
Abstract
Over the past decade a plethora of noncoding RNAs (ncRNAs) have been identified, initiating an explosion in RNA research. Although RNA sequencing methods provide unsurpassed insights into ncRNA distribution and expression, detailed information on structure and processing are harder to extract from sequence data. In contrast, northern blotting methods provide uniquely detailed insights into complex RNA populations but are rarely employed outside specialist RNA research groups. Such techniques are generally considered difficult for nonspecialists, which is unfortunate as substantial technical advances in the past few decades have solved the major challenges. Here we present simple, reproducible and highly robust protocols for separating glyoxylated RNA on agarose gels and heat denatured RNA on polyacrylamide–urea gels using standard laboratory electrophoresis equipment. We also provide reliable transfer and hybridization protocols that do not require optimization for most applications. Together, these should allow any molecular biology lab to elucidate the structure and processing of ncRNAs of interest.
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others


Key words
1 Introduction
Northern blotting methods allow for simultaneous quantification and molecular weight determination of RNA. Although superseded by qPCR and sequencing methods for routine mRNA quantification, northern blotting is the method of choice when complex mixtures of overlapping species are under investigation. This is particularly true when studying RNA processing by complexes such as the exosome, and generally aids in resolving the behavior of differentially expressed RNA isoforms. Historically, northern analysis has been something of a black art; running a high-quality formaldehyde gel required substantial skill and a little luck, while radioactive probing of RNA membranes often resulted in terrible cross-hybridization and invisible signals. Fortunately, technology has moved on such that modern northern analysis methods are simple and robust.
Electrophoretic separation of single-stranded RNA is more complex than double-stranded DNA as RNA forms strong secondary structures that impede separation by molecular weight in a gel matrix. For analysis of high molecular weight RNA in agarose gels, chemical modification of guanine is the preferred method to melt secondary structure, which disrupts C:G base pairing and allows single stranded RNA to migrate according to size. Although formaldehyde has been widely used for this purpose [1], it is not ideal due to issues with sample migration and batch-to-batch variation. Furthermore formaldehyde gels release toxic formaldehyde gas; this did not overly concern early investigators eager to replace the hideously toxic denaturant methyl mercury used in the first northern blotting protocols [2, 3], but is clearly a problem in modern labs. To circumvent these issues glyoxal was long ago suggested as an effective RNA denaturant [4], but originally required technically awkward buffer recirculation. This problem was solved by the introduction of BPTE running buffer, which allows agarose gels of glyoxylated RNA to be run with no more difficulty than a normal DNA agarose gel [5].
High-resolution separation of small RNA fragments (~20–250 bp) is best performed on denaturing acrylamide gels, which rely on heat and urea rather than chemical modification to prevent secondary structure formation. The technique is identical to traditional sequencing gel electrophoresis [6], however, the apparatus used for sequencing is not practical for northern blotting and standard protein gel electrophoresis systems are well-suited for this purpose. Helpfully, many of the complications inherent to running a high quality sequencing gel can be safely ignored unless base-pair resolution is required.
Separated RNA is transferred to a membrane by capillary transfer for agarose gels or using an electroblotting system for acrylamide gels [7,8,9]. Various different membranes and transfer conditions have been described but we find charged nylon membrane best for all standard applications [10], and observe little difference between transfer methods. Similarly, many combinations of probes and hybridization buffers can be used to detect RNA species, each having their own strengths and weaknesses. Here we provide a protocol for using RNA probes transcribed from PCR products; in our hands these are both the most reliable and the most sensitive, and as such are the probe of choice for new users [11,12,13,14]. We also provide probing conditions for use of synthetic oligonucleotides, which are widely used in RNA processing analysis since they provide unparalleled resolution of intermediates, and for random-primed DNA probes.
The source of RNA used for northern blotting is rarely critical as long as it is of high quality (see Note 1), and therefore in this chapter we focus purely on the gel systems and hybridization methods.
2 Materials
2.1 RNA Handling and RNase-Free Technique
-
1.
A source of RNase-free milliQ water.
-
2.
Commercially available nuclease free water.
-
3.
Certified RNase-free filter tips.
-
4.
1.5 mL microfuge tubes specifically designated for RNA work.
-
5.
A set of pipettes that are RNase-free.
-
6.
RNaseZAP (Sigma) or similar.
-
7.
3% H2O2.
2.2 RNA Electrophoresis in Agarose Gels
-
1.
RNA samples (1–10 μg total RNA or 10–1000 ng poly(A)+) in 3 μL total volume or less.
-
2.
Glyoxal mix (10 mL): 6 mL DMSO, 2 mL of 40% glyoxal solution (Sigma), 1.2 mL 10× BPTE, 0.6 mL 80% glycerol. For long-term storage store at −80 °C. For routine use store at −20 °C.
-
3.
10 mg/mL ethidium bromide solution (see Note 2).
-
4.
Molecular weight marker—MassRuler.
-
5.
6× bromophenol blue dye: 30% (v/v) glycerol, 0.25% (w/v) bromophenol blue (see Note 3).
-
6.
10× BPTE (1 L): 30 g PIPES free acid (Melford), 60 g Bis-Tris free base (Melford), 20 mL 0.5 M EDTA pH 8.0. Water to 1 L, stir to dissolve, check pH is around neutral. The components will not dissolve until the EDTA is added.
-
7.
Molecular biology grade agarose.
-
8.
Gel imaging and quantification system.
-
9.
Sub-Cell® GT Horizontal Electrophoresis System, 15 × 15 cm tray, with casting gates (Bio-Rad), or equivalent electrophoresis system.
2.3 Transfer of RNA from Agarose Gels onto Membranes
-
1.
2 plastic boxes and 2 glass plates. Both at least 20 × 20 cm.
-
2.
Chromatography paper (Whatman 3MM or similar).
-
3.
Nylon membrane HYBOND N+ (GE Healthcare).
-
4.
Guillotine paper cutter (see Note 4).
-
5.
Paper towel.
-
6.
Parafilm.
-
7.
0.5 M NaOH.
-
8.
Neutralizing solution: 1.5 M NaCl, 0.5 M Tris pH 7.5.
-
9.
20× SSC: 3 M NaCl, 300 mM trisodium citrate, pH 7.
-
10.
UV cross-linker with a 254-nm light source.
2.4 RNA Electrophoresis in Acrylamide Gels
-
1.
RNA samples (1–10 μg total RNA or 10–1000 ng poly(A)+) in 5 μL total volume or less.
-
2.
30% acrylamide and bisacrylamide solution, 29:1 (v/v) (Bio-Rad).
-
3.
10× and 1× TBE.
-
4.
Urea.
-
5.
10% APS: Dissolve 1 g ammonium persulfate in water to 10 mL final volume. Store at 4 °C for up to 6 months.
-
6.
TEMED.
-
7.
Empty Gel Cassettes, mini, 1.5 mm (Thermo Fisher Scientific).
-
8.
50 bp DNA ladder.
-
9.
2× formamide loading dye: 95% formamide, 0.025% bromophenol blue, 0.025% xylene cyanol, 5 mM EDTA, 0.025% SDS. Store long-term at −20 °C, up to 1 month at room temperature.
-
10.
XCell SureLock™ Mini-Cell Electrophoresis System (Thermo Fisher Scientific) (or equivalent system, see Note 5).
-
11.
Comb with 10 or 15 wells, 1.5 mm.
-
12.
Syringe and needle.
-
13.
0.5× TBE.
-
14.
10 mg/mL Ethidium bromide solution (see Note 6).
-
15.
Imaging and quantification system.
2.5 Transfer of RNA from Acrylamide Gels onto Membranes
-
1.
XCell II™ Blot Module (Thermo Fisher Scientific).
-
2.
0.5× TBE.
-
3.
Chromatography paper (Whatman 3MM or similar).
-
4.
Nylon membrane HYBOND N+ (GE healthcare).
-
5.
Guillotine paper cutter (see Note 4).
-
6.
UV cross-linker with a 254-nm light source.
2.6 Hybridization of RNA Probes
-
1.
Gel purified probe template DNA (see Note 7).
-
2.
100 μM rUTP.
-
3.
10 mM rCTP/rGTP/rATP mix.
-
4.
100 mM DTT.
-
5.
10 mg/mL BSA.
-
6.
20 U/μL T7 RNA polymerase and transcription buffer (Promega) (see Note 8).
-
7.
Radioactive workroom and protective equipment.
-
8.
3000 Ci/mMol [α-32P] UTP, 370 MBq/mL (see Note 9).
-
9.
Hybridisation bottles and rotisserie oven.
-
10.
ULTRAHyb ultrasensitive hybridisation buffer by Ambion (Thermo Fisher Scientific) (see Note 10).
-
11.
mini Quick Spin DNA Columns by Roche or similar.
-
12.
0.1× SSC 0.1% SDS.
-
13.
6× SSC.
-
14.
Typhoon Storage Phosphorimager FLA7000 (GE) or equivalent.
-
15.
Storage phosphor screen and exposure cassette.
-
16.
FLA image eraser (GE) or equivalent.
2.7 Hybridization of DNA Oligonucleotide Probes
-
1.
Desalted oligonucleotide probe (synthesized by Sigma-Genosys or equivalent) (see Note 11).
-
2.
QIAquick Nucleotide Removal Kit (QIAGEN) or similar.
-
3.
10 U/μL T4 polynucleotide kinase and buffer (NEB).
-
4.
100 mM DTT.
-
5.
3000 Ci/mMol [γ-32P] ATP, 185 MBq/mL (see Note 9).
-
6.
Radioactive workroom and protective equipment.
-
7.
mini Quick Spin Oligo Columns by Roche or similar.
-
8.
Hybridization bottles and oven.
-
9.
ULTRAHyb Oligo ultrasensitive hybridization buffer by Ambion (Thermo Fisher Scientific).
-
10.
6× SSC.
-
11.
2× SSC 0.1% SDS.
-
12.
Typhoon Storage Phosphorimager FLA7000 (GE) or equivalent.
-
13.
Storage phosphor screen and exposure cassette.
-
14.
FLA image eraser (GE) or equivalent.
2.8 Hybridization of Random Primed DNA Probes
-
1.
DNA probe template—100–1000 bp gel purified DNA fragment.
-
2.
5 U/μL Klenow (3′-5′ exo-) (NEB).
-
3.
5× Labeling buffer: Mix 33 μg Random 9-mers (NEB) dissolved in 653 μL water, 660 μL NEBuffer 2 (NEB), 2.18 μL each of 100 mM dATP, dGTP, and dTTP. Store frozen in 100 μL aliquots.
-
4.
3000 Ci/mMol [α-32P] dCTP, 370 MBq/mL (see Note 9).
-
5.
Radioactive workroom and protective equipment.
-
6.
mini Quick Spin Columns by Roche or similar.
-
7.
Hybridization bottles and oven.
-
8.
ULTRAHyb ultrasensitive hybridization buffer by Ambion (Thermo Fisher Scientific).
-
9.
6× SSC.
-
10.
0.2× SSC 0.1% SDS.
-
11.
Typhoon Storage Phosphorimager FLA7000 (GE) or equivalent.
-
12.
Storage phosphor screen and exposure cassette.
-
13.
FLA image eraser (GE) or equivalent.
3 Methods
3.1 RNA Handling and RNase-Free Technique
The difficulties in handling RNA are often overemphasized. In our hands, most of the cumbersome procedures recommended for avoiding RNase contamination seem to be dispensable. We use tubes and water specifically designated for RNA work, but take few other precautions. Solutions for gels and blotting are made with milliQ water in normal lab bottles with standard chemicals weighed in disposable weighing boats. We do not DEPC treat water or solutions, or use RNase decontaminating sprays or wipes routinely. It is good to have RNase ZAP in the laboratory to clean occasional contaminations or to wipe for example tissue grinding tools that will be in direct contact with the sample. For the final resuspension of RNA samples, we use commercially available nuclease free water. On first use, electrophoresis tanks can be rendered RNase-free by treating with 3% H2O2 for 10 min, then rinsed with milliQ water, then set aside for RNA work if required. Blotting and hybridization can be carried out in normal laboratory trays and glassware. We use certified RNase-free filter tips and set aside a specific set of pipettes for handling stocks of RNase and RNase-containing solutions like plasmid miniprep resuspension buffer. Our bodies are a good source of RNase contamination, so care should be taken to avoid touching the inside of the lids when handling tubes. After assembling reagents, solutions and equipment for northern blotting, it is advisable to run a test gel using RNA of known quality before handling precious samples; ribosomal RNA bands should be clearly resolved and the higher molecular weight band (28S in mammals, 25S in yeast) should be brighter than the lower band (18S). This will confirm that the reagents are sufficiently RNase free.
Conversely, nonenzymatic mechanisms of RNA degradation need to be considered: RNA hydrolysis is catalyzed by alkaline pH and/or divalent cations, particularly with increasing temperature. Therefore, store RNA in water as opposed to TE (pH 8) or similar and beware of reaction conditions involving divalent cations and heat. DNase treatment can be a problem as all DNase I buffers contain magnesium, and DNase treatment is rarely necessary for northern blots as high molecular weight genomic DNA usually resolves far from the bands of interest.
3.2 RNA Electrophoresis in Agarose Gels
Note that the method described below is for a 15 × 15 cm gel.
-
1.
Add ethidium bromide to an aliquot of glyoxal mix to a final concentration of 20 μg/mL.
-
2.
Mix 15 μL glyoxal/EtBr with 3 μL or less of RNA sample then incubate at 55 °C for 1 h (see Note 12).
-
3.
Mix 3 μL DNA ladder with 15 μL glyoxal mix, add 0.3 μL of 10 mg/mL ethidium bromide and incubate at 55 °C along with the RNA samples.
-
4.
Make a 15 × 15 cm 1.2% agarose gel by mixing 1.8 g of agarose with 150 mL of 1× BPTE (see Note 13).
-
5.
Fill the tank with 1× BPTE.
-
6.
Centrifuge the samples briefly and load on the gel. If a spare lane is available, add 5 μL of bromophenol blue loading dye to track migration.
-
7.
Run the gel at 150 V until bromophenol blue is ~2 cm from the bottom (about 1.5 h). This keeps all RNA on the gel, or migrate further if required. The voltage for this electrophoresis should be no higher than 6 V/cm, based on the distance between the electrodes.
-
8.
Acquire a digital image of the ethidium staining pattern using a trans-illuminator or fluorescence imager to allow quantification and assessment of molecular weight based on the marker.
-
9.
Wash the gel 20 min with 75 mM NaOH (see Note 14).
-
10.
Rinse the gel with milliQ water.
-
11.
Wash twice with neutralization solution for 20 min (see Note 15), all on a shaker.
3.3 Transfer of RNA from Agarose Gels onto Membranes
-
1.
Cut a 15 × 15 cm piece of nylon membrane, two 15 × 15 cm pieces of Whatman paper and a 15 × 46 cm piece of Whatman paper (see Note 16). Cut a corner of the nylon membrane and label the membrane with pencil (see Note 17).
-
2.
Soak the 15 × 15 cm Whatman paper and the membrane in 6× SSC in the same plastic box.
-
3.
To set up the transfer apparatus (see Fig. 1), fill a box with 6× SSC up to about 2 cm deep (see Note 18).
-
4.
Put a glass plate over the top leaving a gap to fit the Whatman paper through into the 6× SSC.
-
5.
Wet the long strip of Whatman paper with 6× SSC and place over the plate such that both ends are in the 6× SSC reservoir.
-
6.
Very carefully flip the gel and place it onto the Whatman paper with the wells facing down (see Note 19).
-
7.
Place the wetted membrane over the gel (see Note 20) using the cut corner to track the orientation of the wells (see Fig. 1b).
-
8.
Place the two square pieces of Whatman paper on top of the membrane. Roll a Stripette over the gel “sandwich” to get rid of bubbles (see Note 21).
-
9.
Place two strips of Parafilm covering the ends of the long strip of paper that are not covered by the gel (see Note 22 and Fig. 1c, d).
-
10.
Place two stacks of paper towel of about 5 cm high next to each other over the “sandwich” (see Note 23).
-
11.
Place another glass plate on top and leave the transfer apparatus overnight (see Fig. 1a, b, e).
-
12.
Pick up the two pieces of Whatman paper and the membrane together and place on the floor of the cross-linker (see Note 24) with the RNA side facing upward, enabling direct irradiation of the RNA by the ultraviolet bulbs. Irradiate using Auto-crosslink mode for a Stratalinker or 120,000 μJ/cm2 for other cross-linkers.
-
13.
Wrap the membrane only in cling film and keep at room temperature for probing on the same day or at −20 °C for long-term storage.

Schematic representation and images of blotting apparatus for agarose and acrylamide gels. (a) Schematic representation of the front and side view of a blotting apparatus for an agarose gel. (b) Intermediate step with the long strip of Whatman paper soaked in 6× SSC, the gel and the membrane with the cut corner for tracking purposes. (c) Intermediate step with the two squares of Whatman paper laid on the membrane and Parafilm covering the excess Whatman paper to avoid short circuiting. (d) Elimination of air bubbles by rolling a Stripette over the gel “sandwich.” (e) Complete blotting apparatus for an agarose gel. (f) Schematic representation of the blotting apparatus for an acrylamide gel
3.4 RNA Electrophoresis in Acrylamide Gels
-
1.
In a 15 mL tube (Falcon/Corning type), make 10 mL of an 8% acrylamide, 1× TBE, 42% urea (w/v) solution, and warm to 50 °C until urea dissolves. Let cool to room temperature.
-
2.
Meanwhile, prepare the empty gel cassette. It is important to have this ready, as the gel will polymerize very rapidly on addition of APS and TEMED.
-
3.
Add 50 μL of 10% APS to the gel, invert to mix, add 10 μL of TEMED and mix again. Pour the gel into the empty gel cassette and push in the comb carefully (at a slight angle works best), avoiding bubbles forming in the wells.
-
4.
Mix samples with 5 μL of 2× formamide loading dye.
-
5.
Also, mix 1 μL of 50 bp DNA molecular weight marker with 5 μL of 2× loading dye.
-
6.
Once the gel has set (20–30 min), denature the samples and ladder at 95 °C for 5 min then chill on ice for 2 min.
-
7.
Meanwhile, heat 750 mL 1× TBE buffer to ~50 °C, assemble the gel running apparatus according to manufacturer instructions and fill the inner chamber with hot buffer. Pour hot buffer in the outer chamber until it is 5 mm from the top of the tank.
-
8.
When the samples are on ice remove the comb from the gel, and use a needle and syringe to clean urea out of the wells (this is critical).
-
9.
Load the samples and immediately run the gel at 300 V (see Note 25).
-
10.
For the example shown in Fig. 2d the gel was migrated until the xylene cyanol was at the bottom of the gel. Tables of dye migration in different gel percentages are readily available on line, for example see http://www.elpisbio.com/brochure/gel%20electrophoresis%20buffer.pdf.
-
11.
Dismantle the apparatus according to the manufacturer’s instructions, put the gel in 0.5× TBE containing ethidium bromide (1 μL per 100 mL) and shake gently for 15 min.
-
12.
Replace the staining solution with fresh 0.5× TBE and destain for 15 min on a shaker.
-
13.
Acquire the gel image using a transilluminator or fluorescence imager.

Example images of northern blots hybridized with RNA probes against exosome substrates. (a) 10 μg total RNA isolated using the hot phenol method from wild-type yeast and TRAMP mutant trf4Δ, grown to mid-log in YPD at 25 °C and treated with methotrexate (M) or copper sulfate (C), separated on a 1.2% agarose gel, transferred and probed for CUP1 upstream cryptic unstable transcript (CUT) in ULTRAHyb at 65 °C using an RNA probe [15]. CUTs have no defined termination site and therefore are of very heterogeneous length [16], note that the signal for the CUT is only visible on CUP1 induction using copper, and some degradation products are also visible in the wild type + copper lane. (b) 10 μg total RNA isolated using the GTC phenol method from a variety of TRAMP mutants along with exosome mutant rrp6Δ, grown to mid log at 25 °C in YPD except trf4Δ PGAL-TRF5 which was grown either in YPGal or shifted for 24 h from YPGal to YPD), separated on a 1.2% agarose gel, transferred and probed for MAL32 antisense in ULTRAHyb at 65 °C using an RNA probe. Multiple low abundance isoforms are readily detected in TRAMP mutants. (c) 1 μg total RNA isolated using a mirVANA kit from wild-type yeast, exosome mutant rrp6Δ, and TRAMP mutants trf4Δ and air1Δ air2Δ grown to mid-log in YPD at 25 °C separated on a 1.2% agarose gel, transferred and probed using 35S2 which binds to a region of the pre-ribosomal RNA. Various aberrant rRNA intermediates are visible in the absence of exosome activity [17]. Due to the high signal from pre-rRNA species, hybridization was performed at 65 °C in Church Hyb. (d) RNA samples from C separated on an 8% acrylamide/urea gel, transferred and probed using U14 3′ which binds immediately downstream of the U14 snoRNA sequence. This is an example of a difficult sequence, being 70% AT in the probe binding region. A weak signal was obtained initially from probing at 65 °C in ULTRAHyb, and the membrane was immediately hybridized with the same probe at 60 °C in ULTRAHyb. Discrete bands are visible in TRAMP mutant samples, but not in rrp6Δ cells where these species are polyadenylated [17]
3.5 Transfer of RNA from Acrylamide Gels onto Membranes
-
1.
Paper and membrane: cut two pieces of Whatman paper and one piece of membrane to the size of the gel, a maximum of 9 × 9 cm. Cut a corner and label the membrane as in Subheading 3.3.
-
2.
Follow the manufacturer’s instructions to assemble the blotting apparatus using 0.5× TBE as transfer buffer (see Fig. 1f). Before laying the membrane on the gel, cut off the gel foot to avoid bubble formation as this will impair the transfer.
-
3.
Transfer for 2 h at 30 V (although 1.5 h seems to be enough in our hands).
-
4.
Crosslink the RNA to the membrane following the instructions in step 12 of Subheading 3.3.
3.6 Hybridization of RNA Probes
IMPORTANT: Make sure you have appropriate training to work with radioactivity under the local rules and legislation for your institution, and perform all radioactive work in the designated area.
-
1.
Ensure that the hybridization bottles and internal seals are in good condition to avoid radioactive buffer leaking during hybridization.
-
2.
Prewarm a bottle of ULTRAHyb to 68 °C until the SDS fully dissolves (15–20 min).
-
3.
Wet the membrane with 0.1× SSC 0.1% SDS. Roll it up with the RNA facing inward, drop it into the hybridization bottle and slowly turn the bottle to unroll the membrane against the side of the bottle (see Note 27).
-
4.
Prehybridize for 1 h at 65 °C with 7.5 mL ULTRAHyb (see Notes 28 and 29).
-
5.
To make the probe, mix the following reagents in the given order at room temperature in a 1.5 mL screw cap tube:
-
6.
Mix well and incubate 1–2 h at 37 °C (see Note 32).
-
7.
Flick a mini Quick Spin column inverted and then correct way up to get the sepharose to the bottom, remove top then bottom cap (see product instructions).
-
8.
Place the column in a 2 mL microcentrifuge tube, spin at 1000 × g for 30 s, discard tube and place the column in a new 1.5 mL screw cap tube.
-
9.
Dilute the probe with water to 50 μL and then pipette carefully on to the centre of the sepharose matrix in the column.
-
10.
Centrifuge at 1000 × g for 4 min.
-
11.
Test incorporation by briefly holding the probe and column at equal distances from the Geiger counter. Use tweezers for this. At least 50% of the label should be incorporated, which means the Geiger counter will read the same signal from both column and probe. Discard the column to the radioactive waste.
-
12.
Add the probe to the hybridization bottle while vertical so that it falls directly into the hybridization buffer, and continue to rotate at 65 °C overnight.
-
13.
Pour the probe into a 50 mL tube (Falcon/Corning type) and store at −20 °C (see Note 33).
-
14.
Pour ~50 mL of 6× SSC (see Note 34) in the bottle, close tightly and shake gently then pour off the wash solution.
-
15.
Repeat wash in step 2 once.
-
16.
Add 50 mL of 6× SSC, return the bottle to the hybridization oven and rotate at 65 °C for at least 20 min. Make sure the bottle is balanced by another also containing 50 mL of liquid.
-
17.
Pour off the 6× SSC and add 50 mL of prewarmed (65 °C) 0.1× SSC 0.1% SDS, return to hybridization oven and rotate at 65 °C for 20 min (see Note 35).
-
18.
Repeat the wash in step 5 once.
-
19.
Pour off the remaining buffer, and use tweezers to pull the membrane out of the bottle onto a paper towel (see Note 36).
-
20.
Pat lightly to leave semidry. It is wise to do this behind a Perspex shield as membranes sometimes carry a substantial amount of radioactivity.
-
21.
Wrap the membrane in Saran Wrap.
-
22.
Check the signal with a Geiger counter to obtain an estimated exposure time (see Table 1 and Note 37).
-
23.
Expose the membrane to a storage phosphor screen in a cassette. The screen should be blanked just before use as it will expose slowly with time.
3.7 Hybridization of DNA Oligonucleotide Probes
Oligonucleotide probes are normally 20–45 nt, 40% GC, this protocol is designed for 30–45 nt DNA probes (see Note 38). They are very good for detecting abundant targets by northern blot and allow precise dissection of processing intermediates, but not all probes label or hybridize well for reasons that remain unclear. Oligonucleotide probes can be stripped off the membrane very easily which is useful. Probes should not be designed with a C at the 5′ end as this inhibits labeling. Clean the oligonucleotide probe using a QIAquick Nucleotide Removal Kit before labeling.
-
1.
Ensure that the hybridization bottles and internal seals are in good condition to avoid radioactive buffer leaking during hybridization.
-
2.
Prewarm a bottle of ULTRAHyb Oligo to 68 °C until the SDS fully dissolves (15–20 min).
-
3.
Wet the membrane with 0.1× SSC 0.1% SDS. Roll it up with the RNA facing inward, drop it into the hybridization bottle and slowly turn the bottle to unroll the membrane against the side of the bottle (see Note 27).
-
4.
Prehybridize for 1 h at 42 °C with 7.5 mL UltraHyb Oligo (see Note 28).
-
5.
Mix in this order:
-
5 pMoles oligonucleotide (see Note 39).
-
1.5 μL 10× T4 polynucleotide kinase buffer.
-
1.5 μL 100 mM DTT.
-
Water to 9 μL.
-
1 μL T4 polynucleotide kinase.
-
5 μL [γ-32P] ATP 3000 Ci/mmol 185 MBq/mL.
-
-
6.
Incubate 1 h at 37 °C (see Note 32).
-
7.
Clean the probe through a column as for an RNA probe (see Subheading 3.6, steps 7–11), but use a mini Quick Spin Oligo column as oligonucleotides are retained in normal mini Quick spin columns. Incorporation can be variable.
-
8.
Add the probe to the hybridization bottle while vertical so that it falls directly into the hybridization buffer and incubate overnight at 42 °C.
-
9.
Pour the probe into a 50 mL tube (Falcon/Corning type) and store at −20 °C (see Note 33).
-
10.
See Subheading 3.6 for washing and exposing directions (steps 14–23). Perform a single low stringency wash with 6× SSC for 10 min at 42 °C and a single high stringency wash with preheated 2× SSC 0.1% SDS at 42 °C for 10 min. Perform one last wash for 10 min with 50 mL of 6× SSC at room temperature (see Note 40).
3.8 Hybridization of Random-Primed DNA Probes
DNA probes lack the strand specificity of RNA or oligonucleotide probes, but are much more sensitive than oligonucleotides and are easier to strip than RNA probes.
-
1.
Ensure that the hybridization bottles and internal seals are in good condition to avoid radioactive buffer leaking during hybridization.
-
2.
Prewarm a bottle of ULTRAHyb to 68 °C until the SDS fully dissolves (15–20 min).
-
3.
Wet the membrane with 0.1× SSC 0.1% SDS. Roll it up with the RNA facing inward, drop it into the hybridization bottle and slowly turn the bottle to unroll the membrane against the side of the bottle (see Note 27).
-
4.
Prehybridize for 1 h at 42 °C with 7.5 mL ULTRAHyb (see Note 28).
-
5.
Dilute 25 ng DNA template to 37 μL with water in a screw-cap tube.
-
6.
Heat at 100° for 5 min.
-
7.
Snap-chill on ice water for 2–3 min, centrifuge briefly.
-
8.
Add 10 μL 5× labeling buffer and 1 μL or Klenow (exo-) enzyme, followed by 2 μL [α-32P] dCTP.
-
9.
Mix well by pipetting and incubate 1–3 hr. at 37 °C.
-
10.
Clean the probe through a column as for an RNA probe (see Subheading 3.6, steps 7–11).
-
11.
Heat probe at 100° for 5 min.
-
12.
Snap chill on ice water for 2–3 min, centrifuge briefly.
-
13.
Add the probe to the hybridization bottle while vertical so that it falls directly into the hybridization buffer and incubate overnight at 42 °C.
-
14.
Pour the probe into a 50 mL tube (Falcon/Corning type) and dispose. Reuse of random primed probes is not recommended.
-
15.
See Subheading 3.6 for washing and exposing directions (steps 14–23). Perform the low stringency rinse and wash with 6× SSC at 55 °C and the high stringency washes with preheated 0.2× SSC 0.1% SDS at 55 °C.
3.9 Removing Residual Background
Sometimes membranes have a residual smeary background that interferes with the signal. This is more common with large membranes or when two membranes are probed in one bottle. Wash at room temperature in a plastic box on a rocker with copious 0.1× SSC 0.1% SDS for at least 1 h. If this fails, use a paper towel to scrub the membrane on both sides while it is under the SSC/SDS—repeat until no more counts come off onto the paper towel. This will not damage the RNA or the probe and removes residual radioactive gunk that precipitates out of the hybridisation buffer.
3.10 Stripping Membranes
Wash with boiling 0.1× SSC 0.1% SDS in a plastic box on a rocker, check residual signal with a Geiger counter and repeat if necessary.
4 Notes
-
1.
For mammalian cells, QIAQuick RNA columns, mirVANA kits, TRIreagent, and TRIzol all provide high-quality RNA. There are a few limitations to be aware of particularly that many column-based methods do not isolate RNA of less than ~200 nt. For budding yeast, kits are less widely used but GTC/phenol and hot phenol methods work well, and the mirVANA protocol can be readily adapted using glass bead lysis (see, e.g., [18] for all three methods). RNA can be further manipulated for example by poly(A)+ purification or RNase H cleavage.
-
2.
After purchasing a bottle of 10 mg/mL ethidium bromide, split into aliquots and freeze. An aliquot can be freeze-thawed but discard if the DNA molecular weight marker stops resolving. In our experience, old ethidium solutions compromise glyoxylation reactions.
-
3.
Xylene cyanol comigrates with rRNA and can interfere with quantification, so it is best to omit from this tracking dye.
-
4.
If Northern and Southern blotting is going to become a regular technique, we recommend investment in a good guillotine paper cutter.
-
5.
We have also used Bio-Rad minigel systems to good effect with the same protocol.
-
6.
If the concentration of the samples is very low, stain with SYBR gold (Thermo) after migration for higher sensitivity. Follow the manufacturer’s instructions.
-
7.
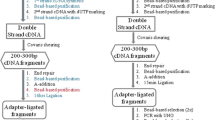
Antisense RNA probes should be 100–500 bp, and are transcribed from PCR products with a T7 promoter at the 5′ end (Fig. 3 and Table 2). To include the T7 promoter in the PCR product, simply fuse this sequence to the 5′ end of the forward primer GGATCCTAATACGACTCACTATAGGGAGAGGA (see Fig. 3). It is critical to amplify the probe template using a polymerase such as Phusion that does not leave 3′ A overhangs. The probe should be gel purified from a 50 μL PCR product and eluted in 30 μL.
-
8.
T7 tolerates low concentration labelled nucleotides better than T3 or SP6.
-
9.
32P sources do not need to be particularly fresh. We use sources up to 6 weeks past the activity date with no problem, or even more for high signals. The exposure time required obviously increases, but the output is similar.
-
10.
It is good practice to split the ULTRAHyb buffer into 4 aliquots and store at 4 °C. For highly abundant transcripts, a lower sensitivity hybridization is advisable using Church Hybridization buffer instead. Church Hybridization buffer: 0.17 M Na2HPO4, 0.079 M NaH2PO4 (moles not g given as hydration state of stocks varies widely), 35 g SDS, 1 mL 0.5 M EDTA, milliQ H2O to 400 mL. Warm to dissolve, and then cool to room temperature. pH should be ~7.2. Dissolve 5 g of BSA in 100 mL of milliQ H2O and add slowly to the previous solution while stirring. Aliquot and store at −20 °C.
-
11.
We have occasionally observed that desalted oligonucleotides do not label well due to unknown contaminants from the manufacturing process. This seems occur at random. Because of this, we routinely purify new oligonucleotide batches using a Nucleotide Removal Kit (Qiagen). Other oligonucleotide manufacturers may not have similar problems, and ordering HPLC-purified oligonucleotides may also solve this problem.
-
12.
The ratio of glyoxal mix to RNA should be at least 5:1, but varying the ratio does not alter the result so samples do not all need to be at the same concentration. Glyoxylation at 55 °C can be left for longer than an hour if the gel is not ready to be loaded. Do not take the samples out and let them sit at room temperature. In our experience, if samples are left for too long at room temperature migration is compromised.
-
13.
Making a thicker gel to be able to fit more sample in is not a good idea as RNA transfer can be compromised. Also, make sure the agarose is fully melted as agarose lumps impair the transfer.
-
14.
It is critical that the NaOH wash is done at the specified concentration and length of time for the RNA to break into the right size fragments. This is to ensure good transfer and good hybridization of the probe.
-
15.
The neutralization solution washes can be longer. Up to 1.5 h works fine in our hands.
-
16.
46 cm is just the width of our paper. This piece has to be just long enough to reach the SSC on both sides.
-
17.
Cutting a corner of the membrane helps to keep track of the orientation of the gel once the blotting apparatus is disassembled. Labeling in pencil is important as ink is removed by some hybridization buffers, and should always be on the same side of the membrane as the RNA to aid orientation.
-
18.
If the SSC runs out, the transfer will be compromised, though this is not terminal. Transfer over the weekend is not a problem though more 6× SSC is required.
-
19.
Flipping the gel, although not absolutely necessary, aids transfer.
-
20.
Before laying the membrane over the gel make sure there are no dry patches on the membrane as these impair transfer.
-
21.
This is important as bubbles completely inhibit the transfer and leave a blank patch in the signal.
-
22.
The Parafilm is placed to avoid contact between paper towel and Whatman paper to prevent a short circuit that would compromise the transfer.
-
23.
It is important to have sufficient paper towel. When the blotting apparatus is disassembled there should still be some dry paper towels at the top of the stack. If all the paper towels are soaked in SSC your transfer might not be complete. However, over-weekend transfers always completely soak the paper towels.
-
24.
Stratagene suggest that cross-linking works best if the paper and membrane are lightly damp, not dripping or dry after the transfer.
-
25.
This high voltage keeps the gel hot, which is important for obtaining the best resolution. It is not disastrous if this cannot be attained with the power supplies available, most RNA species will resolve on much slower runs, but aberrant migration of the DNA ladder is often observed.
-
26.
RNA probes are hard to strip, so if probing a blot sequentially, start with the weakest expected signal. Be aware that a weak signal from a new hybridization may be unstripped probe from the last hybridization.
-
27.
A Stripette can be used to help with this. Push out any obvious air bubbles. Put bottles in the oven so that the visible edge of the membrane is moving with the direction of rotation; if the membrane rolls up into a thin cigar during hybridization or washing then it was the wrong way round. When using a hybridization oven with rotisserie, always be sure to balance the bottles, including the volume of liquid inside. Two membranes can be hybridized in the same bottle, one facing the glass and the other facing the inside of the bottle.
-
28.
This is an important step so do not feel tempted to skip it or shorten it. It allows blocking agents in the hybridization buffer to occupy nonspecific binding sites.
-
29.
In our hands, 65 °C is the best temperature for almost all RNA probes. However, if cross-hybridization to ribosomal RNA is observed, increase the prehybridization, hybridization, and washing temperatures to 68 °C. Beware that this is often caused by the oven temperature calibration drifting such that the oven is not actually at the set temperature of 65 °C. Leave a beaker of water overnight in the oven and check the temperature with a trustworthy thermometer to be sure. In contrast, if the probe is unavoidably AT rich (such as the probe used in Fig. 2d) and a very low signal is obtained, reduce the prehybridization, hybridization and washing temperature to 60 °C and hybridize again with the same probe. Further temperature optimization is unlikely to improve the signal.
-
30.
Some cold UTP is important otherwise the in vitro transcription is inefficient. However, more cold UTP means that the radioactivity per probe molecule is lower, reducing the signal. This amount is a compromise, try not to change unless absolutely necessary.
-
31.
It is critical that the polymerase is added after the radioactive rUTP, then mixed well.
-
32.
In our hands 2 h or more of labeling is fine.
-
33.
RNA probes and oligonucleotide probes can be reused up to 2 times (a total use of 3 times) but only over a week or two taking into account the half-life of the isotope. To reuse a probe, prehybridize the membrane with half the normal quantity of hybridization buffer, meanwhile defrost the probe by letting the tube stand in a beaker of hot water for 20 min then pour the probe into the hybridization bottle.
-
34.
The washes with 6× SSC are the low stringency washes. After the probe is poured off, the blot is washed repeatedly with low stringency wash buffer (high salt) to remove unbound probe.
-
35.
The washes with 0.1× SSC 0.1% SDS are the high stringency washes. The blot is washed 2–3 times with high stringency wash buffer (low salt) at high temperature to remove nonspecifically bound probe.
-
36.
To clean the bottle, simply rinse with water and give it a wipe with a paper towel to remove residual counts. The lids are harder to clean: run water into the lid (hopefully this will dislodge the rubber seal), and wipe inside with a paper towel. Do not attempt to prise the seal out as this can damage the seal, leading to leaks during future hybridization. It is often not possible to completely decontaminate hybridization bottle lids, so we always treat them as if contaminated. With careful handling, the bottles, lids, and seals last for many years.
-
37.
If the exposure time needs to be adjusted, the phosphor screens can be blanked and reexposed. For very intense signals (thousands of counts per second) the phosphor screen may need to be exposed to natural UV light for several days to fully blank the signal (i.e., leave it on the windowsill!).
-
38.
RNA or LNA oligos can also be employed to improve specificity but the hybridization conditions will need to be optimised.
-
39.
The amount of oligonucleotide is important. The molarity of most radionucleotides is very low (this can be calculated for any source as the activity/mmol and the activity/unit volume are provided—for the recommended [γ-32P] ATP, the molarity is 1.67 pM/μL on the activity date). Aim to have less oligonucleotide than [γ-32P] ATP—increasing the amount of oligo will seem to improve the incorporation, but will increase the amount of unlabeled oligonucleotide that competes for hybridization much more. In other words, this protocol produces a small amount of very highly labelled probe which is best for hybridization.
-
40.
Washing twice for 30 min each with 2× SSC 0.5% SDS at 42 °C can improve signal-to-noise but can also reduce the signal for some probes. The easiest approach when using a new probe is to follow the low stringency washing protocol, expose the membrane and if background is unacceptable rewash using these more stringent conditions.

RNA probe design. (a) Schematic representation of the oligonucleotides (primers) used to make an RNA probe template showing the orientation and position of the primers and T7 promoter sequence with respect to the probe target (ncRNA). (b) T7 promoter sequence, position and orientation respect to the forward (Fwd) primer
Change history
01 December 2020
A correction has been published.
References
Lehrach H, Diamond D, Wozney JM, Boedtker H (1977) RNA molecular weight determinations by gel electrophoresis under denaturing conditions, a critical reexamination. Biochemistry 16(21):4743–4751
Bailey JM, Davidson N (1976) Methylmercury as a reversible denaturing agent for agarose gel electrophoresis. Anal Biochem 70(1):75–85
Cummins JE, Nesbitt BE (1978) Methyl mercury and safety. Nature 273(5658):96–96
McMaster GK, Carmichael GG (1977) Analysis of single- and double-stranded nucleic acids on polyacrylamide and agarose gels by using glyoxal and acridine orange. PNAS 74(11):4835–4838
Burnett WV (1997) Northern blotting of RNA denatured in glyoxal without buffer recirculation. BioTechniques 22(4):668–671
Sambrook J, Russell DW (2001) Molecular cloning: a laboratory manual, 3rd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY
Littlehales WJ (1989) Electroblotting technique for transferring specimens from a polyacrylamide electrophoresis or like gel onto a membrane. Google Patents
Rave N, Crkvenjakov R, Boedtker H (1979) Identification of procollagen mRNAs transferred to diazobenzyloxymethyl paper from formaldehyde agarose gels. Nucleic Acids Res 6(11):3559–3567
Alwine JC, Kemp DJ, Stark GR (1977) Method for detection of specific RNAs in agarose gels by transfer to diazobenzyloxymethyl-paper and hybridization with DNA probes. PNAS 74(12):5350–5354
Reed KC, Mann DA (1985) Rapid transfer of DNA from agarose gels to nylon membranes. Nucleic Acids Res 13(20):7207–7221
Melton DA, Krieg PA, Rebagliati MR, Maniatis T, Zinn K, Green MR (1984) Efficient in vitro synthesis of biologically active RNA and RNA hybridization probes from plasmids containing a bacteriophage SP6 promoter. Nucleic Acids Res 12(18):7035–7056
Logel J, Dill D, Leonard S (1992) Synthesis of cRNA probes from PCR-generated DNA. BioTechniques 13(4):604–610
Church GM, Gilbert W (1984) Genomic sequencing. PNAS 81(7):1991–1995
ThermoFisher (2017) ThermoFisher TechNotes 14(2). https://www.thermofisher.com/uk/en/home/references/ambion-tech-support/rna-buffers-chemicals/tech-notes/get-more-from-your-blots.html. Accessed 21 Dec 2017
Hull RM, Cruz C, Jack CV, Houseley J (2017) Environmental change drives accelerated adaptation through stimulated copy number variation. PLoS Biol 15(6):e2001333
Wyers F, Rougemaille M, Badis G, Rousselle JC, Dufour ME, Boulay J, Regnault B, Devaux F, Namane A, Seraphin B, Libri D, Jacquier A (2005) Cryptic pol II transcripts are degraded by a nuclear quality control pathway involving a new poly(A) polymerase. Cell 121(5):725–737
LaCava J, Houseley J, Saveanu C, Petfalski E, Thompson E, Jacquier A, Tollervey D (2005) RNA degradation by the exosome is promoted by a nuclear polyadenylation complex. Cell 121(5):713–724
Cruz C, Houseley J (2014) Endogenous RNA interference is driven by copy number. eLife 3:e01581
Acknowledgments
We thank Courtney Hanna for critical reading of the manuscript and David Tollervey for sharing unpublished data. Funding was from the Wellcome Trust [110216] and the BBSRC [BI Epigenetics ISP: BBS/E/B/000C0423].
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2020 The Author(s)
About this protocol

Cite this protocol
Cruz, C., Houseley, J. (2020). Protocols for Northern Analysis of Exosome Substrates and Other Noncoding RNAs. In: LaCava, J., Vaňáčová, Š. (eds) The Eukaryotic RNA Exosome. Methods in Molecular Biology, vol 2062. Humana, New York, NY. https://doi.org/10.1007/978-1-4939-9822-7_5
Download citation
DOI: https://doi.org/10.1007/978-1-4939-9822-7_5
Published:
Publisher Name: Humana, New York, NY
Print ISBN: 978-1-4939-9821-0
Online ISBN: 978-1-4939-9822-7
eBook Packages: Springer Protocols