
Abstract
Microarray technology can rapidly generate large databases of gene expression profiles. Our laboratory has applied these techniques to analyze differential gene expression in cardiac tissue and cells based on mouse heart transplantation. We have analyzed the different gene expression profiles such as stress or injury including ischemia following transplantation. We also have investigated the role of infiltrating inflammatory cells during graft rejection by purifying subsets of infiltrating cells using GFP transgenic mice and detailed all technical experiences and issues. The purpose of this study is to assist researchers to simplify the process of analyzing large database using microarray technology.
Similar content being viewed by others


Key Words
1 Introduction
The analysis of gene expression profiles using microarray technology is a powerful approach to investigate the functions of specific tissues or cells. Our laboratory has applied these techniques to analyze differential gene expression in cardiac tissue and cells in a model of murine heart transplantation (1–4). Specifically, we have analyzed the response by cardiac cells to various forms of stress or injury including ischemia following transplantation (5). In addition, we have investigated the role of infiltrating inflammatory cells during graft rejection by purifying subsets of infiltrating cells. Using current microarray technology, it is possible to analyze approx 45,000 probe sets representing known mouse genes or expressed sequence tags (ESTs). The ability to perform global analyses of gene expression creates the potential to analyze complex biological systems. These methods could be applied to other questions of cardiac development or disease.
2 Materials
-
1.
Collagenase II (Gibco) and pancreatin (Sigma).
-
2.
D-phosphate-buffered saline (PBS; Gibco).
-
3.
Tri Reagent (Gibco-BRL Life Technologies, Rockville, MD).
-
4.
Dnase I (Invitrogen).
-
5.
SuperScript II (Invitrogen).
-
6.
ALTRA flow cytometer (Beckman Coulter).
-
7.
SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA).
-
8.
GeneAmp 5700 Sequence Detection System (Applied Biosystems).
-
9.
SuperScript Choice system (Gibco-BRL Life Technologies) and T7-(dT) polymerase (Gensetoligos, La Jolla, CA).
-
10.
BioArray High Yield RNA Transcript Labeling Kit (Enzo Diagnostics, Farmingdale, NY).
-
11.
RNeasy mini kit (Qiagen, Valencia, CA).
-
12.
Affymetrix GeneChip Software.
3 Methods
3.1 Vascularized Heterotopic Cardiac Transplantation
-
1.
Murine hearts are transplanted as previously described (6,7).
-
2.
Briefly, hearts are harvested from freshly sacrificed donors and immediately transplanted into 8- to 12-wk-old recipients that are anesthesized via intraperitoneal injection with 100 mg/kg of ketamine and 20 mg/kg of xylazine.
-
3.
The donor aorta is attached to the recipient abdominal aorta by end-to-side anastamosis, and the donor pulmonary artery is attached to the recipient vena cava by end-to-side anastomosis.
-
4.
All surgical procedures should be completed in less than 45 min from the time that the donor heart is harvested to ensure similar ischemia times. Donor hearts that do not beat immediately after reperfusion or that stop within 1 d following transplantation should be excluded (>98% of all grafts function at 1 d following transplantation).
3.2 Single Cell Suspension
-
1.
Donor grafts are harvested at the indicated time following transplantation and processed to prepare a single-cell suspension using collagenase and pancreatin digestion.
-
2.
The graft heart is harvested following cold saline perfusion.
-
3.
Hearts are minced to fine fragments with a scalpel or razor blade.
-
4.
The heart tissue is digested four times with 0.5% collagenase II (Gibco) and 2.5% pancreatin (Sigma) in 37°C water for 7 min (see Note 1 ).
-
5.
The cell suspension should be filtered and washed twice with 2% FCS D-PBS solution.
-
6.
Resuspend cells, add 2 mL 2% FAS solution, and perform flow cytometry analysis.
3.3 Cell Sorting
Graft infiltrating cells have been shown to play important roles in triggering immune responses during graft rejection after transplantation and other inflammatory diseases such as myocarditis. To determine whether gene expression differences were expressed in infiltrating inflammatory or stromal cells, we used microarray technology to analyze the gene expression profile. To purify cell populations of infiltrating or stromal cells, we purified cell subsets by fluorescence-activated cell sorting (FACS) based on expression of green fluorescent protein (GFP) or fluorescent labeled monoclonal antibodies (see Note 2 ). Gene expression can be analyzed by DNA microarrays or real-time polymerase chain reaction (PCR) in the purified cell populations.
3.3.1 Analysis of Graft Infiltrating Cells
Because of technical difficulties, methods of purifying infiltrating cells often isolate a small percentage of the total population of infiltrating cells. To improve specificity and yield, we have developed a protocol using donor or recipient mice containing a transgene that constitutively expresses the GFP in all cells. These cells have greater than three logs of green fluorescence, making purification by FACS efficient and quantitative. As previously reported, we can purify sufficient infiltrating cells to perform microarray analysis from small numbers of mice. For example, our typical yield is from 106 (at early time points) to 107 (at late time points) infiltrating cells per graft (see Note 3 ). Thus, we can harvest sufficient cells from a single mouse at d 7 following transplantation to obtain sufficient RNA for microarray analysis. An advantage of this approach is that infiltrating cells can be analyzed without requiring amplification of RNA.
3.4 RNA Extraction
Total RNA is isolated from tissues or purified cell populations using TRIZOL reagent (Gibco-BRL Life Technologies). RNA purity is determined initially by 260/280 = 1.85 to 2.01 and by scanning with an Agilent 2100 Bioanalyzer using the RNA 6000 Nano LabChip®. RNA samples not meeting these basic parameters of quality should be excluded from the study.
3.5 DNA Microarrays
-
1.
The initial step of cDNA synthesis is performed using Affymetrix protocols with the T7 dT Primer (100 pM) 5′-GGCCAGTGAATTGTAATACGACTCACTATAGGGAGGCGG-(dT) 24-3′.
-
2.
In vitro transcription and preparation of labeled RNA is performed using the Enzo BioArray High Yield RNA Transcription Labeling Kit.
-
3.
The in vitro transcription sample is cleaned with standard Affymetrix protocols and quantified on a Bio-Tek UV Plate Reader.
-
4.
Twenty micrograms of in vitro transcription material is the nominal amount hybridized to the GeneChip® arrays, an amount easily obtainable from graft tissue.
-
5.
The arrays are incubated in a model 320 (Affymetrix) hybridization oven at a constant temperature of 45°C overnight.
-
6.
Preparation of the microarray for scanning is performed with Affymetrix wash protocols on a model 450 Fluidics station.
-
7.
Scanning is performed on an Affymetrix model 3000 scanner with an autoloader.
-
8.
Chip library files specific to each array and necessary for scan interpretation are stored on the computer workstation controlling the scanner.
3.6 Real-Time Quantitative PCR
Quantification of differentially expressed genes detected by DNA microarrays can be confirmed by real-time PCR. RNA is prepared from each sample of tissue or purified cells and analyzed by real-time PCR.
-
1.
Briefly, isolated RNA is reverse transcribed using SuperScript II RNase Reverse Transcriptase (Gibco, Carlsbad, CA).
-
2.
All primer pairs are designed with the Primer Express software (Applied Biosystems).
-
3.
During primer testing, nontemplate controls and dissociation curves are used to detect primer-dimer conformation and nonspecific amplification.
-
4.
Direct detection of the PCR product is monitored by fluorescence of SYBR Green induced by binding to double-stranded DNA.
-
5.
Reactions are performed in a MicroAmp Optical 96-well reaction plate (Applied Biosystems) using each primer pair in 5 µL of cDNA mix, 5 µL of primer, and 10 µL of SYBR Green Master Mix (Applied Biosystems) per well.
-
6.
The gene-specific PCR products are continuously measured by means of the GeneAmp 5700 Sequence Detection System (Applied Biosystems) during 40 cycles (see Note 4 ).
-
7.
The threshold cycle (which equals the PCR cycle at which an increase in reporter fluorescence first exceeds a baseline signal) of each target product is determined and normalized to the amplification plot of GAPDH.
-
8.
All experiments are run in triplicate, and the thermal cycling parameters are maintained at constant values.
-
9.
Fold change is calculated relative to control cycle threshold (C T). The C T value is defined as the number of PCR cycles required for the fluorescence signal to exceed the detection threshold value. With a PCR efficiency of 100%, the C T values of two separate genes can be compared (ΔC T); the fold difference =
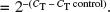 .
.
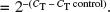 .
.3.7 Bioinformatics
3.7.1 Data Management
-
1.
For each microarray, we store the expression level of approx 45,000 probe sets linked with the experimental group as the class label.
-
2.
Experimental data are catalogued in a manner consistent with the Minimum Information About a Microarray Experiment (MIAME) checklist published by the Microarray Gene Expression Data Society (MGED) (8). Documentation should include the experimental design, samples used, sample preparation and labeling, hybridization procedures and parameters, measurement data and specifications, and array design as described in the checklist on the MGED website (http://www.mged.org/Workgroups/MIAME/miame_checklist.html).
-
3.
All data are available upon request. All samples from experiments are assigned a sequential number with each individual aliquot/sample given an extension of this number. Therefore, each aliquot/sample can be individually tracked and linked with our microarray data. Experimental data including date, mouse strains, date of birth, weight, and sex for both the donor and recipient, as well as time of harvest, are stored for each experiment.
-
4.
All microarray data including .dat files should be backed up and accessible by password-protected Internet access.
3.7.2 Low-Level Data Processing
-
1.
Raw microarray data are normalized and processed by the Affymetrix Microarray Data Analysis Suite (MAS5.0), GCRMA, or dChip software.
-
2.
The quantitative RNA level is computed from the signal strength of the 11 pairs of perfect match (PM) and mismatch (MM) probe pairs representing each gene, where MM probes act as specificity controls that allow the direct subtraction of background and cross-hybridization signals.
-
3.
In the analysis with MAS5, each array is normalized to a standard of 2500 units per probe set. To determine the quantitative RNA level, the averages of the differences (avg diff) representing PM − MM for each gene-specific probe set are calculated. The expression of each probe set is categorized as present (P), marginal (M), or absent (A).
-
4.
We also tried to use the rank invariant set normalization and model-based expression algorithm by dChip software as well as the GCRMA algorithm implemented in BioConductor (http://www.bioconductor.org/) to perform the normalization and calculate the expression levels. As previously reported, our comparison of dChip and GCRMA showed that GCRMA identified a greater number of significantly modulated genes.
-
5.
To eliminate noise and facilitate future gene selection procedures, a filtering process based on a coefficient of variation (CV) is applied to the whole data set of 45,000 probe sets. Nondifferentially expressed genes with a low CV and nonexpressing genes with low expression levels across all the microarrays are considered noninformative and are excluded in the subsequent analyses. The class labels are masked during the gene filtering step to prevent bias in the gene selection process.
3.8 Algorithms to Cluster Gene Expression Profiles
There are multiple algorithms and software that perform clustering analysis of gene expression data. Two examples that we have used in our laboratory are hierarchical clustering and self-organizing maps (SOMs).
3.8.1 Hierarchical Clustering Dendrograms
Hierarchical clustering analysis can be performed using either commercial or free access software, such as Cluster and TreeView (9) (courtesy of M. Eisen, Lawrence Livermore Radiation Laboratory, Berkeley, CA) and GeneCluster (courtesy of Whitehead Institute for Biomedical Institute, Cambridge, MA) software. Briefly, dissimilarity between groups is determined by calculation of a difference metric, such as Euclidean distance or the Pearson correlation coefficient, between each series of values.
3.8.2 Self-Organizing Maps
-
1.
SOMs can be generated by GeneCluster among the experimental groups.
-
2.
The number of maps is selected empirically to eliminate clusters with few genes or large standard deviations. The number of epochs (iterations) of the algorithms is selected to minimize the standard deviations (SDs) of the groupings and is limited only by computer time. For example, we commonly used between 100 and 5000 epochs.
-
3.
Using multiple heuristic observations, the goal is to generate maps in which increased number of nodes produced clusters with low number of genes, whereas decreased number of nodes produced larger SDs. Also, increasing the number of epochs (= 500) should not produce substantial changes in the number of clusters or SDs.
3.9 Statistics
For analysis of gene expression data between two experimental groups, the correlation or regression analysis between the response variable and the expression level is calculated.
-
1.
The expression levels of each gene, under different experimental conditions or different groups of samples, are compared by the statistical methods just given.
-
2.
The p-values are calculated and adjusted by false discovery rate (FDR) control. Calculation of the p-value and FDR adjustment is conducted via R, using functions provided by the packages from BioConductor (http://www.bioconductor.org/). Genes whose FDR adjusted p-values below a specified level are selected as differentially regulated and are used in follow-up studies.
-
3.
Alternatively, for the two-class problem, genes can be selected by building a classification model, e.g., the support vector machine (SVM) model, estimating the prediction error by cross-validation, and also selecting important genes by evaluating their relative contribution to the classification model. The RSVM algorithm was developed by Zhang and Lu (10) and has been used successfully in protein marker identification problems (http://www.stanford.edu/group/wonglab/RSVMpage/R-SVM.html).
3.9.1 Selection of Significantly Differentially Expressed Genes
Our previous studies compared the power to detect significantly modulated genes in duplicate versus quadruplicate microarrays, analyzing independent samples. These results established that quadruplicates allowed detection of greater numbers of modulated genes. Based on the number of significantly regulated genes detected, a cost analysis indicated that the most efficient approach would be to analyze quadruplicate samples. Therefore, quadruplicate microarrays are recommended to estimate the individual and replicate variations, to ensure the maintenance of quality control standards, and to determine whether the experimental variation exceeds the technical variation for our data.
-
1.
The data can be classified according to the categories of the experimental groups, and an initial two-way comparison can be performed between groups. In these studies the expression of each gene in the experimental group is compared with the corresponding expression samples in the control group.
-
2.
We can identify differentially expressed genes by applying FDR adjustment to the raw p-values of all genes, and we can select genes that are differentially expressed, with the FDR controlled below tuned criterion, e.g., 5%.
-
3.
Using this approach, we can select subsets of genes that are significantly differentially expressed in the experimental and control groups.
3.9.2 Randomization Test of the Gene Selection Procedure
-
1.
The gene selection procedure should be validated by a randomization test.
-
2.
The class labels of the microarray are randomly permuted, and the same gene selection procedure and Gene Ontology (GO) annotation analysis are implemented.
-
3.
This randomization is performed multiple times and the number of genes, as well as highly concentrated GO terms, if any, is compared with the discoveries based on true class label.
3.9.3 Statistical Validation
-
1.
The predictive power of a certain subset of genes, with a certain prediction model, can be estimated by cross-validation.
-
2.
In cross-validation, we will leave a small number of samples out, e.g., leave one out when the sample size is very small, or leave 10 to 20% out when we have a moderate number of samples; then we build a predictive model based on the selected genes of the training set and predict the class labels of the left out samples.
-
3.
This is equivalent to testing the model using another independent test set, but the cross-validation procedure is performed multiple times rather than only one splitting of the whole data set. In this way, we can get a better estimation of the predicting error rates.
-
4.
When doing leave-one-out cross-validation, the number of iterations typically is the same as the sample size, i.e., leave out one sample each time. When doing cross-validation by leaving out a portion of samples each time, the number of iterations is no less than the sample size; the only limitation is the computational power available.
3.10 Biological Interpretation
Microarray technology can rapidly generate large databases of gene expression profiles. The challenge in array studies is to link the descriptive expression profiles with relevant biological processes or clinical diagnoses. A common analytical approach has been to select a few genes with the greatest change in expression and correlate them with a specific disease or biological phenotype. However, this approach eliminates >99% of the data from subsequent analysis and may ignore important biological observations. For example, is a gene upregulated early, but to a low level, less important (or a less effective therapeutic target) than a gene upregulated late, but to a high level? Thus, many studies have used arbitrary criteria, such as the ratio of expression, to focus on only a few of the observed changes.
3.10.1 Biological Validation
In addition to the statistical validation of our results, we also assess the biological interpretation of our selected genes based on determination of the biological processes, molecular functions, and cellular components, as defined by the GO database. In association with each gene-based classification, we validate the biological significance of our candidate lists of differentially expressed genes by GO annotation.
-
1.
The list of genes identified as differentially regulated are analyzed by GO annotations to find highly concentrated biological processes, cellular components, and molecular functions.
-
2.
The GO analysis is performed by GeneMerge (http://www.oeb.harvard.edu/hartl/lab/publications/GeneMerge/GeneMerge.html) (11) and GeneNotes (http://combio.cs.brandeis.edu/GeneNotes) software.
-
3.
The pathways that evolved in the regulated genes are found by matching the gene list with a pathway database, e.g., the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway database.
-
4.
Information from other public databases will also be integrated whenever necessary by GeneNotes software. The databases include, but are not limited to, chromosome mapping, gene annotation, homologous genes, unigenes, RefSeq, protein sequences, protein-protein interaction, and PubMed literature.
-
5.
For regulatory motif finding, we cluster genes by their expression profile using GeneCluster, GenePattern (http://www.broad.mit.edu/cancer/software/software.html), and TightCluster (http://www.pitt.edu/~ctseng/research/tightClust_download.html) (12).
-
6.
Motifs are found from the clustered genes by de novo motif-finding algorithms, including BioProspector (13), MDscan (14), and CompareProspector (15); they are validated by the TransFac database.
3.11 Quality Control
-
1.
Various sources of experimental noise will inevitably be introduced into the data set; thus we must assess the noise before interpreting the data with classification information.
-
2.
The level of experimental noise should be estimated by replicates of microarrays on samples that are processed independently (including RNA processing and microarray hybridization).
-
3.
With this approach, the within-replicate experimental variation level can be estimated and compared with individual variations and between-group variations. Only when the replicate variation is significantly smaller than the individual variation and in turn the individual variation is significantly smaller than the between-group variations, can any differences observed by comparing different groups be considered significant.
-
4.
Another source of variation is the batch effect. Because of the ongoing experimental design, it is not feasible to collect all samples and perform the microarray studies simultaneously. Therefore, it is essential to monitor possible batch effects to avoid/correct any bias between batches.
4 Notes
-
1.
Enzyme digestion methods vary. This modified method contributes to a higher yield of viable cardiac cells during our previous experiments.
-
2.
Each gate can clearly show the two populations of GFP+ and GFP− cells. The purity of each isolated cell fraction was >99% by FACS.
-
3.
Every purified cell population should be 10,000 or more to get enough RNA. During RNA extraction, you may not be able to see the pellet at the final step.
-
4.
Because of the lower concentration of cDNA (≤1.0 µg/µL), we used 50 cycles for the primer pair amplification to obtain good production, instead of the 40 cycles we usually use for real-time PCR.
References
Christopher, K., Mueller, T. F., Ma, C., Liang, Y., and Perkins, D. L. (2002) Analysis of the innate and adaptive phases of allograft rejection by cluster analysis of transcriptional profiles. J. Immunol. 169, 522–530.
Christopher, K., Mueller, T. F., DeFina, R., et al. (2003) The graft response to transplantation: a gene expression profile analysis. Physiol. Genomics 15, 52–64.
DeFina, R. A., Liang, Y., He, H., et al. (2004) Analysis of immunoglobulin and T-cell receptor gene deficiency in graft rejection by gene expression profiles. Transplantation 77, 580–586.
Liang, Y., Christopher, K., DeFina, R., et al. (2003) Analysis of cytokine functions in graft rejection by gene expression profiles. Transplantation 76, 1749–1758.
He, H., Stone, J. R., and Perkins, D. L. (2002) Analysis of robust innate immune response after transplantation in the absence of adaptive immunity. Transplantation 73, 853–861.
Corry, R. J., Winn, H. J., and Russell, P. S. (1973) Heart transplantation in congenic strains of mice. Transplant. Proc. 5, 733–735.
McKee, C. M., Defina, R., He, H., Haley, K. J., Stone, J. R., and Perkins, D. L. (2002) Prolonged allograft survival in TNF receptor 1-deficient recipients is due to immunoregulatory effects, not to inhibition of direct antigraft cytotoxicity. J. Immunol. 168, 483–489.
Brazma, A., Hingamp, P., Quackenbush, J., et al. (2001) Minimum information about a microarray experiment (MIAME)—toward standards for microarray data. Nat. Genet. 29, 365–371.
Eisen, J. S. (1998) Genetic and molecular analyses of motoneuron development. Curr. Opin. Neurobiol. 8, 697–704.
Zhang, X., Lu, X., Shi, Q., et al. (2006) Recursive SVM feature selection and sample classification for mass-spectrometry and micrarray data. BMC Bioinformatics 7, 197.
Castillo-Davis, C. I. and Hartl, D. L. (2003) Gene Merge—post-genomic analysis, data mining, and hypothesis testing. Bioinformatics 19, 891–892.
Tseng, G. C. and Wong, W. H. (2005) Tight clustering: a resampling-based approach for identifying stable and tight patterns in data. Biometrics 61, 10–16.
Liu, X., Brutlag, D. L., and Liu, J. S. (2001) BioProspector: discovering conserved DNA motifs in upstream regulatory regions of co-expressed genes. Pac. Symp. Biocomput. 127–138.
Liu, X. S., Brutlag, and Liu, J. S. (2002) An algorithm for finding protein-DNA binding sites with applications to chromatin-immunoprecipitation microarray experiments. Nat. Biotechnol. 20, 835–839.
Liu, Y., Wei, L., Batzoglou, S., Brutlag, D. L., Liu, J. S., and Liu, X. S. (2004) A suite of web-based programs to search for transcriptional regulatory motifs. Nucleic Acids Res. 32, W204–207.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2007 Humana Press Inc.
About this protocol
Cite this protocol
Liang, Y., Lu, X., Perkins, D.L. (2007). Microarray Analysis of Gene Expression in Murine Cardiac Graft Infiltrating Cells. In: Zhang, J., Rokosh, G. (eds) Cardiac Gene Expression. Methods in Molecular Biology, vol 366. Humana Press. https://doi.org/10.1007/978-1-59745-030-0_1
Download citation
DOI: https://doi.org/10.1007/978-1-59745-030-0_1
Publisher Name: Humana Press
Print ISBN: 978-1-58829-352-7
Online ISBN: 978-1-59745-030-0
eBook Packages: Springer Protocols