for only 3 months when stored at this temperature.
-
7.
Stacking gel buffer (4X): 0.5 M Tris-HCl [pH 6.8] in dH2O. Store at room temperature.
-
8.
Layering buffer: 0.2% (w/v) SDS in dH2O. Store at room temperature.
-
9.
Running buffer (5X): 125 mM Tris, 1 M glycine, and 0.5% (w/v) SDS in dH2O. Store at room temperature.
-
10.
Loading sample buffer (LSB) (4X): 200 mM Tris-HCl [pH 6.8], 8% (w/v) SDS, 60% (v/v) glycerol, and 0.02% (w/v) bromophenol blue in dH2O. Store at 4°C. Since the buffer solidifies at 4°C, it has to be heated briefly at 65°C before use.
-
11.
2-mercaptoethanol (BioRad). Store at room temperature.
-
12.
Molecular weight markers: Precision Plus ProteinTM All Blue Standards for immunoblotting (BioRad) or [14C] molecular weight markers (Amersham Pharmacia Biotech, Inc., Piscataway, NJ) for radioactive gels. Store at ––20°C.
2.14 Immunoblotting
-
1.
Cooled transfer buffer: 25 mM Tris, 0.2 M glycine, 15% (v/v) MeOH in dH2O. Store at 4°C. Buffer can be used six times before being discarded.
-
2.
3 MM Chr chromatography paper (Whatman Inc., Florham Park, NJ).
-
3.
Polyvinylidene fluoride (PVDF) membrane (Millipore Corporation, Billerica, MA) (see Note 7).
-
4.
Methanol: Store at room temperature in a flammables cabinet.
-
5.
TTBS: 150 mM NaCl, 0.05% (v/v) Tween-20, and 10 mM Tris-HCl [pH 7.4] in dH2O. Store at 4°C.
-
6.
Blocking buffer: 5% (w/v) nonfat dry milk in TTBS. Make fresh before each use.
-
7.
Primary antibody dilution buffer: 1% (w/v) nonfat dry milk in TTBS. Make fresh before each use.
-
8.
Secondary antibodies: 1:10,000 dilution of HRP-conjugated donkey anti-rabbit IgG antibodies (Amersham) in blocking buffer. Make fresh before each use.
-
9.
HRP substrate: ECLTM Western Blotting Detection Reagents (Amersham). Store at 4°C.
3 Methods
3.1 Preparing Peptides for Injection into Rabbits
-
1.
Protein sequence must be analyzed to determine likely antigenic epitopes. First, hydropathy plots should be created to identify putative transmembrane segments in the protein. Peptides corresponding to transmembrane regions should not be used for immunizing rabbits since they may not allow the antibody access to the protein in some assays. One helpful website for creating hydropathy plots is found at http://ca.expasy.org/tools/protscale.html. Proteins should also be analyzed for the presence of domains that are highly conserved in many different proteins, such as coiled-coil, protein targeting, or DNA-binding motifs. Peptides corresponding to these types of motifs should not be used for immunizing rabbits as they may generate antibodies that recognize several different proteins. A list of helpful websites for predicting these types of motifs is found at http://ca.expasy.org/tools/. We have had success using peptides (around 14 amino acids in length) that correspond to the unique C- or N-terminal regions of the IBV gene 3 proteins. However, the antigenicity of these different peptides has varied, as shown in Fig. 1A.
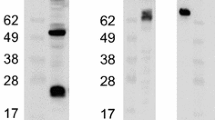
Fig. 1. 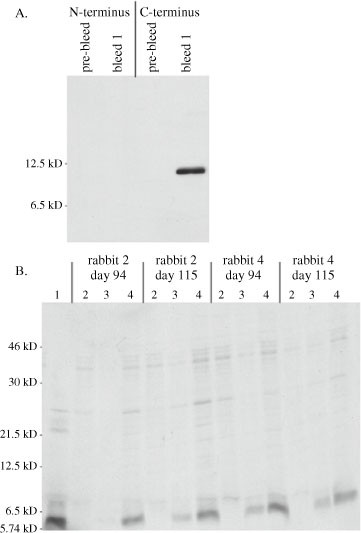
Screening sera from immunized rabbits: (A) IBV-infected Vero cells (Section 3.7) were lysed at 15 h postinfection according to the protocol in Section 3.3, step 2. Sera from rabbits immunized with peptides corresponding to either the N-terminus or the C-terminus of IBV E were then screened using the immunoblotting protocol in Section 3.5.2. Results from a 1:1000 dilution of sera harvested on day 52 (bleed 1) are shown. The C-terminal peptide of IBV E was much more immunogenic than the N-terminal peptide. Although rabbits immunized with the N-terminal IBV E peptide eventually produced antibodies specific to IBV E, the titers were always lower than those of rabbits immunized with the C-terminal IBV E peptide. (B) Sera from rabbits immunized with GST-IBV 3b were screened using the protocol in Section 3.5.1. The responses of two different rabbits at two different times are shown. Lane 1: 10% input of in vitro transcribed and translated IBV 3b. Lane 2: immunoprecipitation of in vitro transcribed and translated IBV 3b using preimmune rabbit sera. Lane 3: immunoprecipitation of in vitro transcribed and translated IBV 3b using 1 μl of immunized rabbit sera. Lane 4: immunoprecipitation of in vitro transcribed and translated IBV 3b using 3 μl of immunized rabbit sera.
-
2.
Peptides are synthesized using an automated synthesizer and purified to ≥90% purity (see Note 8). If the peptide region does not contain a cysteine, then a cysteine must be added during the synthesis process so that it can be coupled to keyhole limpet hemocyanin (see Note 9). A minimum of 1.75 mg of peptide is required for injection into rabbits in Section 3.4. We order commercially synthesized peptides from Boston Biomolecules, Inc. (Boston, MA), precoupled to keyhole limpet hemocyanin through a cysteine added to one end of the peptide. However, coupling can also be done using commercially available kits from companies such as Pierce Biotechnology, Inc. (see Note 10).

3.2 Preparing Fusion Proteins for Injection into Rabbits
-
1.
GST fusion proteins are expressed and purified using the GST gene fusion system (Amersham). A minimum of 875 μg of protein with a concentration of no less than 0.25 mg/ml is required for injection into rabbits in Section 3.4 (see Note 10).
-
2.
50 μl of competent BL21 E. coli bacteria is thawed on ice before being incubated for 20 min on ice with 10 ng of plasmids encoding either GST (pGEX-2T) or GST fused to the protein of interest. Bacteria are then heated at 42°C for 1 min, placed on ice for 2 min, and plated on YT-amp plates overnight at 37°C.
-
3.
One individual bacterial colony carrying the pGEX-2T plasmid is picked and grown overnight in 2 ml of YT-amp broth. Twelve individual bacterial colonies carrying the plasmid encoding the GST fusion protein are picked and grown overnight in 2 ml of YT-amp broth (see Note 11).
-
4.
The next day each individual culture is diluted 1:10 in 2 ml of YT-amp broth and grown to an OD600 of approximately 0.6.
-
5.
GST protein expression is induced by adding 2 μl of inducing solution to cultures and growing them for 48 h at 13.5°C in a shaking water bath (see Notes 12 and 13).
-
6.
Bacteria are pelleted by centrifugation at 20,800 × g for 1 min and resuspended in microcentrifuge tubes in 200 μl of PBSP.
-
7.
Bacteria are lysed by sonication (three 30-sec pulses on setting 4) using a Sonic Dismembrator Model 100 (Fisher) (see Note 14).
-
8.
Cellular debris is pelleted by centrifugation at 20,800 × g for 5 min at 4°C. Samples should be kept at 4°C throughout the rest of the protocol to minimize protein degradation.
-
9.
25 μl of glutathioine sepharose beads per 200 μl of sonicated supernatant are prepared by washing beads twice in 500 μl of PBS to 25 μl of beads in microcentrifuge tubes. Beads are centrifuged at 1020 × g for 5 min at 4°C.
-
10.
The supernatant protein is bound to glutathione sepharose beads by rotation overnight at 4°C.
-
11.
Beads are washed three times with PBSP at 4°C, followed by centrifugation at 1020 × g for 5 min at 4°C after each wash.
-
12.
GST proteins are eluted by rotating beads at 4°C for 24 h in the elution solution. This elution step is repeated and GST or GSTfusion protein eluates are pooled (see Note 15).
-
13.
20 μl of eluate is mixed with 2X LSB supplemented with 5% (v/v) 2-mercapto- ethanol, heated at 95°C for 5 min, and subjected to SDS-PAGE as described in Section 3.13. Then 10, 5, and 1 μg of purified bovine serum albumin (BSA) is also run onto gels as standards for visually assessing eluate yield.
-
14.
Gels are stained with Coomassie solution for 1 h at room temperature. Gels are then rinsed once with destain solution and incubated overnight at room temperature without rocking in destain solution.
-
15.
Gels are soaked for 10 min in dH2O and dried for 1.25 h at 80°C on a gel dryer. Representative data using this procedure with the GST-IBV3b fusion protein are shown in Fig. 14.2.

Expression and purification of the GST-IBV 3b fusion protein. GST and GST-IBV 3b were expressed in bacteria and purified according to the protocol in Section 3.2, with 20 μl of the final eluate being subjected to SDS-PAGE and Coomassie staining. Lane 1 shows purified GST eluate. Lane 2 shows eluate from uninduced cultures containing plasmids that encoded GST-IBV 3b. Lane 3 shows purified GST-IBV 3b eluate.
3.3 Prescreening Rabbit Sera before Injection of Peptides or Proteins
-
1.
We have found that sera from some nonimmunized rabbits may recognize cell or viral proteins. Therefore, we prescreen the sera from several nonimmunized rabbits (generally twice as many rabbits as we use for immunization procedures) before immunizing them with peptides or fusion proteins. Any rabbits whose preimmune sera recognize proteins from infected or transiently transfected cells should not be used for generating antibodies.
-
2.
For prescreening by immunoblotting, IBV-infected, vTF7-3 infected, or transiently transfected cells are washed once with PBS and lysed directly in their dishes with 2X LSB plus 5% (v/v) 2-mercaptoethanol. Noninfected or untransfected cells are also lysed. Approximately 100 μl of the LSB mixture is used per 35-mm dish. Once this mixture is added, a pipette tip is used to gently swirl the mixture across the cells. As the cells are lysed, the mixture becomes viscous owing to DNA and is then transferred to a microcentrifuge tube (samples can be stored at this point at –20°C for several weeks). Samples are then heated for 5 min at 95°C, until they are no longer viscous when the side of the tube is tapped. Samples are then subjected to SDS-PAGE (Section 3.13) and immunoblotting (Section 3.14). The primary antibodies used are a 1:1000 dilution of nonimmunized rabbit sera in the primary antibody dilution buffer. An example of the results produced from this protocol is shown in Fig. 141.3.
Fig. 3. 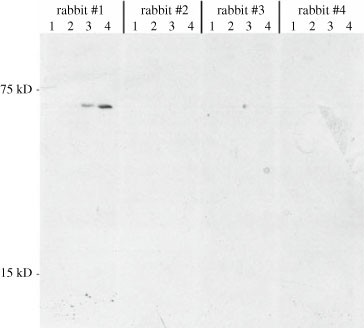
Prescreening rabbit sera before injection of peptides or proteins. Before immunizing rabbits with the GST-IBV 3b fusion protein, four rabbit sera were tested for nonspecific recognition of proteins by SDS-PAGE and immunoblotting according to the protocol in Section 3.3. Lane 1: noninfected Vero cells. Lane 2: IBV-infected Vero cells (Section 3.7). Lane 3: vTF7-3 infected cells transfected with an empty plasmid. Lane 4: vTF7-3-infected cells transfected with a plasmid encoding IBV 3b behind the T7 RNA polymerase promoter (Section 3.8). Rabbits 2 and 4 were subsequently immunized with GST-IBV 3b. Rabbit 1 contains antibodies that recognize a vaccinia virus protein, a problem we have encountered occasionally.
-
3.
For prescreening by indirect immunofluorescence microscopy, IBV-infected, vTF7-3 infected, or transiently transfected cells can be processed according to Section 3.11. The primary antibodies used are a 1:500 dilution of nonimmunized rabbit sera.

3.4 Rabbit Immunization
-
1.
Following prescreening in Section 3.10, two rabbits were chosen for immunization with peptides or fusion proteins. We use two rabbits per immunogen, since individual rabbit immunization responses may differ. The following procedure can be done commercially by a company such as Covance Research Products, Inc. (Denver, PA) (see Notes 16 and 17).
-
2.
21-day-old NZW rabbits are injected intradermally in the back with 250 μg of protein per rabbit combined with Freund’s complete adjuvant (FCA).
-
3.
Subsequent injections were done with 125 μg of protein combined with Freund’s incomplete adjuvant (FIA) on the following days: 42 (subcutaneous nodal area injection), 63 (subcutaneous neck injection), 84 (subcutaneous and intramuscular injection), 105 (subcutaneous dorsal injection), 135 (subcutaneous nodal area injection), and 156 (subcutaneous dorsal injection) (see Note 18).
-
4.
Approximately 5 ml of serum was harvested from rabbits on days 52 and 73 and approximately 20 ml of serum on days 94, 115, and 145. The terminal rabbit bleed on day 167 yielded approximately 50 ml. All sera are stored at –80°C.
3.5 Screening Sera from Immunized Rabbits
3.5.1 Screening Sera from Immunized Rabbits Using in vitro Transcribed and Translated Protein
-
1.
Transcribe and translate the protein of interest in vitro as described in Section 3.10. An empty vector should be used as a control for the in vitro transcription and translation reaction (see Note 19).
-
2.
Samples are diluted in 500 μl of detergent solution.
-
3.
Samples are immunoprecipitated as in Section 3.12 starting with step 3. The primary antibodies used are 3 μl of undiluted sera from each harvest day from each rabbit. Always use preimmune sera (sera from the rabbit before immunization began) as a negative control. A representative result obtained from the method is shown in Fig. 14.1B.
3.5.2 Screening Sera from Immunized Rabbits Using Other Methods
-
1.
Vero or DF1 cells are infected with IBV (Section 3.7). Noninfected cells should be used as a negative control. Alternatively, cells that are transiently transfected with plasmids encoding the protein of interest can be used (Section 3.8 or 3.9). In this case, cells transiently transfected with an empty vector should be used as a negative control (see Note 19).
-
2.
Cells can then be subjected to immunoblotting (Section 3.14), immunoprecipitation (Section 3.12), or indirect immunofluorescence microscopy (Section 3.11) (see Note 20).
-
3.
If screening by immunoblotting, primary antibodies are 1:1000, 1:2000, and 1:5000 dilutions of sera from each harvest day from each rabbit. Always use similar dilutions of preimmune sera as a negative control during screening.
-
4.
If screening by immunoprecipitation, the primary antibodies used are 3 μl of undiluted sera from each harvest day from each rabbit. Always use preimmune sera as a negative control (see Note 21).
-
5.
If screening by indirect immunofluorescence microscopy, primary antibodies are 1:50, 1:100, 1:200, 1:500, and 1:1000 dilutions of sera from each harvest day from each rabbit. Always use preimmune sera as a negative control during screening (see Note 22).
3.6 Affinity Purification of Sera
-
1.
Occasionally, sera may show high background staining in negative control samples, and when this occurs, affinity purification of the sera may help reduce it. Affinity purification can be done with either the peptides or the GST fusion proteins that were used to immunize rabbits in Section 3.4. The protocol for affinity purification of sera using peptides is given below. Changes that must be made to this protocol when using fusion proteins rather than peptides are given in the Notes.
-
2.
First, the cysteines in the purified peptides from Section 3.1 are reduced. To do this, we used the commercially available Reduce-Imm Immobilized Reductant Kit according to the manufacturer’s instructions.
-
a.
The kit reductant column is washed with 10 ml of kit equilibration buffer #1 and activated by adding 10 ml of DTT solution.
-
b.
The column is then washed again in 20 ml of kit equilibration buffer #1 (see Note 23).
-
c.
1 ml of diluted peptide is then added to the column (see Note 24).
-
d.
The column is eluted with 9 ml of kit equilibration buffer #1, and 1-ml fractions are collected (see Note 25).
-
a.
-
3.
Fractions are tested for the presence of free sulfhydryls using Ellman’s Reagent.
-
a.
50 μl of each fraction is transferred to a microcentrifuge tube containing 950 μl of kit equilibration buffer #1.
-
b.
100 μl of Ellman’s Reagent is then added to each tube.
-
c.
Tubes are incubated for 15 min at room temperature, and free sulfhydryl concentration is determined by absorbance measured at 412 nm.
-
d.
Fractions with similarly high absorbance measurements are pooled for use in subsequent steps.
-
e.
Additionally, the absorbance at 280 nm of the pooled fractions is measured, in order to calculate coupling efficiency during step 3.
-
a.
-
4.
To couple peptides to agarose beads, we used the commercially available SulfoLink kit according to the manufacturer’s instructions.
-
a.
First, the coupling column is brought to room temperature, and the storage buffer is allowed to drain from the column after removing both top and bottom caps.
-
b.
The column is then equilibrated with 12 ml of SulfoLink equilibration buffer.
-
c.
The pooled peptide fractions from step 3d are then added to the column and the top cap of the column is replaced.
-
d.
The column is then rotated for 15 min at room temperature.
-
e.
Column-peptide mixtures are then incubated for an additional 30 min at room temperature without rotation.
-
f.
The buffer is allowed to drain and the columns are washed with 6 ml of coupling buffer.
-
g.
The absorbance at 280 nm of the column eluate is measured (see Note 26).
-
h.
Unbound column sites are then blocked by the addition of 2 ml of cysteine buffer.
-
i.
The column is then rotated for 15 min at room temperature, before being allowed to incubate without rotation for an additional 30 min at room temperature.
-
j.
All liquid is then drained from the column and the column is washed four times in 4 ml of wash buffer and three times in 4 ml of 0.05% NaN3.
-
k.
If needed, the column can now be stored at 4°C. Usually, an additional 4–6 ml of 0.05% degassed NaN3 is added to the column before storage to ensure that the column will not dry out during storage.
-
a.
-
5.
Protein-specific antibodies in the rabbit sera are then bound to the column, washed, and eluted.
-
a.
The storage liquid is removed from the column, and 2.5 ml of serum is added to the column and rotated overnight at 4°C.
-
b.
The column is allowed to drain and this flow-through is saved as it may contain antibodies that did not bind to the column (see Note 27).
-
c.
The column is then washed with 25 ml of ice-cold WB 1 and 25 ml of ice-cold WB 2. The column should be kept at 4°C during these washes.
-
d.
The column is eluted by adding 5 ml of elution solution. Three 1.5-ml fractions are collected and 5 μl of 30% BSA is immediately added.
-
e.
Fractions are transferred into dialysis bags and dialyzed three times for 24 h at 4°C in 1 liter of PBS (see Note 28).
-
f.
Fractions are separated into 100-μl aliquots and screened using procedures from Section 3.5. Aliquots should be stored at –80°C.
-
a.
3.7 IBV Infection
-
1.
IBV is brought to a total volume of 200 μl in serum-free DMEM per 35-mm dish such that the multiplicity of infection (MOI) is 1 (see Note 29).
-
2.
Cells are washed once in serum-free DMEM. This virus dilution is then added to cells for 1 h at 37°C with rocking every 10 min. Virus is then removed and cells are incubated in normal growth medium for the appropriate amount of time postinfection (p.i.).
3.8 vTF7-3 Infection/Transfection
-
1.
The vTF7-3 transient transfection system is used to express large amounts of proteins in a short period of time. In this system, cells are infected with a vaccinia virus that encodes the T7 RNA polymerase. Cells are then transfected with plasmids in which the gene of interest is cloned behind the T7 RNA polymerase promoter (see Note 29).
-
2.
vTF7-3 is brought to a total volume of 200 μl in serum-free DMEM per 35-mm dish such that the MOI is 10.
-
3.
This virus dilution is added to cells for 1 h at 37°C with rocking every 10 min.
-
4.
While the virus is adsorbing to the cells, the transfection mixture is prepared. For each 35-mm dish, 6 μl of Fugene 6 is added to 94 μl of Opti-MEM. 2 μg of plasmid DNA is then added to this mixture and incubated at room temperature for 15 min (see Note 3).
-
5.
At 1 h p.i., the virus dilution is removed from cells and 1 ml of regular growth medium is added to the cells.
-
6.
The transfection mixture is added dropwise with swirling to the infected cells.
-
7.
Cells are lysed for SDS-PAGE or processed for indirect immunofluorescence at approximately 4 h p.i.
3.9 Transient Transfection of Cells
-
1.
For each 35-mm dish, 6 μl of Fugene 6 is added to 94 μl of Opti-MEM. 2 μg of plasmid DNA is then added to this mixture and incubated at room temperature for 15 min (see Notes 3 and 29)
-
2.
The transfection mixture is then added dropwise to the cells with swirling.
-
3.
Generally, cells are lysed for SDS-PAGE or processed for indirect immunofluorescence microscopy at approximately 24 h posttransfection.
3.10 in vitro Transcription and Translation
-
1.
The TNT quick-coupled transcription/translation T7 system (Promega) is used for in vitro transcription and translation. This system utilizes the T7 RNA polymerase to drive gene expression; therefore, genes must be cloned into plasmids behind a T7 RNA polymerase promoter.
-
2.
0.5 μg of a plasmid DNA is incubated for 90 min at 30°C with 10 μl of the TNT master mix and 1 μl of [35S]-Redivue-methionine. This reaction mix is usually used immediately. However, it can be stored at –20°C for 2 or 3 days if necessary.
3.11 Indirect Immunofluorescence Microscopy
-
1.
Adherent cells that will be analyzed by indirect immunofluorescence microscopy are plated on sterilized, No.1, 22-mm square, precleaned, corrosive resistant, borosilicate coverslips (Fisher) in 35-mm dishes.
-
2.
Cells are washed once in 1X PBS before being fixed in 3% paraformaldehyde solution for 10 min at room temperature (see Note 22).
-
3.
Cells are washed quickly once with PBS/Gly before being washed a second time in PBS/Gly for 5 min at room temperature.
-
4.
Cells are permeabilized in permeabilization solution for 3 min at room temperature (see Note 22).
-
5.
Step 2 is repeated.
-
6.
40 μl of diluted primary antibodies is pipetted onto parafilm. Coverslips are then placed onto the primary antibodies, cell side down, for 20 min at room temperature (see Note 30).
-
7.
Coverslips are placed back into the 35-mm dish cell side up and washed as in step 2.
-
8.
Coverslips are incubated with diluted secondary antibodies as in step 6, except that they should be protected from light during this incubation.
-
9.
Coverslips are put back into the 35-mm dish and washed as in step 2, except that they should be protected from light during washing.
-
10.
A small drop of mounting solution is placed onto glass slides, and coverslips are placed, cell side down, onto this mounting solution. Excess mounting solution is removed by aspiration. Slides can be stored at 4°C, protected from light for several weeks. Slides are visualized on an Axioskop microscope (Zeiss, Thornwood, NY) with an attached Sensys charge-coupled device camera (Photometrics, Tucson, AZ) using IP Lab imaging software (Signal Analytics, Vienna, VA) (see Note 31).
3.12 Immunoprecipitation
-
1.
Cells grown in 35-mm dishes are incubated with 0.085 mCi of [35S]-methionine-cysteine in 500 μl of methionine and cysteine-free DMEM for 30 min at 37°C (see Notes 32 and 33)
-
2.
Cells are rinsed once in 0°C PBS and then incubated for 10 min on ice in 500 μl of detergent solution.
-
3.
Cell lysates are transferred to microcentrifuge tubes and centrifuged at full speed (20,800 ×g) for 20 min at 4°C.
-
4.
Cell supernatants are transferred to fresh microcentrifuge tubes, supplemented with 10 μl of 10% SDS, and precleared by adding 20 μl of washed pansorbin and rotating at 4°C for 15 min.
-
5.
Samples are centrifuged at 20,800 ×g for 1 min at room temperature, and supernatants are transferred to fresh microcentrifuge tubes containing primary antibodies (see Note 21). Samples are then rotated overnight at 4°C.
-
6.
15 μl of washed pansorbin is then added to samples and rotated for 20 min at 4°C.
-
7.
Samples are centrifuged at 20,800 ×g for 30 sec at room temperature to pellet the pansorbin with antigen-antibody complexes. Pellets are then washed three times in RIPA buffer. Following the final wash, the last drop of any remaining RIPA buffer is removed. Samples can be stored at this point for several weeks at –20°C.
-
8.
Washed pansorbin pellets are resuspended in 20 μl of 2X LSB plus 5% (v/v) 2-mercaptoethanol, heated for 3 min at 95°C, and centrifuged at 20,800 ×g for 1 min at room temperature.
-
9.
The supernatants are subjected to SDS-PAGE as in Section 3.13.
-
10.
Following SDS-PAGE, gels are washed for 10 min in dH2O (see Note 34) and dried for 1.25 h on a gel dryer.
-
11.
Gels are exposed either to film or to a K-HD imaging screen (Bio-Rad). Exposure to a K-HD imaging screen is followed by scanning with a personal molecular imager FX (Bio-Rad). Pixel intensity can then be determined with Quantity One software (Bio-Rad).
3.13 SDS Polyacrylamide Gel Electrophoresis (SDS-PAGE)
-
1.
This protocol is for use with 16-cm gel plate systems similar to the Hoefer SE 400 or Hoefer SE 600 system; however, it can be easily adapted to other gel systems. Gel plates, spacers, and combs should be wiped thoroughly clean with ethanol before they are assembled.
-
2.
Prepare a 1.5-mm-thick, 17.5% low-bis polyacrylamide separating gel by mixing together 13.1 ml of 40% acrylamide, 1.5 ml of 2% bis-acrylamide, 7.5 ml of 4X separating gel buffer, 7.9 ml of dH2O, and 300 μl of 10% SDS in a side-armed Erlenmeyer flask. Swirl gently to mix (see Note 35).
-
3.
To pour a plug for the gel, remove a small volume (∼2 ml) of the polyacrylamide mixture and place it in a disposable borosilicate glass tube (Fisher Scientific Research, Pittsburg, PA). Add 5 μl of TEMED and 5 μl of 30% APS. Vortex briefly. Using a glass Pasteur pipette, immediately pour this mixture between the gel plates.
-
4.
Deaerate the remainder of the polyacrylamide mixture that is left in the side-armed Erlenmeyer flask for approximately 5 min.
-
5.
Once the small volume from step 3 is polymerized, remove the side-armed flask from the vacuum and add 20 μl of TEMED and 50 μl of 30% APS to the polyacrylamide mixture. Swirl gently to mix. Immediately pour this mixture between the gel plates, leaving room for the stacking gel.
-
6.
Gently layer approximately 2 ml of layering buffer on top of the separating gel. The separating gel should take approximately 15 min to polymerize.
-
7.
Pour off the layering buffer and remove any excess with a paper towel.
-
8.
Prepare a stacking gel by mixing together 1.5 ml of 30% polyacrylamide:bis, 2.5 ml of 4X stacking gel buffer, 6 ml of dH2O, and 100 μl of 10% SDS. Swirl gently to mix. Add 10 μl TEMED and 15 μl of 30% APS and immediately pour the polyacrylamide mixture between the gel plates. Immediately insert a comb. The stacking gel should polymerize within approximately 15 min.
-
9.
Remove the comb, add 1X running buffer to the top and bottom chambers of the gel apparatus and, using a 20-gauge needle, rinse out the stacking gel wells, removing any bubbles caught between the plates in the bottom chamber.
-
10.
Add 2X LSB supplemented with 5% (v/v) 2-mercaptoethanol to the samples. Heat the samples for 5 min at 95°C and load them into the stacking gel wells. One well should also contain molecular weight markers.
-
11.
Run the gel on the continuous voltage setting at 50 mA for approximately 1.5 to 2 h until the dye front is about 1 in. from the bottom of the gel (see Note 36).
3.14 Immunoblotting
-
1.
Experimental samples are subjected to SDS-PAGE according to Section 3.13.
-
2.
A transfer cassette is assembled by first disassembling the gel apparatus. Two pieces of chromatography paper are layered in dH2O onto the transfer cassette. A gel-sized piece of PVDF membrane (see Note 7) is briefly wetted in methanol and then layered on top of the chromatography paper. The gel is then layered on top of the PVDF membrane, followed by two additional pieces of chromatography paper. A 10-ml plastic pipette is then rolled across these layers to remove any bubbles trapped between them before closing the transfer cassette.
-
3.
The transfer cassette is put into a transfer apparatus kept in a 4°C cold room, filled with enough cooled transfer buffer to cover the top of the PVDF membrane. The transfer cassette should be oriented so that the gel is closest to the cathode and the PVDF membrane is closest to the anode. Transfer then proceeds for 1 h at 75 V with constant stirring from a stir bar at the bottom of the transfer apparatus.
-
4.
The membrane is then removed from the transfer apparatus and cassette and blocked for 30 min at room temperature in blocking buffer with rocking.
-
5.
The membrane is then rinsed briefly twice in TTBS at room temperature and incubated with primary antibodies overnight at 4°C.
-
6.
The membrane is washed twice for 15 min each in TTBS at room temperature, followed by two additional washes for 5 min each in TTBS at room temperature.
-
7.
Membranes are incubated in secondary antibodies for 1 h at room temperature.
-
8.
Step 6 is repeated.
-
9.
The membrane is incubated for 1 min with HRP substrate. Excess substrate is removed by briefly patting the membrane with chromatography paper. The membrane is then wrapped in a small amount of saran wrap.
-
10.
The membrane is either exposed to film for an appropriate amount of time or imaged using a VersaDoc Model 5000 imaging system (BioRad) with an attached cooled CCD AF Nikkor camera (Nikon, Inc., Melville, NY). Quantitation of images taken with the VersaDoc imaging system can then be performed using Quantity One software (BioRad).
4 Notes
-
1.
We have noted the commercial source of our chemicals throughout this chapter. However, unless specifically stated otherwise, chemicals from other commercial sources are usually acceptable substitutes.
-
2.
dH2O: water having a resistivity of 18.2 MΩ-cm
-
3.
Other cell types can be used with this transient transfection protocol. However, the protocol listed here is optimized for Vero and DF1 cells. To use this protocol with other cell types, the total amount of DNA and the DNA:Fugene ratio will have to be optimized. Additionally, other transfection reagents may give higher transfection efficiencies with different cell types.
-
4.
Paraformaldehyde solutions will corrode pH electrodes; therefore, pH paper should always be used when testing the pH of paraformaldehyde solutions.
-
5.
Other types of mounting solutions that contain fluorescence stabilizing chemicals can generally be used with this protocol. However, if secondary antibodies are conjugated to cyanine dyes (such as Cy2, Cy3, or Cy5), then mounting solutions that contain aromatic amines (such as phenylenediamine) should not be used.
-
6.
As an alternative to using washed pansorbin, protein A-sepharose can be used to precipitate antigen-antibody complexes.
-
7.
The IBV 3a and IBV 3b proteins do not bind well to nitrocellulose membranes. The IBV E protein binds to both nitrocellulose and PVDF membranes equally well.
-
8.
Peptide purities greater than 90% can be obtained from commercial companies for an increased price. However, in our experience, 90%-pure peptides are sufficient for obtaining rabbit sera specific to the protein of interest.
-
9.
If a peptide corresponding to the C-terminus of a protein is synthesized, then the extra cysteine should be added to the N-terminus of the peptide. On the other hand, if a peptide corresponding to the N-terminus of a protein is synthesized, then the extra cysteine should be added to the C-terminus of the peptide.
-
10.
In our experience, ordering IBV 3a, IBV 3b, and IBV E peptides for injecting rabbits has been much more cost- and time-effective than expressing and purifying fusion proteins from bacteria. Expressing and purifying the IBV 3a and IBV 3b fusion proteins in bacteria have been very difficult, requiring optimization of many steps in the protocol listed in Section 3.2. Moreover, high yields of soluble IBV 3a fusion protein have not yet been obtained, likely owing to toxicity to bacteria.
-
11.
In generating the GST-IBV 3b fusion protein, problems were consistently encountered during attempts to scale up protein expression and purification, so multiple small-scale productions were performed instead.
-
12.
Multiple 1000X inducing solution concentrations (1 M, 0.1 M, 0.01 M, and 0.001 M IPTG) were tested for their ability to produce soluble GST-IBV 3b.
-
13.
Multiple inducing temperatures and times were tested for their ability to produce soluble GST-IBV 3b, including 37°C for 2 h, 30°C for 2 h, 25°C for 2 h, 16°C for 24 h, 16°C for 48 h, 13.5°C for 24 h, 13.5°C for 48 h, 10°C for 24 h, and 10°C for 48 h. The temperature and time that best induced soluble protein was 13.5°C for 48 h. Additionally, one-half of the volume of water in the shaking water bath was added immediately before tubes were added, so that the water temperature gradually decreased over several minutes to 13.5°C.
-
14.
Any frothing of samples during the sonication process increases the likelihood of denaturing GST fusion proteins so that they are unable to bind to glutathione sepharose beads in subsequent steps.
-
15.
Multiple elution times were tested for their ability to yield high GST-IBV 3b concentrations, including 2 h, 4 h, 10 h, 24 h, and 48 h.
-
16.
The same protocol is used for injecting rabbits with peptides, except that the micrograms of peptide used for each injection is doubled.
-
17.
Rats can also be immunized to generate sera for co-localization experiments with rabbit sera. This protocol is also used for immunizing rats, except that 200 μg of protein is required for the initial injection and 100 μg of protein is required for all subsequent injections.
-
18.
This injection schedule can be terminated early if rabbit sera show high enough titers following early injections.
-
19.
In our experience, screening sera by in vitro transcription and translation of protein is essential. We first obtained anti-IBV 3b sera from injecting rabbits with IBV 3b peptides. However, we screened these sera only by immunoblotting and indirect immunofluorescence microscopy of transiently transfected and infected Vero cells. No specific IBV 3b staining was seen during this screening; therefore, we assumed that anti-IBV 3b antibodies were not generated. This result led us to inject rabbits with purified GST-IBV 3b fusion proteins. Screening of these new sera using in vitro transcribed and translated IBV 3b demonstrated that high titers of anti-IBV 3b antibodies had been generated (2 (9). However, IBV 3b could still not be detected in Vero cells. Further experiments demonstrated that the extremely short half-life of IBV 3b in transiently transfected or infected Vero cells hampers its visualization in these cells. However, IBV 3b is readily detected in chicken cells since it is turned over differently in these cells. Subsequent experiments demonstrated that the antibody production using IBV 3b peptides showed similar staining patterns in chicken cells as the antibodies generated from GST-IBV 3b fusion proteins. IBV 3b was eventually detected by immunoprecipitation from Vero cells by increasing the dish size and the amount of radioactive label. Representative results are shown in Fig. 14.4.
Fig. 4. 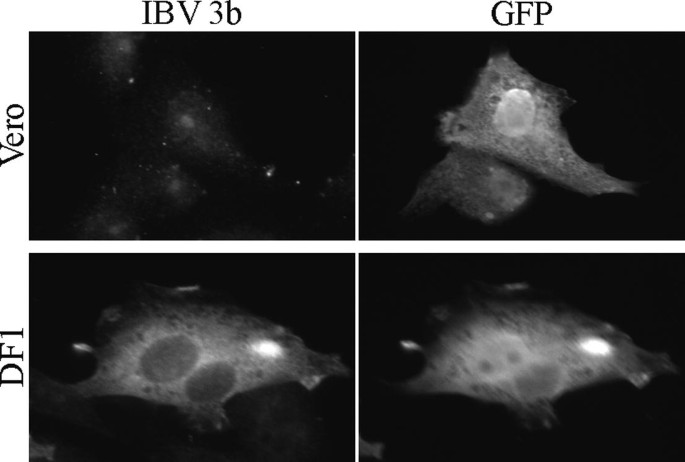
Cell type may affect the expression of coronavirus accessory proteins. Plasmids encoding IBV 3b and green fluorescent protein (GFP) were transiently transfected into Vero and DF1 cells according to the protocol in Section 3.9. At 24 h posttransfection, cells were processed for indirect immunofluorescence microscopy according to the protocol in Section 3.11 with rabbit anti-IBV 3b and mouse anti-GFP primary antibodies. IBV 3b could not be detected in Vero cells, but was readily detected in DF1 cells. Subsequent experiments demonstrated that a greatly decreased IBV 3b half-life in Vero cells was responsible for the inability to visualize the protein in Vero cells (2 (9).
-
20.
Enzyme-linked immunosorbent assay (ELISA) methods can be used for screening sera. However, antibodies may show different binding affinities depending on the methods used. Therefore, screening sera using methods that will be used in future experiments is generally more informative.
-
21.
The amount of primary antibody needed for immunoprecipitation reactions must be titered to ensure that antibodies are saturating. Therefore, immunoprecipitation procedures are repeated with the supernatant to confirm that antibodies are in excess to antigen. We generally use 2 or 3 μl of serum for each immunoprecipitation.
-
22.
Antibodies are often able to access their epitopes differently under different immunofluorescence microscopy fixation and permeabilization conditions. Therefore, alternate fixation and permeabilization conditions may have to be employed when screening antibodies: (i) concurrent fixation and permeabilization in PBS supplemented with 4% paraformaldehyde and 0.18% TritonX-100 for 10 min at room temperature; (ii) concurrent fixation and permeabilization in methanol for 5 min at –20°C; (iii) fixation in 3% paraformaldehyde for 10 min at room temperature followed by permeabilization in methanol of 5 min at –20°C; (iv) concurrent fixation and permeabilization in a 75% methanol, 25% acetic acid solution for 10 min at room temperature; (v) fixation in 3% paraformaldehyde for 10 min at room temperature followed by permeabilization in PBS supplemented with 0.05% saponin and 1% bovine serum albumin. For alternate protocol (v) all subsequent washes and incubations with antibodies should be done in PBS/Gly supplemented with 0.05% saponin and 1% bovine serum albumin.
-
23.
Affinity purification using GST fusion proteins: wash the column with 10 ml of kit equilibration buffer #1, followed by an additional wash with 10 ml of kit equilibration buffer #2.
-
24.
Affinity purification using GST fusion proteins: add 1 ml of the diluted protein to the column and incubate them together for 60 min at room temperature.
-
25.
The column is eluted with 9 ml of kit equilibration buffer #2. 2-ml fractions are collected.
-
26.
If the absorbance of the eluate is similar (given dilution factors) to the absorbance measured at the end of step 2, then the eluate should be reincubated with the coupling column and reeluted.
-
27.
Affinity purification using GST fusion proteins: In order to remove antibodies that recognize GST, the entire procedure found in Section 3.6 can first be done using a column containing GST-coupled beads. The flow-through from this step is saved (it contains antibodies specific to the viral protein) and incubated with a different column that has the GST fusion protein coupled to it.
-
28.
After fractions are collected, the column can be stored for reuse. To store the column it must first be washed with 10 ml of 0.02% (w/v) NaN3 in PBS. 2 ml of storage buffer is then added and the column is stored at 4°C. Before reuse, the column should be washed once in elution buffer.
-
29.
To obtain quality images by indirect immunofluorescence microscopy, cells should be infected or transfected when they are approximately 50% confluent. However, to obtain higher levels of protein expression when immunoblotting or immunoprecipitating, cells can be infected or transfected when they are as high as 70–90% confluent.
-
30.
As a control for antibody specificity, sera from rabbits immunized with peptides can be preincubated with purified peptide before use in indirect immunofluorescence microscopy. 20 μl of serum is mixed with 20 μl of a solution containing 10 mg of peptide per 10 μl of dH2O and rotated for 1 h at 4°C. 160 μl of a solution containing 1% (v/v) bovine serum albumin in PBS/Gly is then added to the antibody mixture and rotated overnight at 4°C. This antibody mixture is then diluted approximately 1:10 and used as the primary antibody in Section 3.11. Representative results of peptide-blocked versus unblocked antibody are shown in Fig. 14.5.
Fig. 5. 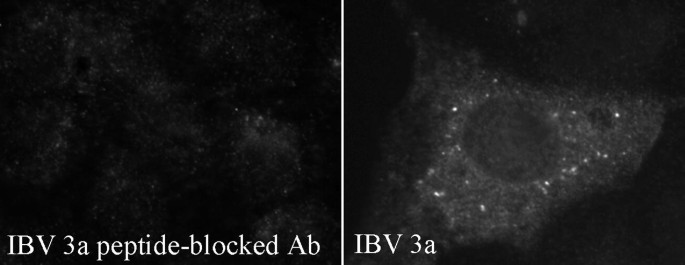
Preincubation of sera with the peptides used to immunize rabbits can test antibody specificity. IBV-infected Vero cells (Section 3.7) were subjected to indirect immunofluorescence microscopy (Section 3.11) at 15 h postinfection. Sera from rabbits immunized with IBV 3a peptides were preincubated with either the IBV 3a peptide or dH2O, as detailed in Note 30.
-
31.
For visualization on an inverted microscope, coverslips can be sealed onto the slides by brushing boat sealer or nail polish around the edges of the coverslip.
-
32.
For visualizing immunoprecipitated, unlabeled proteins, start the Section 3.12 protocol at step 2 and continue through step 9. Then perform the entire immuno- blotting protocol. Rabbit primary antibodies can be used if the protein of interest is less than 20 kDa in size (approximately the size of the rabbit IgG light chains). Alternatively, primary antibodies from a different species can be used for immunoblotting.
-
33.
Proteins with half-lives shorter than 30 min, such as IBV 3b when expressed in Vero cells (29), may not be easily visualized in certain cell types. Therefore, cells may have to be plated in larger dishes and incubated with increased amounts of radioactivity.
-
34.
For reproducible results, gels must be washed no longer than 10 min in dH2O. Longer wash times allow IBV 3a and IBV 3b to diffuse out of the gel. Fixation of gels with 10% (w/v) trichloroacetic acid did not prevent diffusion of these proteins out of the gel in longer wash times.
-
35.
Clear visualization of the IBV 3a and IBV 3b proteins is best achieved on 17.5% low-bis polyacrylamide gels. However, the slightly larger IBV E protein is easily visualized on 12 or 15% low-bis polyacrylamide gels. The same ratio of polyacrylamide:bis-polyacrylamide used when making 17.5% low-bis gels should also be used when making 12 or 15% low-bis gels.
-
36.
If the gel becomes too hot while electrophoresing, then the lanes at the edge of the gel run slower than those in the middle, causing a “smiling” effect. To minimize this possibility, fans and/or cold packs can be used to help cool gels.


References
Navas-Martin, S.R., and Weiss, S. (2004) Coronavirus replication and pathogenesis: Implications for the recent outbreak of severe acute respiratory syndrome (SARS), and the challenge for vaccine development. J. Neurovirol. 10, 75–85.
Baudoux, P., Carrat, C., Besnardeau, L., Charley, B., and Laude, H. (1998) Coronavirus pseudoparticles formed with recombinant M and E proteins induce alpha interferon synthesis by leukocytes. J. Virol. 72, 8636–8643.
Corse, E., and Machamer, C. E. (2000) Infectious bronchitis virus E protein is targeted to the Golgi complex and directs release of virus-like particles. J. Virol. 74, 4319–4326.
Maeda, J., Maeda, A., and Makino S. (1999) Release of coronavirus E protein in membrane vesicles from virus-infected cells and E protein-expressing cells. Virology 263, 265–272.
Vennema, H., Godeke, G. J., Rossen, J.W., et al. (1996) Nucleocapsid-independent assembly of coronavirus-like particles by co-expression of viral envelope protein genes. EMBO J. 15, 2020–2028.
Fischer, F., Stegen, C.F., Masters, P.S., and Samsonoff ,W.A. (1998) Analysis of constructed E gene mutants of mouse hepatitis virus confirms a pivotal role for E protein in coronavirus assembly. J. Virol. 72, 7885–7894.
Arbely, E., Khattari, Z., Brotons, G., Akkawi, M., Salditt, and T., Arkin, I. T. (2004) A highly unusual palindromic transmembrane helical hairpin formed by SARS coronavirus E protein. J. Mol. Biol. 34, 769–779.
Raamsman, M.J., Locker, J.K., de Hooge, A., et al. (2000) Characterization of the coronavirus mouse hepatitis virus strain A59 small membrane protein E. J. Virol. 74, 2333–2342.
Ortego, J., Escors, D., Laude, H., and Enjuanes, L. (2002) Generation of a replication-competent, propagation-deficient virus vector based on the transmissible gastroenteritis coronavirus genome. J. Virol. 76, 11518–11529.
Kuo, L., and Masters, P. S. (2003) The small envelope protein E is not essential for murine coronavirus replication. J. Virol. 77, 4597–4608.
Liao, Y., Lescar, J., Tam, J.P., and Liu, D.X. (2004) Expression of SARS-coronavirus envelope protein in Escherichia coli cells alters membrane permeability. Biochem. Biophys. Res. Commun. 325, 374–380.
Liao, Y., Yuan, Q., Torres, J., Tam, J. P., and Liu, D. X. (2006) Biochemical and functional characterization of the membrane association and membrane permeabilizing activity of the severe acute respiratory syndrome coronavirus envelope protein. Virology 349, 264–275.
Wilson, L., McKinlay, C., Gage, P., and Ewart, G. (2004) SARS coronavirus E protein forms cation-selective ion channels. Virology 330, 322–331.
Madan, V., Garcia, Mde J., Sanz, M.A., and Carrasco, L. (2005) Viroporin activity of murine hepatitis virus E protein. FEBS Lett. 579, 3607–3612.
Brown, T. D. K., and Brierley, I. (1995) The coronavirus nonstructural proteins. In: Siddell, S.G. (ed.) The Coronaviridae. New York: Plenum Press: pp. 191–217.
Youn, S., Leibowitz, J.L., and Collisson, E. W. (2005) In vitro assembled recombinant infectious bronchitis viruses demonstrate that the 5a open reading frame is not essential for replication. Virology 332, 206–215.
Casais, R., Davies, M., Cavanagh, D., Britton, P. (2005) Gene 5 of the avian coronavirus infectious bronchitis virus is not essential for replication. J. Virol. 79, 8065–8078.
Shen, S., Wen, Z. L., and Liu, D. X. (2003) Emergence of a coronavirus infectious bronchitis virus mutant with a truncated 3b gene: functional characterization of the 3b protein in pathogenesis and replication. Virology 311, 16–27.
de Haan, C. A. , Masters, P.S., Shen, X., Weiss, S., and Rottier, P. J. (2002) The group-specific murine coronavirus genes are not essential, but their deletion, by reverse genetics, is attenuating in the natural host. Virology 296, 177–189.
Haijema, B.J., Volders, H., and Rottier, P. J. (2004) Live, attenuated coronavirus vaccines through the directed deletion of group-specific genes provide protection against feline infectious peritonitis. J. Virol. 78, 3863–3871.
Curtis, K.M., Yount, B., and Baric, R. S. (2002) Heterologous gene expression from transmissible gastroenteritis virus replicon particles. J. Virol. 76, 422–434.
Ortego, J., Sola, I., Almazan, F., et al. (2003) Transmissible gastroenteritis coronavirus gene 7 is not essential but influences in vivo virus replication and virulence. Virology 308, 13–22.
Sola, I., Alonso, S., Zuniga, S., Balasch, M., Plana-Duran, J., and Enjuanes, L.. (2003) Engineering the transmissible gastroenteritis virus genome as an expression vector inducing lactogenic immunity. J. Virol. 77, 4357–4369.
Liu, D. X., Cavanagh, D, Green, P., and Inglis, S. C. (1991) A polycistronic mRNA specified by the coronavirus infectious bronchitis virus. Virology 184, 531–544.
Liu, D. X., and Inglis, S.C. (1992) Internal entry of ribosomes on a tricistronic mRNA encoded by infectious bronchitis virus. J. Virol. 66, 6143–6154.
Corse, E., and Machamer, C. E. (2002) The cytoplasmic tail of infectious bronchitis virus E protein directs Golgi targeting. J. Virol. 76, 1273–1284.
Corse, E., and Machamer, C. E. (2003) The cytoplasmic tails of infectious bronchitis virus E and M proteins mediate their interaction. Virology 312, 25–34.
Pendleton, A. R, and Machamer, C. E. (2005) Infectious bronchitis virus 3a protein localizes to a novel domain of the smooth endoplasmic reticulum. J. Virol. 79, 6142–6151.
Pendleton, A. R., and Machamer, C. E. (2006) Differential localization and turnover of infectious bronchitis virus 3b protein in mammalian versus avian cells. Virology 345, 337–345.
Machamer, C. E., and Rose, J. K. (1987) A specific transmembrane domain of a coronavirus E1 glycoprotein is required for its retention in the Golgi region. J. Cell. Biol. 105, 1205–1214.
Fuerst, T.R., Niles, E. G., Studier, F. W., and Moss B. (1986) Eukaryotic transient-expression system based on recombinant vaccinia virus that synthesizes bacteriophage T7 RNA polymerase. Proc. Natl. Acad. Sci. USA 83, 8122–8126.
Acknowledgments
We thank Emily Corse for the following: (i) designing the IBV 3a, IBV 3b, and IBV E peptides, (ii) prescreening rabbits before immunization with the IBV 3a, IBV 3b, and IBV E peptides, and (iii) screening anti-IBV E sera. This work was supported by National Institutes of Health grants GM42522 and GM64647.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2008 Humana Press, a part of Springer Science+Business Media, LLC
About this protocol
Cite this protocol
Pendleton, A.R., Machamer, C.E. (2008). Generating Antibodies to the Gene 3 Proteins of Infectious Bronchitis Virus. In: Cavanagh, D. (eds) SARS- and Other Coronaviruses. Methods in Molecular Biology, vol 454. Humana Press, Totowa, NJ. https://doi.org/10.1007/978-1-59745-181-9_14
Download citation
DOI: https://doi.org/10.1007/978-1-59745-181-9_14
Published:
Publisher Name: Humana Press, Totowa, NJ
Print ISBN: 978-1-58829-867-6
Online ISBN: 978-1-59745-181-9
eBook Packages: Springer Protocols