
Abstract
Metastasis is the main cause of cancer patient mortality. Local tumor invasion is a key step in metastatic dissemination whereby cancer cells dislodge from primary tumors, migrate through the peritumoral stroma and reach the circulation. This is a highly dynamic process occurring in three dimensions that involves interactions between tumor, stromal cells, and the extracellular matrix. Here we describe the organotypic culture system and its utility to study breast cancer cell invasion induced by cancer-associated fibroblasts. This is a three-dimensional model that reproduces the biochemical and physiological properties of real tissue and allows for investigating the molecular and cellular mechanisms involving tumor and its microenvironment, and their contribution to cancer cell invasion . This system provides a robust, accurate, and reproducible method for measuring cancer cell invasion and represents a valuable tool to improve the mechanistic understanding of the initial steps in metastasis .
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others


Key words
1 Introduction
Metastatic dissemination is the major clinical complication in most types of cancer and the cause of 90% of cancer-related deaths [1]. Invasion of cancer cells into the peritumoral stroma is a key step in metastasis [2, 3]. Thus, understanding the mechanisms regulating the invasive abilities of cancer cells is imperative to inform the development of therapeutic modalities to minimize cancer dissemination and improve patient survival [4]. Local cancer cell invasion is a multifunctional and dynamic process occurring in three dimensions that is actively modulated by the peritumoral stroma [5,6,7]. Cancer-associated fibroblasts (CAFs ) are particularly relevant as they can produce soluble factors that promote cancer cell invasion [8]. In addition, CAF-dependent matrix remodeling via focalized proteolysis activity [9] or actomyosin-dependent force generation [10, 11] can lead to the formation of tracks through the extracellular matrix (ECM ) that enable subsequent cancer cell invasion . Accordingly, three-dimensional (3D) models of cancer cell invasion that incorporate stromal components such as fibroblasts and physiologically relevant ECMs recapitulate more closely the in vivo situation. These models provide a platform for investigating the complex interactions between tumor and its microenvironment that are more likely to lead to novel insights of clinical relevance.
Here, we describe a 3D organotypic invasion assay adapted for the robust and accurate assessment of breast cancer cell invasion induced by CAFs [11,12,13]. This approach was originally developed as a 3D coculture model by Fusenig and colleagues [14], and further developed by other groups to study squamous cell carcinoma invasion [10, 15]. Briefly, CAFs are embedded in a dense gel composed of fibrillar collagen I and basement membrane matrix (termed Matrigel ®, Cultrex®, or Engelbroth-Holm-Swarm matrix), which contains laminins, collagen IV , proteoglycans , and a broad spectrum of growth factors (see Fig. 1). A thin layer of gel is used to cover the breast cancer cells seeded on the surface of the CAF-containing gel to mimic the physiological condition of breast tissue. Gels are subsequently laid on a grid and maintained partially immersed in cell medium. Organotypic gels are then fixed and processed by standard histopathological procedures followed by quantitative and qualitative analysis of breast cancer cell invasion using standard image processing and analysis software.

Schematic representation of the workflow of an organotypic invasion assay. Step 1: Embed fibroblasts in gel and seed in 24-well dish. Step 2: Cancer cells are seeded in a single-cell suspension on top of the gel. Step 3: Once the cells have adhered, remove the medium and lift the remodeled gel onto gel-coated nylon filter on a metal bridge. Coat the cancer cells with a thin layer of gel. Step 4: Feed with complete medium up to the nylon filter . Incubate at 37 °C, 5% CO2 for 5 days to allow for cancer cell invasion . Step 5: Terminate assay by fixing organotypic gels. Process gels for H&E staining
2 Materials
2.1 Tissue Culture
-
1.
Cancer-associated fibroblasts (CAFs ):
-
2.
Normal fibroblasts (NFs):
-
3.
Breast cancer cells:
-
4.
Fibroblast culture medium: 10% fetal bovine serum (FBS), 1× GlutaMax™ (Gibco®) and 1× insulin–transferrin–selenium (ITS, Gibco®) in Dulbecco’s modified Eagle’s medium (DMEM). Store at 4 °C. Warm up before use.
-
5.
Cancer cell culture medium: 10% FBS and 1× GlutaMax™ in DMEM (see Note 3 ). Store at 4 °C. Warm up before use.
-
6.
Sterile phosphate buffered saline (PBS): 3.2 mM Na2HPO4, 0.5 mM KH2PO4, 1.3 mM KCl, 135 mM NaCl, pH 7.4. Warm up before use.
-
7.
Sterile 0.05% trypsin–0.02% EDTA. Store at 4 °C. Warm up before use.
-
8.
If a Killing Assay is performed: selective compounds such as puromycin (puromycin dihydrochloride from Streptomyces alboniger, Sigma) (see Note 4 ).
2.2 Gel Preparation
-
1.
5× DMEM: 5% αDMEM powder (Gibco®), 2% NaHCO3 (0.24 M NaHCO3), 0.1 M Hepes pH 7.5. Store at 4 °C (see Note 5 ).
-
2.
Fibroblast culture medium.
-
3.
FBS.
-
4.
Rat-tail collagen type I, high concentration (BD Biosciences). Store at 4 °C (see Note 6 ).
-
5.
Matrigel ® (BD Biosciences). Store at −80 °C in 1 mL aliquots (see Note 7 ).
2.3 Gel Manipulation
-
1.
Nylon NET filters 120 μm (Merck Millipore).
-
2.
Sterile stainless steel metal bridges with a grid size of 2–3 mm. Approximate dimensions: 21 × 21 × 5 mm (Fig. 2a).
Fig. 2 
Gel preparation and processing steps. (a) Metal bridges with approximate dimensions indicated. (b) Useful tools for handling the organotypic cultures, nylon filters and metal bridges. (c) Cell-free or fibroblast-containing gels are plated in a 24-well plate on ice. (d) The ability of fibroblasts to contract the collagen:Matrigel gel (a measure of their matrix remodeling capacity) can be documented prior to further processing. Images show duplicate samples of organotypic cultures free of cells and with different human CAFs that have low (“l”), medium (“m”), and high (“h”) contractility, as indicated. (e, f) Nylon filters are soaked in gel (e) and then separated in a culture dish for setting and fixing (f). (g–k) Setting up the organotypic gel on the metal bridges. Sterile bridges are placed in a 6-well plate (g) and covered by a nylon filter (h). The organotypic cultures are lifted from the 24-well plate and placed over the filter:bridge using a spatula (i); a thin gel layer is added on the top to cover the cancer cell monolayer (j); and complete medium is added to the 6-well dish until soaking the nylon filter underneath the organotypic gel (k)
-
3.
Sterile forceps and spatula (Fig. 2b).
-
4.
Sterile PBS.
-
5.
Filter fixing solution: 4% paraformaldehyde (PFA) and 0.25% glutaraldehyde in PBS.

2.4 Post Processing and Analysis
-
1.
Gel fixing solution: 4% PFA and 1% glutaraldehyde in PBS.
-
2.
Scalpel and forceps (Fig. 2b).
-
3.
70% ethanol.
-
4.
Standard materials for paraffin embedding and hematoxylin and eosin (H&E) staining.
-
5.
Bright field microscope with 20× (or 10×) objective and camera.
-
6.
Image analysis software (Image J—http://imagej.nih.gov/ij—or similar).
3 Methods
This protocol is an adapted version of a previously described method for SCC12 carcinoma cells [10] (see Fig. 1 for a schematic representation). We have optimized the organotypic invasion assay for murine and human breast cancer cells (410.4/4T1 and MDA-MB-231) in combination with murine or human fibroblasts, respectively (see Note 8 ). On a general basis, cancer cells are not invasive in this setting and rely on CAF activities to invade. It is recommended to test the behavior of alternative cancer cell types in the assay by plating them on top of a fibroblast-free gel matrix. Heterogeneity is also observed in CAFs in terms of their ability to contract and remodel gel matrices. The amount of fibroblasts and cancer cells to be used, as well as the length of the protocol, need to be optimized if alternative models are used (see Note 9 ).
3.1 Gel Preparation
All components of the gel mix must be kept on ice. Here we describe the protocol for 1 mL gel mix containing fibroblasts or without fibroblasts. This is the volume required for one sample in one well of a 24-well plate. The total volume of gel mix needs to be scaled up according to sample size. Generally, it is recommended to prepare gel in excess to avoid pipetting errors due to the viscous nature of the components.
-
1.
Prechill all components for the gel preparation on ice.
-
2.
Mix 100 μL FBS, 80 μL 5× DMEM, and 120 μL of fibroblast culture medium.
-
3.
Add 200 μL of Matrigel ® and 400 μL of collagen I (see Notes 10 and 11 ). Keep the mixture on ice (see Note 12 ).
-
4.
Trypsinize fibroblasts of interest (NFs/CAFs ) from a monolayer to a single cell suspension. Count the cells, centrifuge (400 × g for 5 min), and resuspend the pellet at a concentration of 107 cells/mL in fibroblast culture medium (see Notes 13 and 14 ).
-
5.
Avoiding bubble formation, add 100 μL of the cell suspension to the gel mix. If a gel without fibroblasts is required, add 100 μL of fibroblast culture medium instead of cell suspension (see Note 15 ).
-
6.
Add 900 μL of the mixture in a 24 well-plate well (Fig. 2c) (see Note 16 ).
-
7.
Place the plate at 37 °C and 5% CO2 for 1 h.
-
8.
When the gel is set, add 1 mL of appropriate medium on top and incubate overnight at 37 °C and 5% CO2 (see Note 17 ).
3.2 Breast Cancer Cell Preparation
Depending on the number and matrix-remodeling ability of the fibroblasts, some of the gels may have already contracted at this point. The plate can be scanned to document this, as gel contraction can serve as an additional read-out of CAF function (Fig. 2d).
-
1.
Trypsinize a monolayer of breast carcinoma cells (4T1, 410.4 or MBA-MD-231), count the cells and prepare a single cell suspension at 5 × 106 cells/mL in cancer cell culture medium.
-
2.
Aspirate the medium carefully from the 24-well plate containing the gels.
-
3.
Apply 100 μL of the cancer cell suspension on the top of each of the gels (see Notes 18 and 19 ).
-
4.
Incubate at 37 °C and 5% CO2. Leave the cells to adhere to the matrix for 6–8 h.
3.3 Coating of Nylon Filters
Given the softness of the gels, direct contact with the metal can damage the gels. Before placing the gels on the bridges, nylon filters have to be prepared and placed in-between the gels and the metal grid.
-
1.
Prepare the adequate number of nylon filters (1 per organotypic condition) with a sterile forceps on top of each other in a culture dish.
-
2.
Prepare 1 mL of gel without fibroblasts according to the recipe described in Subheading 3.1.
-
3.
Coat the nylon filters using 1 mL of cell-free gel (Fig. 2e).
-
4.
Separate the coated filters in the culture dish (Fig. 2f).
-
5.
Incubate the coated nylon filters at 37 °C for 1 h.
-
6.
Fix the coated nylon, using filter fixing solution for at least 2 h at room temperature (RT) or overnight at 4 °C.
3.4 Lifting the Gel and Covering the Cancer Cells
The gels are placed on the metal bridges and medium is fed from underneath to impose a direction of invasion . The breast cancer cell layer is covered with a thin layer of gel for improved simulation of in vivo conditions (see Note 20 ).
-
1.
Wash the gel-coated nylon filters with sterile PBS for 10 min (×3) to remove all traces of fixing solution.
-
2.
Add cancer cell culture medium to the filters and incubate for 30 min at 37 °C.
-
3.
Place the sterile metal grid bridges in a 6-well plate with sterile forceps (Fig. 2g).
-
4.
Add a coated, washed and medium-adapted nylon filter on top of each metal bridge with sterile forceps (Fig. 2h).
-
5.
Prepare an appropriate amount of cell-free gel according to the recipe in Subheading 3.1 and keep the mixture on ice (100 μL per organotypic gel).
-
6.
Remove the medium on the top of the gels carefully in order not to disturb the organotypic cultures.
-
7.
Lift the gels with a sterile spatula from the 24-well and place it on a coated filter on top of a metal bridge. The cancer cell layer must be facing up (Fig. 2i, j).
-
8.
Add medium under the bridge until it is in contact with the nylon filter . Avoid air bubbles between the interphase medium/nylon filter (Fig. 2k).
-
9.
Add 100 μL of the cell-free gel mixture on top of the lifted gels and spread over the surface.
-
10.
Incubate the culture for 5 days (37 °C, 5% CO2) and change the medium daily (see Note 21 ).
3.5 Gel Fixing and Processing
-
1.
After culturing for 5 days (see Note 9 ), remove the medium from the plate and wash with PBS or transfer the organotypic cultures to a new 6-well plate by lifting the filter and gel. Turn the gel upside-down for fixing (cancer cell layer on the bottom, Fig. 1) (see Note 22 ).
-
2.
Fix the gels in 2 mL of gel fixing solution at 4 °C overnight.
-
3.
Next day, wash the gels with PBS (2 mL) for 10 min (×3).
-
4.
Using the forceps and the scalpel cut the gel in two halves and store one of them in 70% ethanol at 4 °C (see Note 23 ).
-
5.
Embed the other half of the gel in paraffin blocks and perform standard H&E staining on sections (see Notes 24 and 25 ).
3.6 Data Analysis
-
1.
Take 5–7 pictures per gel using a bright field microscope (20×/10× magnification) (see Note 26 and Fig. 3a–e).
Fig. 3 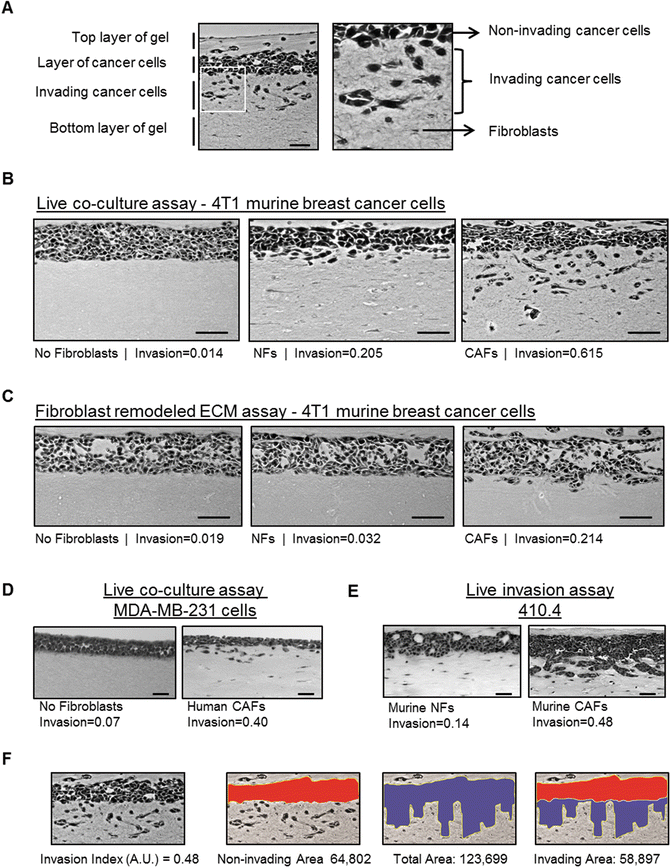
Analysis of cancer cell invasion using carcinoma cells in an organotypic invasion assay. (a) Representative image of H&E staining of an organotypic invasion assay of 4T1 cells cultured in the presence of CAFs . The different parts of the organotypic gel are indicated. Right panel is a zoom up region that shows both collective and single cell invasion of 4T1 cells. Fibroblasts and ECM fibers are also observed. (b) Representative images of H&E staining of organotypic invasion assays of 4T1 murine breast cancer cells cultured in the absence and presence of either murine normal fibroblasts (NFs) or PyMT-CAFs . Invasion indexes of 4T1 cells for eachFig. 3 (continued) experimental setup are also indicated. Scale bar, 50 μm. (c) Representative images of H&E staining of “killing” organotypic invasion assays of 4T1 murine breast cancer cells cultured in gels previously remodeled by murine normal fibroblasts, PyMT-CAFs or mock-remodeled (no fibroblasts). Invasion indexes of 4T1 cells for each experimental set-up are also indicated. Scale bar, 50 μm. (d) Representative images of H&E staining of organotypic invasion assays of MDA-MB-231 human breast cancer cells cultured in the absence and presence of human breast cancer CAFs . The invasion index is indicated. Scale bar, 50 μm. (e) Representative images of H&E staining of organotypic invasion assays of 410.4 murine breast cancer cells with murine NFs and CAFs . Note that 410.4 cells present a “collective” mode of invasion . Scale bar, 50 μm. (f) Exemplars for image analysis of a representative image of H&E staining of an organotypic invasion assay of 4T1 cells cultured in the presence of CAFs (shown in panel a). First, the non-invaded area (red) and the total area (blue) are measured using Image J (http://imagej.nih.gov/ij/). The invading area is then calculated by subtracting the non-invading area from the total area. The invasion index is the ratio of invaded area vs. total area. Invasion index and areas are also indicated
-
2.
Measure the area of noninvasive cancer cells and the total area of cancer cells (noninvasive and invasive) using Image J or a similar software (Fig. 3f).
-
3.
The Invasion Index is calculated by dividing the invading area (total area − invading area) by the total area (see Note 27 ).

3.7 Killing Assay
CAFs have been shown to form tracks in the ECM , which has been associated with increased invasiveness of cancer cells [10]. However, CAFs can also produce soluble factors that can promote cancer cell invasion [8]. The basic organotypic invasion assay described above does not allow discriminating between these two abilities.
In order to specifically study the capacity of CAFs to form tracks in the gel that allow subsequent cancer cell invasion , as well as the potential of cancer cells to invade a gel previously remodeled by CAFs , a variation of the basic organotypic culture assay can be performed: the “killing” assay . Fibroblasts are seeded as described earlier and are allowed to remodel the gel matrix for 5 days before they are removed. When cancer cells are seeded on top of the gel, they will invade into the tracks formed by the fibroblasts (Fig. 3c). This modified version also allows for discriminating the selective effect of chemical compounds on fibroblasts/cancer cells as both compartments are never cultured together.
-
1.
Follow the protocol as described in Subheading 3.1 (steps 1–8). Once the gel is set, add 1 mL of fibroblast culture medium on top.
-
2.
Incubate at 37 °C, 5% CO2 for 5 days. Change the medium daily (see Notes 17 and 28 ).
-
3.
Fibroblast removal (“killing” step): Remove the medium from the gels and add 1 mL of fibroblast culture medium containing an appropriate selection compound (e.g., 10 μg/mL puromycin) (see Note 29 ).
-
4.
Incubate at 37 °C, 5% CO2 for 48 h.
-
5.
Wash the gels for 1 h in fibroblast culture medium (×3). Incubate at 37 °C, 5% CO2 overnight in fibroblast culture medium to remove all traces of selection compound (see Note 30 ).
-
6.
Continue with the procedure as described in Subheadings 3.2–3.6.
4 Notes
-
1.
4T1 cells are a metastatic subpopulation of the original 410.4 murine breast cancer cell line [17].
-
2.
For perturbation studies (i.e., RNAi), NFs/CAFs or cancer cells need to be modified before starting the protocol.
-
3.
ATCC and other studies may suggest different media for culturing the breast cancer cell lines. However, we recommend using DMEM as the organotypic gels are based on it and it does not affect cell behavior or viability.
-
4.
For puromycin, make aliquots of 10 mg/mL and store at −20 °C.
-
5.
Preparation of 5× αDMEM (250 mL): dissolve 12.5 g DMEM powder (Gibco®) and 5 g NaHCO3 in 50 mL of sterile MilliQ water. Add 25 mL of sterile 1 M Hepes pH 7.5 and sterile MilliQ water up to 250 mL. Mix and filter the solution using a 0.2 mm filter in sterile conditions. Aliquot in 50 mL and 10 mL aliquots and store at −20 °C. Thaw fresh aliquots for gel preparation and do not store them longer than 30 days at 4 °C.
-
6.
Rat tail collagen type I, high concentration is supplied as a liquid in 0.02 N acetic acid with a concentration range of 8–11 mg/mL. Note the actual concentration of each particular batch when calculating the volumes to be used during gel preparation (see Note 10 ).
-
7.
Matrigel ® is a viscous substance at 4 °C that solidifies at RT. Store Matrigel® in sterile 1 mL aliquots at −80 °C and thaw the amount required for an experiment the night before at 4 °C. Note the actual concentration of each particular batch, as it will be required to calculate the volumes for gel preparation (see Note 10 ). As an alternative to Matrigel® we have successfully used Engelbroth-Holm-Swarm matrix from Sigma-Aldrich in our organotypics.
-
8.
We have used both murine and human CAFs in this assay. In our experience, murine CAFs require higher cell numbers than human CAFs , as the latter tend to be larger in size and have higher ECM remodeling abilities (see Note 13 ). For comparative studies, make sure an equal amount of NFs/CAFs are used.
-
9.
To optimize the approach, it is recommended to empirically determine the effect of two variables: (1) the amount of NFs/CAFs to be embedded on the gel matrix; and (2) the length of the protocol (i.e., how long the fibroblasts are allowed to remodel the gels and/or the cancer cells are allowed to invade).
-
10.
The final concentrations of collagen and Matrigel ® in the gel are set to 4 mg/mL and 2 mg/mL, respectively. The recipe was optimized to have almost physiological concentrations of collagen I and Matrigel®. In order to prepare reproducible gel solutions the volumes of the components have to be adapted accordingly, depending on the stock concentrations of Matrigel® and collagen I (see Notes 6 and 7 ). For this protocol we base our volume calculations on stock concentrations of 10 mg/mL for both collagen I and Matrigel®. The volume of 5× DMEM can be adapted if larger volumes of the acidic collagen are added to retain optimal buffering.
-
11.
Matrigel ® and collagen I are very viscous and need to be pipetted with care. Avoid bubble formation (as bubbles can be detrimental to the quality of the gel) and retention of gel in the tips (as they may affect the final concentration).
-
12.
Avoid leaving the gel mixture on ice for long periods of time as it may affect its properties.
-
13.
For human NFs/CAFs , resuspend the pellet to a concentration of 2.5 × 106 (see Note 8 ).
-
14.
To reduce the activating effect of FBS in fibroblasts, NFs/CAFs can be cultured for 5–7 days in fibroblast culture medium supplemented with 0.5% FBS (instead of 10% FBS) prior to the use in the organotypic invasion assay.
-
15.
Cell-free gel will also be used for the nylon filter coating procedure (Subheading 3.3) and for covering the cancer cell layer on the top of the organotypic gel (Subheading 3.3).
-
16.
Do not plate the total volume of the mix in order to avoid pipetting errors as the gel is very viscous and sticks to pipette tips. Gels can set at RT; these gels will not have the exact same properties as the ones set at 37 °C. We recommend keeping the 24-well plate on ice while adding the gel, especially when a larger number of samples are handled. This will allow the gel in all wells to set at the same time.
-
17.
At this step, appropriate growth factors , cytokines or drugs can be added to the medium. Cell medium will buffer some of the acidity of the gels and may turn orange. For optimal results, replace with fresh medium after 2–3 h.
-
18.
Seeding 100 μL of cell suspension on the top of highly contracted gels can be problematic. If needed, in these particular cases the empty space in the well can be covered up with fresh cell-free gel; this will allow cancer cells to be seeded on larger volumes (1 mL of 5 × 105 cell/mL suspension). However, the new gel only attaches loosely to the original gel and can sometimes rip off during the lifting process.
-
19.
If gels have not contracted at all, cancer cells can also be applied in a volume of 1 mL per well (at 5 × 105 cell/mL).
-
20.
Alternatively, depending on the type of carcinoma cells used in the assay, the cells can also be grown in an air–cells–liquid interface. The cells are prepared and added on top of the fibroblast-containing organotypic culture as described in Subheading 3.2. However, the cells will not be covered with a layer of gel.
-
21.
At this step, appropriate growth factors , cytokines or drugs can be added to the medium, bearing in mind that they will affect both compartments (i.e., fibroblasts and cancer cells). The gels will become flatter during the incubation on the metal bridges.
-
22.
Alternatively, gels can be snap-frozen for immunofluorescence analysis. Gels are placed in a plastic cuvette, covered by OCT buffer (Tissue-Tek) and immersed in liquid nitrogen. Store at −80 °C until further processing (see Note 25 ).
-
23.
Keep the second half as backup until good H&E staining is obtained or use it to perform additional analysis (see Notes 22 , 24 and 25 ).
-
24.
To allow for invasion analysis, gels must be embedded in paraffin in the correct orientation. Tissue sections need to be obtained from the cut side of the organotypic gel and not on the top or bottom sides. Sectioning the gel within the paraffin block can be challenging as the noninvasive cell layer can easily come off during processing. We recommend very slow trimming of the paraffin block at the first 500 μm to level the gel. Generate two to three cuts (50 μm apart) per organotypic gel per slide for analysis.
-
25.
As an alternative to H&E staining, gels can also be processed for immunohistochemistry or immunofluorescence detection of specific cancer cell or CAF markers according to standard protocols (see Note 22 ). This will ascertain the cell identity of the invading cells. Alternatively, fluorescence-labeled cells can be used in the organotypic invasion assay to allow for detection without staining procedures.
-
26.
Fibroblasts tend to concentrate on the borders of the gels, leading to artefacts on cancer cell invasion in those areas. Pictures need to be taken in the central part of the gel.
-
27.
Other analysis can also be performed. For example, Alternative Invasive Index (invading area/non-invading area), number of invading objects, area of invading cells (without measuring gel areas), etc.
-
28.
NFs/CAFs can be allowed to remodel the gel up to 10 days before the cancer cells are seeded on top. The length of this step has to be determined empirically if other models are used.
-
29.
The choice of selection depends on the resistance genes expressed by the cells (e.g., selection-based stable RNAi/overexpression systems previously inserted). We recommend determining the optimal final concentration beforehand.
-
30.
Thorough washing with PBS and medium is important; otherwise cancer cells added subsequently may be affected. If this becomes problematic, we recommend generating stable cancer cells resistant to the selective compound.
References
Weigelt B, Peterse JL, van’t Veer LJ (2005) Breast cancer metastasis: markers and models. Nat Rev Cancer 5(8):591–602
Gupta GP, Massague J (2006) Cancer metastasis: building a framework. Cell 127(4):679–695
Steeg PS (2006) Tumor metastasis: mechanistic insights and clinical challenges. Nat Med 12(8):895–904
Wan LL, Pantel K, Kang YB (2013) Tumor metastasis: moving new biological insights into the clinic. Nat Med 19(11):1450–1464
Liotta LA, Kohn EC (2001) The microenvironment of the tumour-host interface. Nature 411(6835):375–379
De Wever O, Mareel M (2003) Role of tissue stroma in cancer cell invasion. J Pathol 200(4):429–447
McAllister SS, Weinberg RA (2014) The tumour-induced systemic environment as a critical regulator of cancer progression and metastasis. Nat Cell Biol 16(8):717–727
Ohlund D, Elyada E, Tuveson D (2014) Fibroblast heterogeneity in the cancer wound. J Exp Med 211(8):1503–1523
Zigrino P, Kuhn I, Bauerle T et al (2009) Stromal expression of MMP-13 is required for melanoma invasion and metastasis. J Invest Dermatol 129(11):2686–2693
Gaggioli C, Hooper S, Hidalgo-Carcedo C et al (2007) Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat Cell Biol 9(12):1392–1400
Calvo F, Ege N, Grande-Garcia A et al (2013) Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat Cell Biol 15(6):637–646
Avgustinova A, Iravani M, Robertson D et al (2016) Tumour cell-derived Wnt7a recruits and activates fibroblasts to promote tumour aggressiveness. Nat Commun 7:10305
Calvo F, Ranftl R, Hooper S et al (2015) Cdc42EP3/BORG2 and septin network enables mechano-transduction and the emergence of cancer-associated fibroblasts. Cell Rep 13(12):2699–2714
Fusenig NE, Breitkreutz D, Dzarlieva RT et al (1983) Growth and differentiation characteristics of transformed keratinocytes from mouse and human-skin invitro and invivo. J Invest Dermatol 81(1):S168–S175
Nystrom ML, Thomas GL, Stone M et al (2005) Development of a quantitative method to analyse tumour cell invasion in organotypic culture. J Pathol 205(4):468–475
Guy CT, Cardiff RD, Muller WJ (1992) Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol Cell Biol 12(3):954–961
Aslakson CJ, Miller FR (1992) Selective events in the metastatic process defined by analysis of the sequential dissemination of subpopulations of a mouse mammary tumor. Cancer Res 52(6):1399–1405
Acknowledgments
R.R. and F.C. are funded by The Institute of Cancer Research (UK). F.C. is also supported by Worldwide Cancer Research (Grant 15-0273). We thank Dr. Erik Sahai and Steven Hooper for contributing to develop the technique described here. We also thank lab members for help and advice, and for critically reading the manuscript.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.The images or other third party material in this chapter are included in the chapter’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2017 The Author(s)
About this protocol
Cite this protocol
Ranftl, R.E., Calvo, F. (2017). Analysis of Breast Cancer Cell Invasion Using an Organotypic Culture System. In: Koledova, Z. (eds) 3D Cell Culture. Methods in Molecular Biology, vol 1612. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-7021-6_15
Download citation
DOI: https://doi.org/10.1007/978-1-4939-7021-6_15
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-7019-3
Online ISBN: 978-1-4939-7021-6
eBook Packages: Springer Protocols