
Abstract
Some DNA fragments are difficult to clone in Escherichia coli by standard methods. It has been speculated that unintended transcription and translation result in expression of proteins that are toxic to the bacteria. This problem is frequently observed during assembly of infectious full-length virus clones. If the clone is constructed for transcription in vivo, interrupting the virus sequence with an intron can solve the toxicity problem. The AU-rich introns generally contain many stop codons, which interrupt translation in E. coli, while the intron sequence is precisely eliminated from the virus sequence in the plant nucleus. The resulting RNA, which enters the cytoplasm, is identical to the virus sequence and can initiate infection.
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others



Keywords
1 Introduction
Infectious cDNA clones are useful for analysis of various properties of plant viruses. The cDNA clone allows introduction of specific mutations and exchange, deletion, or insertion of genes or gene fragments. Unfortunately, full-length cDNA clones of many RNA viruses have proved difficult or even impossible to clone and amplify in Escherichia coli. Full-length clones have frequently turned out to be noninfectious, possibly due to spontaneous mutations or deletions, which reduces toxicity to E. coli. The cause for the toxicity to E. coli is not known, but it has been observed that artificially introduced deletions; insertions or frame shifts can alleviate the cloning problems (1, 2).
Insertion of an intron into the virus sequence has proven to be an efficient way to facilitate maintenance of the cDNA in E. coli (1, 3, 4, 5). Upon inoculation of the cDNA to the host cell, the intron is precisely spliced from the precursor nuclear messenger RNA (pre-mRNA) in the nucleus, and a true copy of the viral RNA is transported to the cytoplasm where infection can start. The function of clones with introns requires cloning of the viral cDNA between promoter and terminator sequences, which are active in the host cell. The 35S promoter and NOS terminator are frequently used in plants (see Chaps. 32 and 33 for a description of construction of infectious clones of RNA viruses). Cloning of the viral cDNA in this context also allows inoculation by agroinfiltration, if the transcription cassette is moved to a binary vector (see Chap. 38 for a description of agroinoculation).
Construction of infectious clones with introns has been reported for viruses belonging to the genera Potyvirus (1, 4, 6) Tobravirus (7, 8), Coronavirus (3), and Flavivirus (5). In theory, intron insertion should be applicable for clones of viruses, which are replicated from an RNA template in the cytoplasm.
The sequences required for pre-mRNA intron splicing lie mainly within the intron. These include consensus sequences at the 5′ (GU) and 3′ (AG) ends of the intron; the branchpoint located 18–40 nucleotides upstream of the 3′ splice site and AU-rich sequence elements (9). Furthermore, introns are characterized by high U or AU content compared to more GC-rich exons. In the sequence next to the intron, there is a high representation of A and G at the two positions next to the 5′ end of the intron and G at the first positions 3′ to the intron (10). This is shown as a consensus sequence in Fig. 1. Therefore, it is probably safest to insert introns at positions in the virus sequence matching the AG/G consensus.
There are minor differences between monocot and dicot introns (10), and therefore, it is recommended to select introns from the class in which the infectious clone is going to be analyzed. Furthermore, it should be noted that the monocot sequences included in the analysis by (10) almost exclusively represent introns from the order Poalis. Some differences have been observed between species within the dicots (9). However, at present there is no evidence that differences in dicot introns are important for their use in virus clones. For example, Arabidopsis introns were used in Tobacco mosaic virus, which efficiently infected two species of Nicotiana (11), and an intron from Solanum tuberosum is spliced in Pisum sativum and Capsicum annuum (1, 6).
Introns can also be inserted into native PstI restrictions sites as described in detail by (1). Here, we describe a more flexible method based on overlap extension PCR (12).
2 Materials
2.1 Isolation of Genomic DNA as Template for Intron Amplification
-
1.
Plant material of a monocot or a dicot, depending on the host of the virus
-
2.
Liquid nitrogen and safety glasses
-
3.
2X CTAB buffer: 1.4 M NaCl, 2% cetyl trimethylammonium bromide (CTAB), 100 mM Tris pH 8.0, 20 mM EDTA, 2% polyvinylpyrrolidone (PVP), 0.2% β-mercaptoethanol (added just before use)
-
4.
Waterbath at 65°C
-
5.
Chloroform/isoamylalcohol (24:1)
-
6.
Cold isopropanol, 3 M sodium acetate, and 70% cold EtOH
-
7.
Or use a commercial kit for plant DNA purification
2.2 Identification of Suitable Introns
-
1.
Computer access to NCBI and related resources
-
2.
Sequence data of virus region flanking the intron insertion site
2.3 In silico Test of the Intron Insertion
-
1.
Program to assemble virus sequence with the intron. Either just a word processing program or a designated program for management of sequences
-
2.
Computer access to ‘NetPlantGene Server’ http://www.cbs.dtu.dk/services/NetPGene/(13) or another splice site prediction program
2.4 Primers and Design
-
1.
Commercial supplier of oligonucleotide primers
2.5 Amplification of Introns and Virus Sequences Flanking the Introns
-
1.
Overlapping cDNA clones covering the complete genome of the virus. Make a dilution of 10–100 ng μL−1 plasmid DNA as template for PCR
-
2.
A complete nucleotide sequence of the virus
-
3.
PCR primers (3FW and 4RV) annealing to 5′ and 3′ ends of the intron (Fig. 2)
Fig. 1 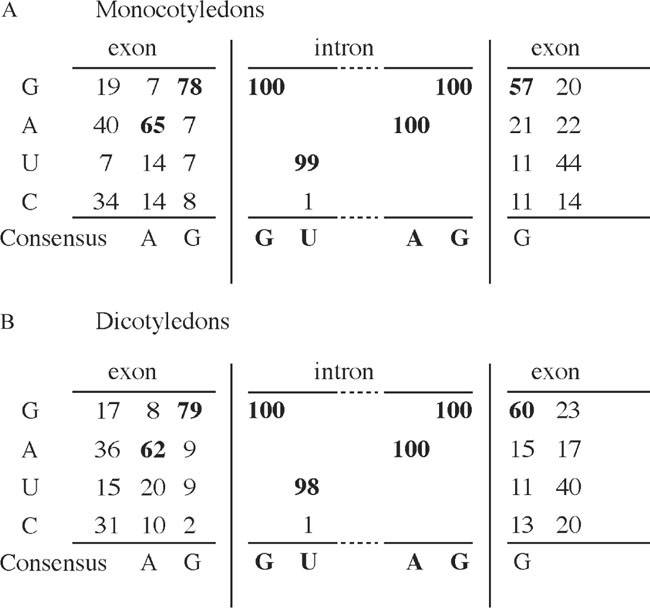
Consensus sequence of plant intron splice sites based on data from (Simpson et al., 1993). (a) Consensus in monocotelydons based on the percentage occurrence of nucleotides at exonintron borders, (b) consensus in dicotelydons based on the percentage occurrence of nucleotides at exon-intron borders
-
4.
PCR primers to amplify the virus sequence 5′ (1FW and 2RV) and 3′ (5RW and 6RV) to the intron insertion site (Fig. 2)
-
5.
Heat stable DNA polymerase, buffer, and 5 mM dNTP mix for PCR
-
6.
Kit to purify DNA from agarose gel fragments

2.6 Sequence Overlap Extension PCR (SOE-PCR)
-
1.
Commercial DNA marker to estimate DNA concentration of the purified PCR fragments
-
2.
Heat stable DNA polymerase, buffer, and 5 mM dNTP mix for PCR
-
3.
Kit for cloning of PCR fragments
3 Methods
3.1 Isolation of Genomic DNA as Template for Intron Amplification
-
1.
Add mercaptoethanol to the 2X CTAB buffer
-
2.
Grind 100 mg plant material in a 1.5-ml microfuge tube immersed in liquid nitrogen. Use safety glasses
-
3.
Add 500 μL 2X CTAB buffer, mix, and incubate at 65°C for 30–60 min
-
4.
Centrifuge at 5000g for 10 min at room temperature
Fig. 2 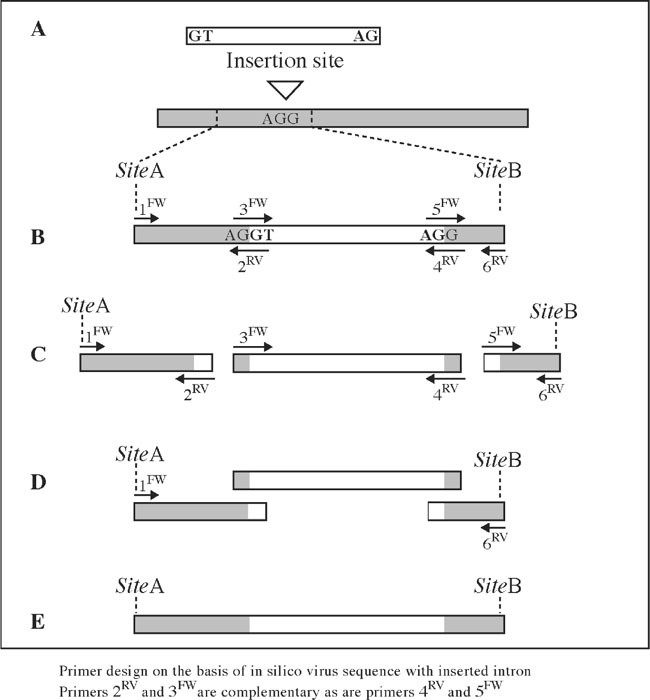
Intron insertion using sequence overlap extension PCR (SOE-PCR). (a) Selection of an intron and intron insertion site matching the consensus. The insertion site should be situated between restriction sites SiteA and SiteB, which are suitable for reinserting the intron containing fragment in the complete virus sequence. (b) Primers are designed on the basis of an in silico construct. Primers indicated as arrows above the construct are virus sense, while primers below are antisense. Primers 1FW and 6RV contain the sequence of SiteA and SiteB, respectively. Complementary primers 3FW and 2RV at the 5′ exon-intron border and 5FW and 4RV at the 3′ intronexon border are approximately 50 nucleotides in length containing approximately 20–25 nucleotides of virus sequence and 20–25 nucleotides of intron sequence. (c) Three separate PCR reactions are performed to amplify overlapping fragments of the region 5′ to the intron (primers 1FW and 2RV), the intron (primers 3FW and 4RV), and the region 3′ to the intron (primers 5FW and 6RV). (d) SOE-PCR is performed on a mixture of the three purified PCR fragments and primers 1FW and 6RV. (e) The PCR fragment of the virus sequence with intron. This fragment is separated from primers and unspecific fragment by gel electrophoresis, cloned, and checked by sequencing before it is reinserted into the complete virus sequence
-
5.
Transfer the supernatant to a clean microfuge tube
-
6.
Add an equal volume of chloroform/isoamylalcohol, shake the tube for 10 min
-
7.
Centrifuge at 5000g for 2 min at room temperature
-
8.
Repeat steps 5–7
-
9.
Add 0.8 volume cold isopropanol and 0.1 volume 3 M sodium acetate
-
10.
Centrifuge at 12000g for 15 min at room temperature
-
11.
Remove the supernatant and add 1 mL 70% cold EtOH
-
12.
Centrifuge at 12000g for 2 min at room temperature
-
13.
Remove the supernatant and dry the DNA
-
14.
Dissolve in 100 μL sterile H2O
-
15.
Or follow the instructions provided with the plant DNA purification kit

3.2 Identification of Suitable Introns
-
1.
Identify introns from complete gene sequences, for example, in the NCBI database http://www.ncbi.nlm.nih.gov/entrez/query.fcgi
-
2.
Select small introns (100–300 basepairs) with a sequence matching the splice site consensus as shown in Fig. 1
-
3.
We have used the second intron (189 basepairs) of the ST-LS1 gene from S. tuberosum (accession X04753 nucleotide 2648–2836) and intron 2 (221 basepairs) from the NiR gene from Phaseolus vulgaris (accession U10419 nucleotide 3536–3756) successfully in N. benthamiana, P. sativum, and C. annuum.
3.3 In silico Test of the Selected Intron Inserted in the virus Sequence
-
1.
If introns are inserted into the virus sequence in order to reduce toxicity, the intron insertion site should be placed in a region, which has been difficult to clone or amplify in E. coli. In addition the insertion site should match the AG/G exon border sequences as indicated in Fig. 1 and Fig. 2a (see note 1)
-
2.
Paste in the intron sequence and run the virus sequence with the intron through a program, which predicts intron splice sites (e.g., NetPlantGene Server).
-
3.
The output will display potential donor sites, acceptor sites, and branchpoints. Only the transcribed virus sense strand is relevant.
-
4.
Check that the intron is recognized in the virus sequence. If the intron is not recognized, then try another insertion site and/or another intron sequence.
-
5.
Also inspect the output for cryptic introns in the virus sequence, which may interfere with correct processing of the virus sequence (see note 1).
3.4 Primers and Design
-
1.
Identify restriction sites, SiteA and SiteB, flanking the intron insertion site, which are suitable for reintroduction of the intron containing subclone into the full-length clone or a larger subclone (see note 7) Choose restriction sites that are no more than 2 kb apart to avoid problems, which may arise with generation of long PCR products.
-
2.
Six primers are needed as shown in Fig. 2b.
-
a.
Primers 1FW and 6RV are forward and reverse primers annealing to the regions with the restriction sites SiteA and SiteB (see note 2). Primers 1FW and 6RV are designed like normal PCR primers and it is advisable to check them for primer dimer formation and proper annealing the target sequence using for example vector NTI, which is available at http://www.invitrogen.com
-
b.
Primers 3FW and 2RV are complementary. Primer 3FW is composed of 20–25 nucleotides matching the 3′ end of the virus sequence upstream the intron and 20–25 nucleotides matching the 5′ end of the intron. Primer 2RV is composed of 20–25 nucleotides complementary to the 5′ end of the intron and 20–25 nucleotides complementary to the virus sequence upstream the intron.
-
c.
Primers 5FW and 4RV are complementary. Primer 5FW is of composed 20–25 nucleotides matching the 3′ end of the intron sequence and 20–25 nucleotides matching the 5′ end of the virus downstream the intron. Primer 4RV is composed of 20–25 nucleotides complementary to the virus sequence downstream the intron and 20–25 nucleotides complementary to the 3′ end of the intron (see note 3).
-
a.
-
3.
Make a 10 μM dilution of each primer in H2O, mix well, and store at −20°C.
3.5 Amplification of Introns and Virus Sequences Flanking the Introns
-
1.
Three separate PCR reactions are set up to amplify overlapping fragments: The virus region 5′ to the intron, the intron and the region 3′ to the intron (Fig. 2c). The virus regions 5′ and 3′ to the intron are amplified from a virus plasmid cDNA clone with primer pairs 1FW/2RV and 5FW/6RV, respectively. The intron is amplified with primers 3FW and 4RV from purified plant DNA. Optimized heatstable high-fidelity DNA polymerases with proofreading are recommended for the PCR. For example, Expand or Phusion, whereas pure Pwo or Pfu are less efficient.
-
2.
The following two protocols are based on the instructions provided by the suppliers. Please refer to these instructions for further information and troubleshooting. Note also that other products can be used.
-
2.1.
PCR reaction in 50 μL with ExpandTM (Roche). Thaw, mix, and centrifuge all solutions except the enzyme before use. Place on ice and assemble reactions on ice. Add in the following order: 38.25 μL H2O; 5 μL 10X Expand High Fidelity with 15 mM MgCl2 buffer; 2 μL 5 mM dNTPs; 1.5 μL 10 μM FW primer; 1.5 μL 10 μM RV primer; 1 μL template DNA (plasmid cDNA clone or genomic DNA); 0.75 μL Expand High Fidelity enzyme mix, mix, and centrifuge. Run the following PCR scheme: Initial denaturation 94°C for 2 min; 30 cycles of denaturation at 94°C for 15 s, annealing at 55°C for 30 s (see note 4), extension at 72°C for 60 s per 1,500 bases; final extension at 72°C for 7 min; 4°C. Go to 3.
-
2.2.
PCR reaction in 50 μL with Phusion™ (Finnzymes). Thaw, mix, and centrifuge all solutions except the enzyme before use. Place on ice and assemble reactions on ice. Add in the following order: 31.5 μL H2O (30 μL if DMSO is included); 10 μL 5X Phusion HF buffer; 2 μL 5 mM dNTPs; 2.5 μL 10 μM FW primer; 2.5 μL 10 μM RV primer; 1 μL template DNA (plasmid cDNA clone or genomic DNA); 1.5 μL DMSO (optional); 0.5 μL Phusion DNA polymerase, mix, and centrifuge. Run the following PCR scheme: Initial denaturation 98°C for 30 s; 30 cycles of denaturation at 98°C for 10 s, annealing at 55°C for 30 s (see note 5), extension at 72°C for 30 s per 1,000 bases; final extension at 72°C for 5–10 min; 4°C. Go to 3.
-
2.1.
-
3.
A small sample (1–5 μL) of each PCR reaction is analyzed by agarose gel electrophoresis to check that PCR products of the expected sizes have been amplified.
-
4.
To purify the PCR products from primers and possible unspecific products, samples of the PCR reactions containing ⩽ 200 ng of the correct PCR product are separated by electrophoresis on an agarose gel. The bands containing the PCR products are cut out of the gel and the DNA is purified from the gel using a gel band purification kit. Spin columns are very efficient and are provided by several commercial suppliers, for example GFX columns (Amersham Biosciences), MinElute and QIAquick (Qiagen), GenElute™ (Sigma), and SpinPrep™ (Novagen). Follow the supplier's instructions and elute the DNA in 20–25 μL sterile H2O. Check again, by agarose gel electrophoresis of 1–2 μL of the eluted sample, that the PCR products have been recovered and estimate the DNA concentration by comparison to the bands in the DNA size marker.
3.6 Splicing by Overlap Extension PCR (SOE-PCR)
-
1.
SOE-PCR reaction in 50 μL with Expand High Fidelity PCR system (Roche). Thaw, mix, and centrifuge all solutions except the enzyme before use. Place on ice and assemble reactions on ice. Add in the following order: H2O to a total reaction volume of 50 μL; 5 μL 10X Expand High Fidelity with 15 mM MgCl2 buffer; 2 μL 5 mM dNTPs; 1.5 μL 10 μM primer 1FW; 1.5 μL 10 μM primer 6RV; approximately 50 ng of each PCR product from the initial reactions; 0.75 μL Expand High Fidelity enzyme mix, mix, and centrifuge. Run the following PCR scheme: Initial denaturation 94°C for 2 min; 30 cycles of denaturation at 94°C for 15 s, annealing at 55°C for 30 s, extension at 72°C for 60 s per 1,500 bases; final extension at 72°C for 7 min; 4°C. Note that Expand High Fidelity enzyme mix produces PCR products with 3′ dA overhangs. Go to step 2.
SOE-PCR reaction in 50 μL with Phusion™ (Finnzymes). Thaw, mix, and centrifuge all solutions except the enzyme before use. Place on ice and assemble reactions on ice. Add in the following order: H2O to a total reaction volume of 50 μL; 10 μL 5X Phusion HF buffer; 2 μL 5 mM dNTPs; 2.5 μL 10 μM primer 1FW; 2.5 μL 10 μM primer 6RV; approximately 50 ng of each PCR product from the initial reactions; 1.5 μL DMSO (optional); 0.5 μL Phusion DNA polymerase, mix, and centrifuge. Run the following PCR scheme: Initial denaturation 98°C for 30 s; 30 cycles of denaturation at 98°C for 10 s, annealing at 55°C for 30 s, extension at 72°C for 30 s per 1,000 bases; final extension at 72°C for 5–10 min; 4°C. Note that Phusion™ produces blunt end PCR products. Go to step 2.
-
2.
A small sample (1–5 μL) is analyzed by agarose gel electrophoresis to check that a PCR product of the expected size has been amplified.
-
3.
The PCR product of the expected size is gel purified and the concentration determined. It is recommended to clone the fragment cloned using a T/A PCR cloning cloning kit for products with 3′dA overhangs (Expand) or blunt ends (Phusion™) (see note 6). The cloned fragment should be sequenced to check for errors before it is reinserted into the virus cDNA using restriction sites SiteA and SiteB (see note 7).
4 Notes
-
1.
It is likely that sequences surrounding the intron may affect splicing efficiency, and the virus may contain cryptic 5′ and or 3′ splice sites, which can interfere with splicing of the true intron. The presence of cryptic splice sites in the virus sequence can be identified using, for example, the ‘NetPlantGene Server’ at http://www.cbs.dtu.dk/services/NetPGene/. To prevent cryptic splicing it may be necessary to eliminate either the 5′ or the 3′ splice site. This may be accomplished by silent mutations (Marillonnet et al., 2005). In our experience, cryptic sites do not seem to be a major problem if the goal is to generate an infectious clone because only a fraction of the transcripts need to be correctly spliced to initiate infection. However, insertion of true introns can improve the specific infectivity as demonstrated by Marillonnet et al. (2005). It is also known that sequences in the exon, known as exon splicing enhancers (ESE), are likely to play an important role in plant intron splicing. However, no splicing event in Arabidopsis has yet been shown to be enhancer dependent. For more information see http://www.tigr.org/software/SeeEse/background.html
-
2.
Primers 1FW and 6RV should contain the sequence of the restriction sites SiteA and SiteB, or flank the sites so the restriction sites are contained within the PCR fragments. If the primers contain the restriction sites, the restriction site consensus should be placed at least three nucleotides from the 5′ end of the primer.
-
3.
The length of the annealing sequence will depend on the GC content. It is advisable to use a longer primer for regions with low GC content.
-
4.
Optimal annealing temperature depends on the melting temperature of the primers and the system used. Annealing at 55°C is usually a good starting point unless one of the primers anneal to a short and very AT-rich region. In this case, the annealing temperature should be lower.
-
5.
The optimal annealing temperature will depend on the primer sequences, but in our experience 55°C is a good starting point.
-
6.
PCR fragments are most conveniently cloned using PCR cloning kits adapted for cloning PCR products with dA overhangs or blunt ends. Although the PCR products described here contain restriction sites at least three nucleotides from the ends of the fragment, it is sometimes difficult to clone PCR fragments with the aid of such sites.
-
7.
Multiple fragment ligations can facilitate reinsertion of the fragment containing the intron if the restriction sites SiteA and SiteB are not unique in the full-length clone. We have assembled potyvirus clones from up to five fragments with sticky ends in a single cloning step. The amounts of DNA of each fragment are adjusted when the fragments are cut from the agarose gel and can be purified together on a single spin column
References
1. Johansen, I.E. (1996) Intron insertion facilitates amplification of cloned virus cDNA in Escherichia coli while biological activity is reestablished after transcription in vivo. Proc. Natl. Acad. Sci. USA 93 12400–12405.
2. Satyanarayana, T., Gowda, S., Ayllon, M.A., and Dawson, W.O. (2003) Frameshift mutations in infectious cDNA clones of Citrus tristeza virus: a strategy to minimize the toxicity of viral sequences to Escherichia coli. Virology 313 481–491.
3. Gonzalez, J.M., Penzes, Z., Almazan, F., Calvo, E., and Enjuanes L. (2002) Stabilization of a full-length infectious cDNA clone of transmissible gastroenteritis coronavirus by insertion of an intron. J. Virol. 76 4655–4661.
4. Lopez-Moya, J.J. and Garcia, J.A. (2000) Construction of a stable and highly infectious introncontaining cDNA clone of plum pox potyvirus and its use to infect plants by particle bombardment. Virus Res. 68 99–107.
5. Yamshchikov, V., Mishin, V., and Cominelli, F. (2001) A new strategy in design of (+)RNA virus infectious clones enabling their stable propagation in E. coli. Virology 281 272–280.
6. Moury, B., Morel, C., Johansen, E., Guilbaud, L., Souche, S., Ayme, V., Caranta, C., Palloix, A., and Jacquemond, M. (2004) Mutations in Potato virus Y genome-linked protein determine virulence toward recessive resistances in Capsicum annuum and Lycopersicon hirsutum. Mol. Plant Microbe Interact. 17 322–329.
7. Constantin, G.D., Krath, B.N., MacFarlane, S.A., Nicolaisen, M., Johansen, I.E., and Lund, O.S. (2004) Virus-induced gene silencing as a tool for functional genomics in a legume species. Plant J. 40 622–631
8. Ratcliff, F., Martin-Hernandez, A.M., and Baulcombe, D. (2001) Tobacco rattle virus as a vector for analysis of gene function by silencing. Plant J. 25 237–245.
9. Brown, J.W.S. and Simpson, C.G. (1998) Splice site selection in plant pre-mRNA splicing. Annu. Rev. Plant Physiol. Mol. Biol. 49 77–95.
10. Simpson, C.G., Leader, D.J., and Brown, J.W.S. (1993) Characteristics of plant pre-mRNA introns. In Plant Molecular Biology Labfax, (Croy, R. R. D., ed.), BIOS Sci., Oxford, pp. 183–251.
11. Marillonnet, S., Thoeringer, C., Kandzia, R., Klimyuk, V., and Gleba, Y. (2005) Systemic Agrobacterium tumefaciens-mediated transfection of viral replicons for efficient transient expression in plants. Nat. Biotechnol. 23 718–723.
12. Horton, R.M., Hunt, H.D., Ho, S.N., Pullen, J.K., and Pease, L.R. (1989) Engineering hybrid genes without the use of restriction enzymes: Gene splicing by overlap extension. Gene 77 61–68.
13. Hebsgaard, S.M., Korning, P.G., Tolstrup, N. Engelbrecht, J., Rouze, P., and Brunak, S. (1996) Splice site prediction in Arabidopsis thaliana DNA by combining local and global sequence information. Nucleic Acids Res. 24 3439–3452.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2008 Humana Press, a part of Springer Science + Business Media, LLC
About this protocol
Cite this protocol
Johansen, I.E., Lund, O.S. (2008). Insertion of Introns: A Strategy to Facilitate Assembly of Infectious Full Length Clones. In: Foster, G.D., Johansen, I.E., Hong, Y., Nagy, P.D. (eds) Plant Virology Protocols. Methods in Molecular Biology™, vol 451. Humana Press. https://doi.org/10.1007/978-1-59745-102-4_36
Download citation
DOI: https://doi.org/10.1007/978-1-59745-102-4_36
Publisher Name: Humana Press
Print ISBN: 978-1-58829-827-0
Online ISBN: 978-1-59745-102-4
eBook Packages: Springer Protocols