
Abstract
Translating ribosome affinity purification (TRAP) is a widely used technique to analyze ribosome-bound mRNAs in particular target cells that express a tagged ribosomal protein. We developed axon-TRAP-RiboTag, a TRAP-based method that allows purification and identification of translated mRNAs from distal neuronal axons in mouse, and identified more than 2000 of translated mRNAs in retinal ganglion cell (RGC) axons in vivo. The use of Cre-negative littermate control to filter out false-positive signals allows unbiased detection, and combining TRAP with in vitro ribosome run-off enables identification of actively translated mRNAs. Here, we describe a detailed protocol to identify translated mRNAs in RGC axons in mouse in vivo. This method can be applied to any neurons whose cell bodies and distal axons are anatomically separated.
Similar content being viewed by others


Key words
1 Introduction
Translation is a key step that controls the abundance of proteins in the cell [1], and therefore it is crucial to profile translated mRNAs in a specific cell population in vivo to understand how cells regulate their gene expression within an organism. Translating ribosome affinity purification (TRAP) , affinity purification of ribosomes carrying an epitope-tagged ribosomal protein (RP) from a specific cell population, is a useful approach to obtain cell type-specific profiles of ribosome-bound mRNAs [2,3,4,5,6,7,8]. This technique utilizes a genetically engineered animal that expresses an epitope-tagged RP, such as EGFP (enhanced green fluorescence protein)-Rpl10a and Rpl22-HA (hemagglutinin), in a genetically defined population of cells. Using a cell type-specific promoter that drives the expression of the tagged RP, ribosome–mRNA complexes can be specifically purified from the cells where the promoter is active in a tissue, an organ, or an organism. Compared to RNase protection-based ribosome profiling, TRAP has the advantage that it generates sequence information on the untranslated regions (UTRs) of isoforms, which would be lost by RNase treatment, although the ribosome profiling can reveal the number of ribosomes bound to a single mRNA species which would accurately reflect the translational rate.
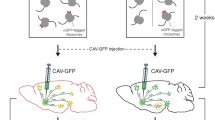
Previously, we developed axon-TRAP, a subcellular TRAP method that allows specific isolation of ribosome-bound mRNAs from the distal RGC axons in Xenopus tadpoles in vivo [4]. We recently extended this technique to mouse, using RiboTag [6], a knockin mouse line in which Cre-mediated recombination labels the 60S subunit ribosomal protein L22 (Rpl22) with hemagglutinin (HA) tags (Rpl22-HA). We crossed this mouse with a Pax6-alpha-Cre [9], in which distal neural retinal progenitors transiently express Cre, leading to permanent HA-labeling of ribosomes in RGCs (Fig. 1a, red area in the eye). This new technique, termed axon-TRAP-RiboTag, involves affinity purification of HA-tagged ribosome–mRNA complexes from the superior colliculus (SCs) in the midbrain, where RGC axons terminate, and enables translational profiling of distal RGC axons at different developmental stages in vivo by deep sequencing [5] (Fig. 1a).


Timeline of the experiments. (a) Axon-TRAP for RGCs . We crossed the RiboTag mouse with the Pax6-alpha-Cre to express HA-tagged ribosomes in RGCs. The affinity purification of HA-tagged ribosome–mRNA complexes from the superior colliculus (SCs), where RGC axons terminate, enables translational profiling of distal RGC axons in vivo by deep sequencing. To exclude false-positive signals, we compared the RNA-seq data from TRAPed mRNAs between the Cre-positive and -negative littermates. (b) In vitro ribosome run-off assay. To distinguish translated mRNAs from translation-stalled mRNAs, we carried out in vitro ribosome run-off assay, which allows translational elongation in vitro in the presence of rabbit reticulocyte lysate (RRL)
Major technical problems of TRAP-based approaches stem from nonspecific binding of mRNAs expressed from cells that do not express the tagged ribosome during the affinity-purification step, which inevitably generates false-positive signals, particularly when combined with sensitive detection methods such as deep sequencing. This becomes particularly problematic when the mRNAs bound to tagged ribosomes represent only a small fraction of the total mRNA in the dissected tissue. For example, the mRNAs translated in distal RGC axons represent a very small fraction of the total mRNAs present in the superior colliculus, where many neurons and glia contain their own mRNAs. We addressed this problem by “differential expression analysis” on biological replicates of Cre-positive and -negative groups, which controls for all potential causes of nonspecific mRNA binding [5] (Fig. 1a).
Another key problem of TRAP-based approaches is that they purify all ribosome-bound mRNAs, not specifically translated mRNAs. As ribosome-bound mRNAs include not only translated mRNAs but also translation-stalled mRNAs, such as those bound to the translational repressor FMRP [10], TRAPed mRNAs do not necessarily represent the translatome (i.e., the entire set of translated mRNAs). To address this point, we carried out in vitro ribosome run-off assay to distinguish translated mRNAs from translation-stalled mRNAs. When allowed to continue translational elongation in vitro in the presence of rabbit reticulocyte lysate and translation initiation inhibitors, only the mRNAs that were being translated at the time of tissue preparation resume translation and finally “run-off” the tagged ribosome (Fig. 1b). Quantitative analysis of TRAPed mRNAs with and without run-off unbiasedly identifies translated mRNAs [5].
Here, we describe a detailed protocol for the axon -TRAP-RiboTag of RGC axons in vivo and a method for in vitro ribosome run-off to validate whether axon-TRAPed mRNAs were being translated. Although the protocol is designed for mouse RGC axons, this approach might be applied to other axons, subcellular compartments and cells.
2 Materials
2.1 Tissue Preparation
-
1.
Mouse embryos or adult mice generated through crosses between a homozygotic RiboTag mouse and a Cre-expressing mouse (see Note 1 ). If a hemizygotic Cre-expressing mouse is used for the crossing, Cre -negative littermates can be used as the negative control for TRAP.
-
2.
Liquid nitrogen in a container (see Note 2 ).
-
3.
Dissection tools: forceps, fine scissors, and springbow dissecting scissors.
2.2 Lysis and Immunoprecipitation
Prepare all solutions using RNase-free water, buffers, and labware.
-
1.
A Dounce homogenizer.
-
2.
CHX-Lysis buffer: 20 mM HEPES–KOH, 5 mM MgCl2, 150 mM KCl, 1 mM DTT, 1 v/v % NP40, 200 U/mL SUPERase In, 100 μg/mL cycloheximide , and Complete EDTA-free Protease Inhibitor Cocktail (see Note 3 ).
-
3.
Wash buffer: 20 mM HEPES–KOH, 5 mM MgCl2, 350 mM KCl, 1 mM DTT, 1 v/v % NP40, and 100 μg/mL cycloheximide.
-
4.
Polyclonal anti-HA antibody (Abcam, ab9110).
-
5.
Magnetic Protein G beads (e.g., Thermo Fisher Scientific Dynabeads).
-
6.
A rotator for Eppendorf tubes in a cold room (4 °C).
-
7.
Magnetic Eppendorf stand (e.g., Thermo Fisher Scientific DynaMag).
-
8.
RNeasy mini kit (Qiagen).
-
9.
2-mercaptoethanol.
2.3 Run-Off Translation
-
1.
A Dounce homogenizer.
-
2.
RO-lysis buffer: 20 mM HEPES-KOH, 5 mM MgCl2, 150 mM KCl, 1 mM DTT, 200 U/mL SUPERase In, Complete EDTA-free Protease Inhibitor Cocktail (Roche, cOmplete), 1% volume of amino acid mix (−leucine), and 1% volume of amino acid mix (−methionine) (amino acids mixes are included in Flexi Rabbit Reticulocyte Lysate System (Promega, L4540).
-
3.
Flexi Rabbit Reticulocyte Lysate System (Promega, L4540).
-
4.
100 μg/mL Harringtonine stock solution.
-
5.
5 mM 4E1RCat stock solution.
-
6.
5 mg/mL cycloheximide .
-
7.
Wash buffer: 20 mM HEPES-KOH, 5 mM MgCl2, 350 mM KCl, 1 mM DTT, 1% NP40, and 100 μg/mL cycloheximide (see Note 4 ).
3 Methods
3.1 Mouse Breeding
-
1.
A Pax6-alpha-Cre hemizygotic male mouse is mated with a RiboTag homozygotic female mouse. Each litter is expected to have roughly equal numbers of Cre-positive (the experimental group) and Cre-negative (the negative control) animals.
3.2 Tissue Preparation
In order to minimize the RNA degradation, prepare the snap-frozen tissue samples as quickly as possible.
-
1.
Dissect the tissues that contain the target cells or subcellular compartments. Dissect another tissue for genotyping (see Note 4). In case of RGC axons, the rostral half of the superior colliculi is quickly dissected.
-
2.
Place the tissue into a prelabeled Eppendorf tube.
-
3.
Secure the lid, and then place the tube directly into the liquid nitrogen.
-
4.
The frozen tissues can be stored in a liquid nitrogen storage tank or −80 °C freezer.
-
5.
Genotype for the Cre transgene for each tissue sample prepared.
3.3 Lysis and Preclearing
-
1.
Prepare the CHX-lysis buffer (lysis buffer with cycloheximide ) immediately before the use.
-
2.
Place the frozen tissues into a Dounce homogenizer containing 500 μL ice-cold CHX-lysis buffer (5–10 w/v % tissues in CHX-lysis buffer). If necessary, pull two to four tissue samples for the same genotype (i.e., pull Cre-positive tissues in one tube, and Cre-negative tissues in another tube). For RGC axons, we used three or four pairs of superior colliculi.
-
3.
On ice, homogenize the tissues gently but thoroughly until lysate becomes clear. If using the same Dounce homogenizer for multiple samples, homogenize Cre-negative tissues first. Wash the homogenizer multiple times with RNase-free water, and once with CHX-lysis buffer.
-
4.
Centrifuge the lysate at 16,000 × g at 4 °C for 10 min. Carefully take the supernatant and transfer to a prechilled tube. The supernatant contains tagged ribosome–mRNA complexes. If necessary, 5 μL of supernatant can be stored as the “input” for TRAP. During this step, prepare the magnetic Protein G beads as in steps 5–7.
-
5.
Resuspend the magnetic Protein G beads in the vial, and transfer 40 μL to a new tube.
-
6.
Place the tube on the magnet to separate the beads from the solution, and remove the supernatant.
-
7.
Resuspend the beads in 40 μL of CHX-lysis buffer and repeat the step 6.
-
8.
Transfer the supernatant after centrifugation (step 4) to the washed beads (step 7).
-
9.
Rotate the tube (head-to-toe rotation) for 1 h in cold room (preclearing step to minimize nonspecific binding of mRNAs to the Protein G beads).
-
10.
Place the tube containing lysate and the beads on the magnet and transfer the supernatant to a prechilled new tube on ice.
3.4 Immunoprecipitation and RNA Purification
-
1.
Add 2.5 μL Rabbit anti-HA antibody to the precleared lysate (step 10 of Subheading 3.3) and rotate the tube (head-to-toe rotation) in cold room overnight.
-
2.
Wash 40 μL Dynabeads with CHX-lysis buffer as steps 5–7 of the Subheading 3.3, and add it to the lysate containing the antibody (step 1). Do not let the beads dry out.
-
3.
After 4 h of head-to-toe rotation in cold room, place the tube on a prechilled magnet on ice and remove the supernatant. Do not let the beads dry out. The beads contain tagged ribosome–mRNA complexes. The supernatant at this step can be saved as the “unbound fraction.”
-
4.
Add 500 μL of wash buffer to the beads and rotate it for 5 min in cold room.
-
5.
Repeat the washing step (steps 3 and 4) at least three times more (total four washing steps are recommended).
-
6.
Add 500 μL of wash buffer to the beads, and transfer the buffer and beads to a new prechilled tube (see Note 5 ).
-
7.
Remove wash buffer from the beads using a DynaMag magnet, and resuspend the beads in 100 μL of CHX-lysis buffer.
-
8.
Add 350 μL of RLT buffer (RNeasy mini kit) containing 2-Mercaptoethanol (see Note 6 ). Vortex vigorously and incubate for 5 min at room temperature. During this step, mRNAs are dissociated from the tagged ribosome.
-
9.
Centrifuge for 30 s using a benchtop centrifuge and collect the supernatant using a DynaMag magnet at room temperature. The supernatant contains axon-TRAPed mRNAs.
-
10.
Add 250 μL of 100% ethanol to the supernatant (step 9).
-
11.
Purify RNA using RNeasy mini kit (Qiagen). Perform on-column DNase digestion to remove potential DNA contamination.
-
12.
Elute RNA from the column in 14 μL of RNase-free water (see Note 7 ).
-
13.
Proceed to cDNA synthesis (see Note 8 ).
3.5 In Vitro Ribosome Run-Off
For the run-off experiments, we use run-off (RO)-lysis buffer that does not contain any detergent and cycloheximide (see Note 9 ).
-
1.
Prepare the RO-lysis buffer immediately before the experiment.
-
2.
Place the frozen tissues into the Dounce homogenizers containing 400 μL of ice-cold RO-lysis buffer (10 w/v % tissues in RO-lysis buffer).
-
3.
Centrifuge the lysate at 16,000 × g at 4 °C for 10 min. During this step, prepare the rabbit reticulocyte lysate (RRL) as following steps 4–6.
-
4.
Add the 8 μL harringtonine (100 μg/mL stock solution) and 4 μL 4E1RCat (5 mM stock solution) to 188 μL RRL, and mix gently. For the “no run-off” control, add 8 μL cycloheximide to the mix (see Note 10 )
-
5.
Preincubate RRL at 30 °C for 5 min
-
6.
Transfer the supernatant (step 3) to the tube and mix it gently.
-
7.
Split the lysate (step 6) into two tubes . One of two tubes can be used for the “no run-off” control.
-
8.
Add 200 μL of RRL mix (step 4) to 200 μL of the lysate (step 7), and incubate at 30 °C for 30 min.
-
9.
Add 800 μL ice-cold CHX-lysis buffer to the run-off reaction and incubate the tube on ice for 5 min to stop the translation elongation.
-
10.
Proceed to immunoprecipitation , following the steps 5–10 of the Subheading 3.3 and then Subheading 3.4.
4 Notes
-
1.
Make sure that no cells in the target tissue (which contains axon terminals of interest and in this case is the rostral half of the superior colliculus) express tagged ribosomes. Histological assays using any Cre reporter mice (e.g., Rosa26-Stop-LoxP-EGFP) is a good start. Confirm this in the RiboTag × Cre mouse to be studied using HA immunohistochemical detection. PCR-based detection of chromosomal DNA that underwent Cre-mediated recombination is a more sensitive method. The “normal” RiboTag allele and “recombined” RiboTag allele can be distinguished by PCR (Fig. 2) [5]. The target tissue should not contain a detectible level of the recombined RiboTag allele. Use the following primers to detect normal and recombined RiboTag alleles: RiboTag fwd, 5′-GGGAGGCTTGCTGGATATG-3′, and HA rev, 5′-ACATCGTATGGGTATAGATCC-3′ (Fig. 2).
Fig. 2 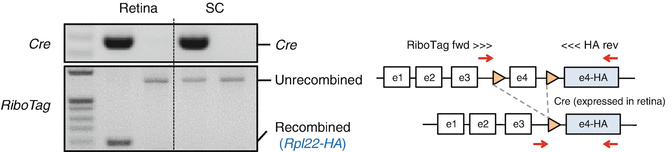
PCR-based detection of Cre -mediated recombination. Cre-mediated recombination converts the normal RiboTag allele to the HA-tagged, recombined allele (see the gene model on the right). These two alleles can be distinguished by the primers depicted in the diagram. Genomic DNAs were extracted from the retina (containing the cell bodies of RGCs) and the superior colliculus (SC, containing the axon terminals of RGCs) of RiboTag mice with or without Pax6-alpha-Cre allele. Cre-mediated “RiboTag” is detectable only in the retina but not in the superior colliculus
-
2.
Wear safety glasses or a face shield when using liquid nitrogen.
-
3.
Prepare all solutions just before the experiment.
-
4.
If genotyping is needed, prepare tail or toe biopsies for genomic DNA PCR experiments. Clean the equipment to avoid carry-over/cross contamination between samples.
-
5.
This step can reduce the false-positive signals due to nonspecific binding of mRNAs to the tube.
-
6.
Alternatively, you can carry out a competitive elution using excessive HA peptides (Sigma, I2149). After step 7 of Subheading 3.4, resuspend the beads containing the tagged ribosome–mRNA complexes in 100 μL of 100 μg/mL HA peptide in CHX-lysis buffer. Vortex vigorously and incubate for 5 min at room temperature. Take the supernatant on a DynaMag magnet and transfer the supernatant to a prechilled tube on ice. The supernatant contains axon-TRAPed mRNAs. Proceed to step 8. Omit step 9 (as the bead is removed). Although the competitive elution reduces the total amount of purified mRNAs, it can improve the signal-to-noise ratio by increasing the specificity.
-
7.
We estimated that approximately 40% of HA-tagged translating ribosomes could be purified using this protocol.
-
8.
If abundant mRNAs are isolated, they can be sequenced by typical RNA sequencing protocols. Alternatively, TRAPed mRNAs can be amplified by a PCR-based method before sequencing. We amplified cDNA by the method developed by Azim Surani and colleagues for single cell transcriptomics , which utilizes PCR-based amplification of polyadenylated RNAs [11]. We followed the detailed protocol up to Step 41 described in this paper [11] to make double-strand DNAs for deep sequencing. Successful axon-TRAP can be validated by agarose gel electrophoresis of cDNAs amplified from Cre-positive and Cre-negative samples. Cre-positive samples should generate stronger bands (Fig. 3).
Fig. 3 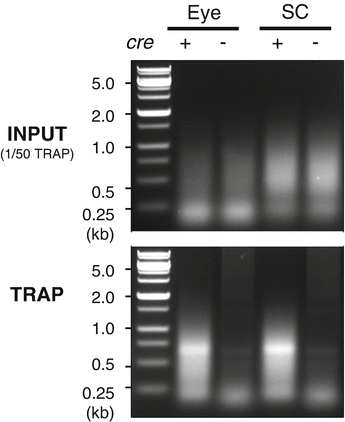
Expected results. Double strand cDNAs made from TRAPed RNAs from neonatal mice (postnatal day 0.5). Three eyes or three pairs of superior colliculi were lysed in ~500 μL CHX-lysis buffer. Total 10 μL of this lysate was used as input (upper), and 500 μL lysate was TRAPed (lower). Polyadenylated RNA was made into double strand DNA using the method described by Tang and colleagues [11]
-
9.
Because the efficiency of the lysis in this condition is significantly lower than in the condition using detergents and cycloheximide , a larger amount of starting materials is needed.
-
10.
Harringtonine and 4E1RCat are added to inhibit the translation initiation [12].




References
Schwanhausser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M (2011) Global quantification of mammalian gene expression control. Nature 473(7347):337–342. doi: nature10098 [pii]10.1038/nature10098
Heiman M, Schaefer A, Gong S, Peterson JD, Day M, Ramsey KE, Suarez-Farinas M, Schwarz C, Stephan DA, Surmeier DJ, Greengard P, Heintz N (2008) A translational profiling approach for the molecular characterization of CNS cell types. Cell 135(4):738–748. doi:10.1016/j.cell.2008.10.028
Dougherty JD, Schmidt EF, Nakajima M, Heintz N (2010) Analytical approaches to RNA profiling data for the identification of genes enriched in specific cells. Nucleic Acids Res 38(13):4218–4230. doi:10.1093/nar/gkq130
Yoon BC, Jung H, Dwivedy A, O’Hare CM, Zivraj KH, Holt CE (2012) Local translation of extranuclear lamin B promotes axon maintenance. Cell 148(4):752–764. doi:10.1016/j.cell.2011.11.064
Shigeoka T, Jung H, Jung J, Turner-Bridger B, Ohk J, Lin JQ, Amieux PS, Holt CE (2016) Dynamic axonal translation in developing and mature visual circuits. Cell 166(1):181–192. doi:10.1016/j.cell.2016.05.029
Sanz E, Yang L, Su T, Morris DR, McKnight GS, Amieux PS (2009) Cell-type-specific isolation of ribosome-associated mRNA from complex tissues. Proc Natl Acad Sci U S A 106(33):13939–13944. doi:10.1073/pnas.0907143106
Doyle JP, Dougherty JD, Heiman M, Schmidt EF, Stevens TR, Ma G, Bupp S, Shrestha P, Shah RD, Doughty ML, Gong S, Greengard P, Heintz N (2008) Application of a translational profiling approach for the comparative analysis of CNS cell types. Cell 135(4):749–762. doi:10.1016/j.cell.2008.10.029
Fang Y, Gupta V, Karra R, Holdway JE, Kikuchi K, Poss KD (2013) Translational profiling of cardiomyocytes identifies an early Jak1/Stat3 injury response required for zebrafish heart regeneration. Proc Natl Acad Sci U S A 110(33):13416–13421. doi:10.1073/pnas.1309810110
Marquardt T, Ashery-Padan R, Andrejewski N, Scardigli R, Guillemot F, Gruss P (2001) Pax6 is required for the multipotent state of retinal progenitor cells. Cell 105(1):43–55
Darnell JC, Van Driesche SJ, Zhang C, Hung KY, Mele A, Fraser CE, Stone EF, Chen C, Fak JJ, Chi SW, Licatalosi DD, Richter JD, Darnell RB (2011) FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146(2):247–261. doi:10.1016/j.cell.2011.06.013
Tang F, Barbacioru C, Nordman E, Li B, Xu N, Bashkirov VI, Lao K, Surani MA (2010) RNA-Seq analysis to capture the transcriptome landscape of a single cell. Nat Protoc 5(3):516–535. doi:10.1038/nprot.2009.236
Cencic R, Hall DR, Robert F, Du Y, Min J, Li L, Qui M, Lewis I, Kurtkaya S, Dingledine R, Fu H, Kozakov D, Vajda S, Pelletier J (2011) Reversing chemoresistance by small molecule inhibition of the translation initiation complex eIF4F. Proc Natl Acad Sci U S A 108(3):1046–1051. doi:10.1073/pnas.1011477108
Acknowledgments
This work was supported by Samsung Science and Technology Foundation under Project Number SSTF-BA1602-13 (to H.J.) and by Wellcome Trust Programme Grant (085314/Z/08/Z) (to C.E.H.).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Science+Business Media LLC
About this protocol
Cite this protocol
Shigeoka, T., Jung, J., Holt, C.E., Jung, H. (2018). Axon-TRAP-RiboTag: Affinity Purification of Translated mRNAs from Neuronal Axons in Mouse In Vivo. In: Gaspar, I. (eds) RNA Detection. Methods in Molecular Biology, vol 1649. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-7213-5_5
Download citation
DOI: https://doi.org/10.1007/978-1-4939-7213-5_5
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-7212-8
Online ISBN: 978-1-4939-7213-5
eBook Packages: Springer Protocols