
Abstract
Since its identification in the 1990s, the RNA interference (RNAi) pathway has proven extremely useful in elucidating the function of proteins in the context of cells and even whole organisms. In particular, this sequence-specific and powerful loss-of-function approach has greatly simplified the study of the role of host cell factors implicated in the life cycle of viruses. Here, we detail the RNAi method we have developed and used to specifically knock down the expression of ezrin, an actin binding protein that was identified by yeast two-hybrid screening to interact with the Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV) spike (S) protein. This method was used to study the role of ezrin, specifically during the entry stage of SARS-CoV infection.
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others



Key words
- RNA interference (RNAi)
- Small interfering RNA (siRNA)
- Ezrin
- Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV)
- Virus–host interactions
1 Introduction
The discovery of RNA interference (RNAi) represents a quantum leap in the fields of molecular and cellular biology [1, 2]. RNAi technologies are powerful tools that are widely used to investigate the biological function of specific proteins either in vitro or in vivo. In particular, RNAi has successfully been used in virology to study the role of specific host proteins in the life cycle and replication of viruses. The introduction into cells of small interfering RNAs (siRNA), 20–25 nucleotide short double-stranded RNAs that are specific to target mRNA sequences and allow for sequence-specific degradation of the mRNA, is a relatively fast, simple and robust method to specifically downregulate protein expression and study their function [3]. In our studies, siRNA has proven very useful to validate the functional relevance of cellular proteins that were identified by yeast two-hybrid screens as binding partners of influenza and coronavirus structural proteins [4, 5]. We describe herein a method to efficiently knock down protein expression of a cellular actin binding protein, ezrin, and measure the knockdown efficiency. This method was successfully used to investigate the role of ezrin during host cell entry and infection of the Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV) [5]. While the method described here is specific to the downregulation of ezrin expression, it can easily be modified and adapted to study the function of other cellular proteins during viral infection. The methodology described also forms the basis for larger scale experiments such as siRNA library screenings (see Note 1 ), which we have successfully established to study host cell factors involved in viral assembly and release [6].
2 Materials
2.1 siRNA Components
-
1.
1× siRNA buffer: 60 mM KCl, 0.2 mM MgCl2, 6 mM HEPES, pH 7.5 using 2 M KOH.
-
2.
20 μM ezrin-specific small interfering RNAs in 1× siRNA buffer (Table 1 and see Notes 2 – 4 ).
Table 1 Forward sequences of siRNA used to knock down ezrin -
3.
20 μM negative control non-targeting siRNA in 1× siRNA buffer.
-
4.
DharmaFECT 1 transfection reagent (GE Dharmacon) or similar.
2.2 Cell Culture Components
-
1.
HeLa-F5 cells (see Note 5 ).
-
2.
96-well cell culture-treated plates (see Note 6 ).
-
3.
Phosphate buffer saline (PBS).
-
4.
Dulbecco’s Modified Eagle Medium (DMEM High Glucose GlutaMax™—Life Technologies) or equivalent.
-
5.
DMEM-C: DMEM, 10 % fetal bovine serum (FBS), 100 U/ml penicillin, 100 μg/mL streptomycin. Pass solution through a 0.22 μm filtration unit before use and store at +4 °C.
-
6.
DMEM-T: DMEM, 10 % FBS. (see Note 7 ).
-
7.
Polystyrene vials.
2.3 Western Blot Components
-
1.
Lysis buffer: RLT buffer (Qiagen). Allows for isolation of proteins as well as nucleic acids.
-
2.
Protein sample loading buffer (LDS sample buffer): 106 mM Tris–HCl, 141 mM Tris Base, 2 % lithium dodecyl sulfate (LDS), 10 % glycerol, 0.51 mM EDTA, 0.22 mM SERVA blue G250, 0.175 mM phenol red.
-
3.
Sample reducing agent (10×): 500 mM dithiothreitol (DTT).
-
4.
Polyacrylamide gel for protein electrophoresis: Novex Bis-Tris 4–12 % gradient gel, ten wells.
-
5.
Gel running buffer (NuPAGE MOPS SDS running buffer): 50 mM MOPS, 50 mM Tris Base, 0.1 % SDS, 1 mM EDTA.
-
6.
Protein ladder: Novex Sharp pre-stained protein standard (Life technologies), or equivalent.
-
7.
Electrophoresis and blotting module: XCell SureLock Mini-Cell (Life technologies), or similar.
-
8.
Transfer buffer (NuPAGE transfer buffer): 25 mM bicine, 25 mM Bis-Tris (free base), 1 mM EDTA, 10 % ethanol.
-
9.
Filter paper.
-
10.
Polyvinylidene fluoride (PVDF) membrane.
-
11.
Blotting sponge pads.
-
12.
Tris-buffer saline (10× TBS): 200 mM Tris base, 1.5 M NaCl, pH to 7.6 with 12 N HCl.
-
13.
TBST: TBS, 0.1 % Tween 20 (see Note 8 ).
-
14.
Blocking solution: 5 % milk in TBST.
-
15.
Rabbit polyclonal anti-ezrin (generous gift from Dr. Monique Arpin, Institut Curie, France).
-
16.
Mouse monoclonal anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH).
-
17.
Horse radish peroxidase (HRP) conjugated goat anti-rabbit.
-
18.
HRP conjugated rabbit anti-mouse IgG.
-
19.
Enhanced chemiluminescence (ECL) compounds.
-
20.
Gel Doc system capable of reading chemiluminescent signals, e.g., Bio-Rad Gel Doc XR system. Alternatively, membranes can be exposed on X-ray films. Exposure times may vary from a few seconds to several minutes.
-
21.
ImageJ or similar software for band quantification.
3 Methods
The following procedures should be performed in a Class II biosafety cabinet, unless otherwise noted. The siRNA transfection method below describes the procedure for transfecting a specific set of siRNAs (ezrin-targeting or non-targeting siRNAs). As siRNAs are fragile, they should be kept on ice as much as possible. Perform each siRNA treatment condition in triplicates. In our experiments, because cells that undergo siRNA treatment will subsequently be virally infected, care should be taken at all steps to ensure the cells being treated are in the best condition and viability assessed as much as possible (see Note 9 ).
3.1 siRNA Transfection (for Each Type of siRNA)
-
1.
Seed 3.6 × 103 cells per well of a 96-well plate. Incubate at 37 °C for 16–18 h.
-
2.
Dilute stock siRNA solution 1/10, to 2 μM, with 1× siRNA buffer.
-
3.
Mix 2 μM siRNA solution with serum-free DMEM to obtain a 1 μM solution using a 1:1 ratio. For mock siRNA condition, replace siRNA solution by DMEM.
-
4.
Perform gentle up–down pipetting and incubate tube at room temperature for 5 min.
-
5.
Dilute to 1/50 DharmaFECT 1 transfection reagent with DMEM in a polystyrene vial.
-
6.
Perform gentle up–down pipetting and incubate tube at room temperature for 5 min.
-
7.
Mix siRNA-DMEM solution with transfection reagent-DMEM solution using a 1:1 ratio, by adding the transfection reagent-DMEM solution to the siRNA DMEM solution.
-
8.
Incubate at room temperature for 20 min.
-
9.
Dilute siRNA-transfection reagent mix with DMEM-T using a 1/5 dilution.
-
10.
Aspirate culture medium from wells.
-
11.
Add 100 μl of transfection mix per well.
-
12.
Incubate at 37 °C cell culture incubator for 48 h.
-
13.
Check for cytotoxicity or cell morphological changes routinely by observing transfected cells under microscope.
-
14.
Optional: Repeat siRNA transfection to increase knockdown efficiency and incubate for another 48 h (see Note 10 ). Check for cytotoxicity or cell morphological changes under microscope (Fig. 1).
Fig. 1 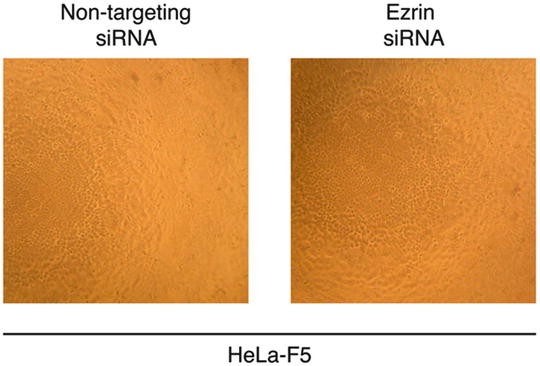
Cell morphology and density after siRNA transfections. 3.6 × 103 HeLa-F5 cells were seeded in wells of a 96-well plate. The cells were then transfected with either non-targeting or ezrin-targeting siRNAs and incubated at 37 °C cell culture incubator for 48 h. A second round of siRNA transfection was then performed and cells were incubated at 37 °C cell culture incubator for 48 h. The cells were then observed under an inverted microscope using at 10× objective and pictures of representative fields were taken

3.2 Measurement of siRNA Knockdown Efficiency
A crucial step in any siRNA transfection experiment is to assess the level of knockdown of expression induced by such treatment. The section below describes how to evaluate siRNA knockdown by measuring the lowering of ezrin protein expression using a Western blot approach (see Note 11 ).
-
1.
Lyse cells by adding 100 μl lysis buffer per well (see Note 12 ), and incubate plate at room temperature for 15 min. Perform gentle up down pipetting to ensure lysis is complete.
-
2.
Add 40 μl 4× Loading buffer, 16 μl 10 × DTT, and 4 μl H2O to each sample.
-
3.
Boil samples at 95 °C for 5 min.
-
4.
Cool samples down on ice and perform a quick centrifugation to bring down condensate. At this point, samples can be run through a gel or stored at −20 °C for later analysis.
-
5.
Place the pre-cast Bis-Tris polyacrylamide gel in electrophoresis module (see Note 13 ).
-
6.
Fill inner and outer compartments of electrophoresis module with the 1× running buffer.
-
7.
Load 10 μl of molecular weight standard ladder and load 10 μl of samples in each lane.
-
8.
Run electrophoresis using following settings: 200 V with constant voltage for 45–60 min.
-
9.
Cut out Whatman paper and PVDF membrane to the gel dimensions.
-
10.
Dehydrate PVDF membrane in 100 % ethanol for 1 min.
-
11.
Rehydrate membrane in H2O for 5 min, and then incubate it in 1× transfer buffer for 5 min.
-
12.
Soak blotting paper and sponge pads in 1× transfer buffer for at least 5 min.
-
13.
Remove gel from plastic encasing and immerse gel in 1× transfer buffer.
-
14.
Prepare transfer stack by layering (from bottom to top) two blotting sponge pads, three blotting paper cutouts, gel, PVDF membrane. Gently roll out bubbles with roller, e.g., a small pipette. Continue stack by adding three blotting paper cutouts and two sponge pads.
-
15.
Place the stack in transfer module and perform transfer inside electrophoresis module filled with 1× transfer buffer using the following settings: 170 mA constant current for 1–2 h.
-
16.
Remove membrane from stack and place in TBST solution.
-
17.
Block membrane in blocking solution for 1 h at room temperature.
-
18.
Prepare primary antibody solutions by diluting them in blocking solution: rabbit polyclonal anti-ezrin: 1/1,000; goat anti-GAPDH 1/10,000.
-
19.
Cut a straight line on the membrane at 50 kDa marker. Incubate top part with anti-ezrin antibody solution and the bottom part with anti-GAPDH antibody solution for 1 h each.
-
20.
Wash membranes three times 10 min in TBST.
-
21.
Prepare secondary antibodies by diluting them in blocking solution: HRP goat anti-rabbit IgG 1/1,000; HRP rabbit anti-goat IgG 1/1,000.
-
22.
Incubate membranes in the corresponding secondary antibody solutions for 45 min.
-
23.
Wash membranes three times 10 min in TBST.
-
24.
Mix ECL solutions using a 1:1 ratio and add 1–2 mL of mixed solution to membrane surface.
-
25.
Incubate for 1 min, remove excess moisture, and perform band detection using gel doc or film and developer (Fig. 2).
Fig. 2 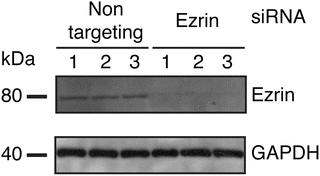
Assessment of ezrin protein expression knockdown induced by siRNA transfections. 3.6 × 103 HeLa-F5 cells were seeded in wells of a 96-well plate. The cells were transfected twice with non-targeting or ezrin-targeting siRNAs. For each condition, cells from one well were lysed and analyzed for ezrin and GAPDH housekeeping protein content by Western blot. The Western blot shown here displays three independent replicates for either non-targeting or ezrin targeting siRNAs
-
26.
Perform band quantification analysis using Gel Analysis module of ImageJ or similar software. For each lane, the GAPDH (housekeeping protein) band serves as a loading control. Normalize ezrin band relative intensity (I ezrin) to GAPDH band relative intensity (I GAPDH):

-
27.
The percentage of ezrin protein expression knockdown by siRNA treatment, compared to non-targeting control is calculated by the following equation (KD, expressed in %, Table 2) :
Table 2 Knockdown analysis after siRNA transfection
4 Notes
-
1.
The method described here focuses on knocking down expression of a single cellular gene, ezrin, to uncover its functional role in SARS-CoV infection. This methodology forms the basis for larger-scale functional analyses as it can easily be scaled up to perform siRNA-based functional screen studies to identify host genes involved in the replicative cycle of viruses. Such siRNA-based screening approach has been successfully developed in our lab using a siRNA library screen of 122 cellular genes involved in membrane trafficking to reveal host factors that are functionally implicated in dengue virus assembly and release [6].
-
2.
siRNAs should be handled with special care. RNA molecules are prone to degradation by RNases. Gloves and RNAse-free pipette tips should be used. Tubes containing siRNAs should be kept on ice as much as possible. Store siRNAs at −20 °C and aliquot stock solutions to avoid multiple freeze–thaw cycles.
-
3.
The ezrin siRNAs used here are in the form of an equimolar mix of four siRNA duplexes. This allows for robust knockdown of expression because it increases the odds of binding to target mRNA sequence and silencing to ensue. It is important to note however that individual siRNAs can also be used to silence specific mRNA expression. This alternative approach has the advantage to minimize potential off-target effects of pooled siRNA mixes. We have validated the use of individual siRNA treatment for ezrin silencing, and successfully used an individual siRNA duplex to silence annexin A6 to investigate its functional role in influenza virus infection [4]. Another consideration when performing siRNA studies is the potential for functional redundancies found in families of closely related proteins. In another dengue virus study from our lab, we have found that, while individually silencing the closely related small GTPases Arf4 or Arf5 had minimal effect on dengue virus secretion from cells, combined silencing of these two GTPases allowed for marked decrease in secretion [7].
-
4.
siRNAs are used at a final concentration of 100 nM for transfection of cells. This concentration was determined to be the best compromise between siRNA knockdown efficiency and cell viability by prior optimization experiments that tested increasing concentrations of siRNAs. As optimal siRNA concentration varies depending on cell type used and target gene, we recommend performing such optimization during the setting up of any siRNA assay.
-
5.
The choice of cell lines for conducting siRNA transfections is an important step during the setup and optimization of the assay. HeLa-F5 cells were chosen in our experiments because they robustly express ACE-2, the SARS-CoV receptor, and have been shown previously to be susceptible to SARS-CoV S-mediated viral entry. Furthermore, we have conducted preliminary siRNA transfection on a panel of cell lines, which included HeLa-F5, and found that those cells could be efficiently transfected, with ezrin protein expression levels significantly reduced after siRNA treatment.
-
6.
In the experiment described herein, siRNA transfections and subsequent assays were performed in 96-well cell culture-treated plates. Depending on the experiment planned using the siRNA-treated cells, the format can be adapted to larger culture plates. In that case, the number of cells and volumes of reagents used will have to be proportionally scaled up.
-
7.
Avoiding addition of penicillin/streptomycin to the transfection medium (DMEM-T) is important because lipid-based transfection reagents, such as DharmaFECT 1 increases the permeability of the cell plasma membrane. If present in the transfection medium, there is a greater risk for cellular uptake of the antibiotics with potentially higher cytotoxicity and lower transfection efficiency.
-
8.
Tween 20 is a viscous solution. To ensure that the correct volume of Tween 20 is added to the TBS buffer, cut a 1,000 μL pipette tip, gently aspirate Tween 20 and add to TBS buffer. Pipette up–down gently and eject tip in buffer. Add stir bar and let solution stir with pipette tip for ~30 min, or until Tween 20 has completely dissolved.
-
9.
The siRNA transfection procedure involves many steps of aspiration of supernatants and addition of solutions on cells. To avoid detaching cells, care should be taken to avoid letting the cells be without medium for more than a few minutes. Also, when adding new medium, solutions should not be pipetted directly on cells, but on the walls of the wells. Media should be pre-warmed at 37 °C as much as possible, as cold solutions can easily detach cells. We performed routine cell viability assays to determine the cytotoxic effects of siRNA treatments, using Trypan blue exclusion assay after treatments.
-
10.
The repeat of the siRNA transfection step 48 h after the first one depends on several parameters including the efficiency of knockdown after 48 h, the turnover of the targeted mRNA, or the half-life of the protein product. We have observed through a series of tests that the optimal conditions for the knockdown of ezrin was to perform two successive siRNA transfections, 48 h apart.
-
11.
If antibodies for the protein of interest are unavailable, an alternative would be to perform a quantitative RT-PCR assay, using specific primers, to measure levels of corresponding messenger RNAs.
-
12.
After HeLa-F5 cells have undergone two successive siRNA transfections, the siRNA efficiency control plate is used to assess the quality of knockdown. Analysis of ezrin or GAPDH (housekeeping protein) protein content from one well of a 96 well plate is sufficient for detection by Western blot.
-
13.
An alternative to the pre-cast Bis-Tris polyacrylamide gel electrophoresis (PAGE) system is the use of gels prepared in the laboratory using gel casters. This allows customizing gels to the most appropriate percentage of polyacrylamide for the protein to be analyzed. 8–12 % polyacrylamide separating gel (8, 10 or 12 % Acrylamide–Bis-Acrylamide, 400 mM Tris–HCl pH 8.8, 0.1 % SDS, 0.04 % ammonium persulfate (APS), 0.07 % tetramethylethylenediamine (TEMED) added last, ddH2O) and 4 % polyacrylamide stacking gel (4 % Acrylamide–Bis-Acrylamide, 100 mM Tris–HCl pH 6.8, 0.1 % SDS, 0.06 % APS, 0.1 % TEMED added last, ddH2O) are commonly used. Laboratory-made running, loading, and transfer buffers can also be prepared in the laboratory following standard Western blotting procedures.
References
Napoli C, Lemieux C, Jorgensen R (1990) Introduction of a chimeric chalcone synthase gene into petunia results in reversible co-suppression of homologous genes in trans. Plant Cell 2:279–289
Fire A, Xu S, Montgomery MK et al (1998) Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391:806–811
Sen GL, Blau HM (2006) A brief history of RNAi: the silence of the genes. FASEB J 20:1293–1299
Ma H, Kien F, Manière M et al (2012) Human annexin A6 interacts with influenza A virus protein M2 and negatively modulates infection. J Virol 86:1789–1801
Millet JK, Kien F, Cheung C-Y et al (2012) Ezrin interacts with the SARS coronavirus spike protein and restrains infection at the entry stage. PLoS One 7:e49566
Wang PG, Kudelko M, Lo J et al (2009) Efficient assembly and secretion of recombinant subviral particles of the four dengue serotypes using native prM and E proteins. PLoS One 4:e8325
Kudelko M, Brault JB, Kwok K et al (2012) Class II ADP-ribosylation factors are required for efficient secretion of dengue viruses. J Biol Chem 287:767–777
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer Science+Business Media New York
About this protocol
Cite this protocol
Millet, J.K., Nal, B. (2015). Investigation of the Functional Roles of Host Cell Proteins Involved in Coronavirus Infection Using Highly Specific and Scalable RNA Interference (RNAi) Approach. In: Maier, H., Bickerton, E., Britton, P. (eds) Coronaviruses. Methods in Molecular Biology, vol 1282. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-2438-7_19
Download citation
DOI: https://doi.org/10.1007/978-1-4939-2438-7_19
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-2437-0
Online ISBN: 978-1-4939-2438-7
eBook Packages: Springer Protocols