
Abstract
Quantitative proteomics, based on stable isotope labeling by amino acids in cell culture (SILAC), can be used to identify host proteins involved in the intracellular interplay with pathogens. This method allows identification of proteins subject to degradation or upregulation in response to intracellular infection. It can also be used to study intracellular dynamics (trafficking) of proteins in response to the infection. Here, we describe the analysis of changes in protein profiles determined in Golgi-enriched fractions isolated from cells that were either mock-infected or infected with Salmonella typhimurium. Using the SILAC approach we were able to identify 105 proteins in Golgi-enriched fractions that were significantly changed in their abundance as a result of Salmonella infection.
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others


Key words
- Host–pathogen interactions
- Salmonella typhimurium
- SILAC
- Cellular fractionation
- Golgi membrane enrichment
1 Introduction
In the last decade a powerful quantitative proteomic approach has been developed in which proteins are marked using stable isotope labeling by amino acids in cell culture (SILAC). This technique was developed by Ong et al. [1] in 2002 and makes use of in vivo incorporation of stable isotopes allowing mass spectrometry (MS)-based quantitative proteomic analyses. Typically, two cell pools are used for SILAC experiments: one with cells grown in normal medium (containing the “light” amino acids) and one with cells in medium in which one or two essential amino acids are replaced by the “heavy” isotope forms of these amino acids, allowing for the identification of peptides from each sample in MS-MS spectra. After a few cell passages, all proteins produced by the cells in the “heavy” medium have incorporated the “heavy” amino acids. Cells in the “light” and “heavy” medium can then be treated differently, e.g., treated versus non-treated or infected versus non-infected cells. The cells from both treatments are then combined in a 1 to 1 ratio and differences in protein abundance due to the treatment can be assessed and quantified by MS (Fig. 1) [2]. SILAC has become a powerful tool to investigate changes in the host cell proteome upon infection with an intracellular pathogen, allowing elucidation of host–pathogen interactions [3–9].

Stable isotope labeling by amino acids in cell culture (SILAC). Schematic representation of the SILAC procedure. Two cell pools incubated under different conditions are used for SILAC experiments; one pool with cells growing in normal media (containing “light” amino acids) and one pool with cells growing in media containing “heavy” amino acids. Cells from the two pools are combined to a 1:1 ratio, proteins are separated and digested in gel with trypsin. Subsequently, the peptides are applied to a mass spectrometer (MS), analyzed and quantified. Ratios are calculated between the “light” and “heavy” peptides, which reflect differences between protein abundance in the two cell pools. Adapted and modified from Oda et al. [2]
Most proteomic approaches published so far deal with the analysis of cellular proteomes of total cell lysates rather than specific cellular compartments [6, 7, 9]. However, a major advantage of using quantitative proteomics to study host–pathogen interactions is the ability to fractionate the samples before analysis. In this way, specific compartments of the host cell, such as the organelles that make up the secretory pathway, can be studied.
In this chapter we describe the procedure recently used in our lab [10] to investigate Salmonella–host interactions in more detail by using a quantitative proteomics approach (SILAC) in combination with cell fractionation. We compared the protein profiles of isolated Golgi-enriched fractions from cells that were either infected with S. typhimurium or mock-infected. After statistical analysis, 105 proteins were identified that were significantly changed in their abundance in the Golgi-enriched fraction upon Salmonella infection. This technique was also previously applied in our laboratory to investigate interactions of coronaviruses with the host secretory pathway [11].
2 Materials
2.1 Cells and Bacteria
-
1.
HeLa cells.
-
2.
Dulbecco’s Modified Eagle medium (DMEM; Cambrex).
-
3.
Fetal calf serum (FCS; Life Technologies (Paisley)).
-
4.
Penicillin and streptomycin (pen/strep); both from Life Technologies (Paisley).
-
5.
PBS.
-
6.
Trypsin.
-
7.
T75, T175 flasks (Corning) and 24-well plates (Corning) for cell culture.
-
8.
Luria–Bertani (LB) broth and agar plates (see Note 1 ).
-
9.
Gentamycin (Gibco).
2.2 13C15N-Arginine- and 13C15N-Lysine-Labeling of HeLa Cells
-
1.
DMEM lacking l-arginine and l-lysine (PAN-biotech cat.no. P04-04510S2).
-
2.
l-arginine-13C6 15N4 hydrochloride (heavy; Spectra Stable Isotopes, cat.no. 548ARG98).
-
3.
l-lysine-13C6 15N2 hydrochloride (heavy; Spectra Stable Isotopes, cat.no. 548LYS98).
-
4.
l-arginine-12C6 14N4 hydrochloride (light; Sigma, cat.no. A5131).
-
5.
l-lysine-12C6 14N2 hydrochloride (light; Sigma, cat.no. L5626).
-
6.
Dialyzed FCS (Invitrogen, cat.no. 26400-044).
-
7.
Lysisbuffer: 20 mM Tris–HCl, pH 7.6; 150 mM NaCl; 1 % nonidet P-40; 0.5 % NaDOC; 0.1 % SDS; 2 μg/ml aprotinin; 2 μg/ml leupeptin; 1 μg/ml pepstatin; 1 mM PMSF.
2.3 Isolation of Golgi-Enriched Fractions
-
1.
Homogenization buffer: 250 mM sucrose in 10 mM Tris–HCl, pH 7.4.
-
2.
Mixtures of protease inhibitors as tablets (Roche). Each tablet is sufficient for a 50 ml solution.
-
3.
PBS buffer prepared as 10× buffer: 1.37 M NaCl, 81 mM Na2HPO4, 15 mM KH2PO4 and 27 mM KCl, pH of the 10× buffer is adjusted to pH 7.4.
-
4.
EDTA stock solution: 100 mM EDTA/KOH, pH 7.1.
-
5.
Sucrose solutions: 29 % w/w, 35 % w/w, and 62 % w/w sucrose in 10 mM Tris–HCl, pH 7.4 (see Note 2 ).
2.4 SDS Polyacrylamide Gel Components
-
1.
Laemmli sample buffer: 50 mM TRIS, pH 6.8; 2.5 % β-Mercaptoethanol; 2 % SDS; 0.02 % Bromophenol; 10 % Glycerol.
-
2.
Bio-Rad Gel electrophoresis system.
-
3.
Ingredients for 5 % stacking gel: 30 % Acrylamide/Bis solution; 1 M Tris–HCl, pH 6.8; Demi water; 10 % SDS solution; 10 % Ammonium Persulfate (APS) solution; TEMED.
-
4.
Ingredients for 12 % running gel: 30 % Acrylamide–Bis solution; 1.5 M Tris–HCl, pH 8.8; Demi water; 10 % SDS solution; 10 % APS solution; TEMED.
-
5.
Fixing solution: 5 % acetic acid/30 % methanol.
-
6.
Coomassie staining (GelCode Blue reagent (Pierce)).
-
7.
Destain solution: 30 mM potassium ferricyanide (K3Fe(CN)6; 9.9 mg/ml), 100 mM sodium thiosulfate (Na2S2O3⋅5H2O; 24.8 mg/ml) (Merck 6516).
2.5 In-Gel Tryptic Digestion
-
1.
50 mM ammonium bicarbonate, pH 8.5 (NH4HCO3; 4 g/L) (Ambic).
-
2.
Acetonitrile (AcN).
-
3.
6.5 mM DTT (1 mg/ml in 50 mM ammonium bicarbonate, pH 8.5) (ICN 194821).
-
4.
54 mM iodoacetamide (10 mg/ml in 50 mM ammonium bicarbonate, pH 8.5) (Sigma 16125).
-
5.
Trypsin: Dissolve 100 μg Trypsin in 1 ml 0.1 M HCl (store in 10–50 μl aliquots at −80 °C). During the experiment, dilute stock 10× with 50 mM bicarbonate (10 ng/μl final concentration) just before addition to gel piece.
3 Methods
Carry out all procedures at room temperature unless otherwise specified.
3.1 Cell Culture
This step describes how the HeLa cells are cultured and kept in culture before performing experiments. Specific requirements for HeLa cells, necessary for the experiments, are described in the paragraphs of the respective experiments.
-
1.
DMEM medium, used for the cell culture, is supplemented with 10 % fetal calf serum (FCS), 100 IU of penicillin/ml, and 100 μg of streptomycin/ml (pen/strep).
-
2.
HeLa cells are grown in T75 flasks and incubated overnight in an incubator at 37 °C/5 % CO2.
-
3.
When 80 % confluency is reached, cells are trypsinized using trypsin (see Note 3 ) and plated in other flasks or plates (see Note 4 ).
3.2 Salmonella Infection
This step describes how an infection of HeLa cells with Salmonella bacteria is performed in general [12]. Specific adjustments made in the experiments are described in the protocols of these experiments.
-
1.
S. typhimurium cultures are grown in LB broth for 16–18 h at 37 °C with continuous shaking. On the day of the experiment, bacteria from the overnight culture are diluted 33 times in LB broth and incubated for another 3.5 h to reach the exponential phase as described before [13] (see Note 5 ).
-
2.
To follow a synchronized population of intracellular bacteria, HeLa cells are infected with Salmonella at a multiplicity of infection (MOI) of 100, for 15 min at 37 °C in DMEM with 10 % FCS (see Note 6 ).
-
3.
Cells are washed thoroughly with PBS and incubated in DMEM with addition of 100 μg/ml gentamycin (Gibco) for 1 h to kill all extracellular bacteria (see Note 7 ).
-
4.
The medium is replaced by DMEM with 10 % FCS and 10 μg/ml gentamycin and the infection is continued for 5 h.
3.3 13C15N-Arginine- and 13C15N-Lysine-Labeling of HeLa Cells
This step describes how labeling of HeLa cells works in general. Before performing SILAC experiments, it must be ensured that cells are labeled completely and how many passages this takes to accomplish. Therefore, this should be tested using the protocol below. Upon complete incorporation of labeled amino acids, the protocol described here results in peptide mass shifts of 8 and 10 amu for each lysine and arginine in the peptide, respectively.
-
1.
For the 13C15N-arginine- and 13C15N-lysine-labeling of HeLa cells, cells are cultured in specialized medium: DMEM lacking l-arginine and l-lysine, which is reconstituted with the heavy amino acids, l-arginine-13C6 15N4 hydrochloride and l-lysine-13C615N2hydrochloride (referred to as heavy medium). As a control for the extent of labeling, cells are also cultured in medium with the normal, light amino acids, l-arginine-12C6 14N4 hydrochloride and l-lysine-12C6 14N2 hydrochloride (referred to as light medium) (see Note 8 ).
-
2.
The heavy and light culture media are supplemented with dialyzed FCS and pen/strep (see Note 9 ).
-
3.
Cells are passaged in fresh medium when 80–90 % confluency is reached. The extent and efficiency of the stable isotope labeling of the HeLa cells are checked using MALDI-TOF-TOF analysis as shown previously [8] (see Note 10 ).
-
4.
When the mass spectrometry analysis shows that the incorporation of the 13C15N amino acids is complete (Fig. 2), the SILAC experiments can be performed. In our case, incorporation was complete after 1 or 2 passages, but for practical reasons, cells that had been passaged six times in the heavy medium were used in the SILAC experiments.
Fig. 2 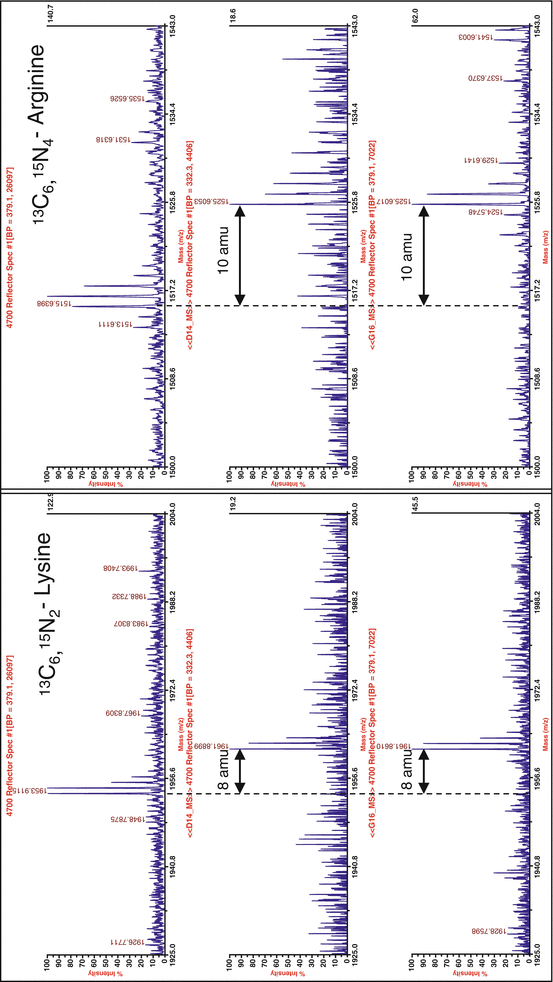
Maldi TOF analysis to investigate incorporation of SILAC labels. To investigate whether incorporation of labeled AA in the protein is complete after 1 (top), 2 (middle), or 3 (bottom panel) cell passages, trypsinized proteins (from a single gel band) were analyzed by Maldi TOF. For both the heavy Lysine (left) as the heavy Arginine, right) complete incorporation can already be observed after 2 passages, indicated by the absence of the original peptide peaks (displayed in upper panels) and the appearance of the peptide peaks corresponding to the labeled peptides (+8 amu for Lysine, +10 amu for Arginine, both indicated). The labeling is maintained in passage 3

3.4 SILAC Experiments; Salmonella Typhimurium Infection
The SILAC procedure, which was described by Ong et al. [1], are performed with certain adjustments as we describe below and as was published previously [10, 11].
-
1.
Two pools of HeLa cells are used that have been passaged six times in either the heavy or the light medium.
-
2.
The HeLa cells are grown in T175 flasks (Corning) until confluency of 80–90 % was reached (approximately 1 × 107 cells per flask).
-
3.
Cells from eight culture flasks are used per experiment, four flasks per labeling condition.
-
4.
In each SILAC experiment, cells cultured either in the light or the heavy medium are mock-infected or infected with WT S. typhimurium at a MOI of 100 for 6 h (see Note 11 ). In our case a 6 h infection is used since that is the time point when Salmonella reaches the Golgi apparatus.
3.5 SILAC Experiments; Isolation of a Golgi-Enriched Fraction
-
1.
Six hours post infection (p.i.), the Salmonella-infected cells are harvested in homogenization buffer and combined in a 1:1 ratio with the mock-infected cells (see Note 12 ).
-
2.
A Golgi-enriched fraction is isolated from the cells using an established method [11, 14, 15].
-
(a)
Cells from 8 culture flasks are trypsinized and harvested at 500 × g for 10 min and washed twice in cold PBS with subsequent centrifugation at 500 × g for 10 min.
-
(b)
The pellet is washed with cold homogenization buffer and the cells are spun down at 500 × g for 10 min (see Note 13 ).
-
(c)
The pellet is resuspended in 5 volumes of cold homogenization buffer (see Note 14 ), followed by homogenization using a Balch homogenizer (Fig. 3) [16].
Fig. 3 
Balch homogenizer. To keep the homogenate at 4 °C, precool the homogenizer for 15 min on ice. Depending of the volume of PNS use a suitable syringe size (from 1 to 10 ml). The volume of the PNS to be homogenized should not exceed half of the maximum volume of the syringe [16]
-
(d)
Cells are homogenized with the Balch homogenizer (gap size 9 μm) with approximately 50–60 strokes (see Note 15 ).
-
(e)
Post nuclear supernatant (PNS) is obtained after centrifugation of the cell homogenate at 600 × g for 10 min at 4 °C.
-
(f)
To 12 ml of PNS, 11 ml of 62 % (w/w) sucrose solution and 250 ml of 100 mM EDTA (pH 7.1) are added to obtain a homogenate with 37 % (w/w) sucrose concentration (see Note 16 ).
-
(g)
Four milliliter of this homogenate is placed into a SW40 tube Beckman) and overlaid with a 5 ml 35 % (w/w) and a 4 ml 29 % (w/w) layer of sucrose solution (in 10 mM Tris, pH 7.4) (see Note 17 ).
-
(h)
This gradient is centrifuged for 2 h and 40 min at 100,000 × g.
-
(i)
Approximately 1 ml of a Golgi-enriched fraction is collected at the interphase of 35–29 % sucrose layers (see Note 18 ).
-
(j)
For further analysis, the collected membranes are pelleted by centrifugation for 30 min at 100,000 × g at 4 °C after the addition of 4 volumes of PBS to 1 volume of the Golgi-enriched fraction. Alternatively, the collected membranes are stored at −80 ºC (see Note 19 ). Golgi-enrichment of the isolated fraction is verified by western blot analysis using established organelle marker proteins.
-
(a)

3.6 SDS-PAGE and Coomassie Staining
-
1.
Golgi-enriched membranes (80 μg protein) are dissolved in Laemmli sample buffer containing 10 mM DTT and heated for 5 min at 95 °C (see Note 20 ).
-
2.
Clean the glass plates of the Bio-Rad Gel Electrophoresis system. Prepare the 12 % running gel by mixing 1.98 ml 30 % Acrylamide/Bis, 1.25 ml 1.5 M Tris–HCl, pH 8.8, 1.7 ml demi water, 50 μl 10 % SDS, 50 μl APS, and 3 μl TEMED and cast the gel between the glass plates into the cassette. Allow space for the stacking gel, gently overlay it with water, and let it solidify for 45 min at room temperature.
-
3.
Prepare the 5 % stacking gel by mixing 170 μl 30 % Acrylamide/Bis, 130 μl 1 M Tris–HCl pH 6.8, 700 μl demi water, 10 μl 10 % SDS, 10 μl APS, and 1 μl TEMED. Remove the water from the solid running gel and cast the stacking gel on top of the running gel and insert a 10-well gel comb immediately without introducing air bubbles. Let it solidify for 30 min at room temperature.
-
4.
Remove the comb and add loading standard to the first lane. Samples are loaded in the other lanes of the gel. Electrophoresis is performed at 100 V (15 mA) for 5 min until the samples have entered the stacking gel. Then continue at 150–200 V (20–30 mA) until the dye front has reached the bottom of the running gel (see Note 21 ).
-
5.
After the electrophoresis run, open the glass plates using a spatula, leaving the gel on one of the glass plates. Carefully remove the gel from the plate by rinsing the plate and the gel in deionized water. Transfer the gel into a container filled with fixing solution and fix the gel for 30–45–60 min.
-
6.
After fixation, rinse the gel with ultrapure Milli-Q (MQ) water and stain using GelCode Blue reagent for 45–60 min. After this, distain the gel using MQ water and incubate for 60 min (for publication quality pictures refresh MQ and distain overnight).
-
7.
Each gel lane is cut into 24 equally sized slices (see Note 22 and Fig. 4) and each slice is transferred into an Eppendorf tube.
Fig. 4 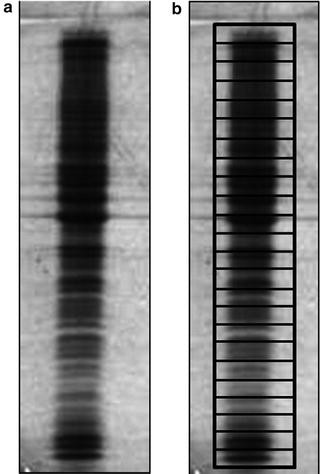
SDS-PAGE protein profile of a Golgi-enriched fraction after a gel electrophoresis. Separation separation of 80 μg of proteins from a purified Golgi fraction. (a) Coomassie GelCode Blue stained gel. (b) Illustration of the size of the 24 gel slices used for MS analysis

3.7 Mass Spectrometry
The 24 gel slices are subjected to in-gel tryptic digestion as described below (see Note 23 ) and before [8]:
-
1.
Add 100 μl AcN per tube, leave it for a few seconds and then remove it from the tube.
-
2.
Reduction of proteins is performed by adding with 100 μl DTT (6.5 mM in 50 mM ambic) and incubation at room temperature for 60 min.
-
3.
Remove the DTT and add 100 μl AcN per tube, leave it for a few seconds and then remove it again from the tube.
-
4.
Alkylate the proteins by adding 100 μl iodoacetamide (54 mM in 50 mM ambic) and incubating at room temperature for 60 min in the dark.
-
5.
Shrink the gel slice by adding (and removing) 100 μl AcN, swell the slice by adding (and removing) 100 μl ambic and shrink again by adding (and removing) 100 μl AcN.
-
6.
Dry the samples in the air (leave cups open).
-
7.
Add 10 μl of diluted trypsin (10 ng/μl final concentration) and incubate on ice for 60 min. Remove supernatant (if there is any) and add 50 μl ambic to cover the gel pieces. Digest the peptides overnight at 37 °C.
-
8.
20–80 % of the supernatants obtained after the digestion is used for LC-MS/MS analysis on a Thermo Finnigan FT-ICR equipped with a 7 Tesla magnet coupled to an Agilent Series 1100 binary pump system (Agilent Technologies).
-
9.
Peptide mixtures are trapped on an in-house packed 5 cm × 100 μm Aqua™ C18 reversed phase column (Phenomenex) at a flow rate of 5 μl/min (see Note 24 ).
-
10.
Peptide separation is achieved on an 15 cm × 75 μm Aqua™ C18 reversed phase column using a gradient of 0–70 % solution B (solution A = 0.1 M acetic acid; solution B = 80 % [v/v] acetonitrile, 0.1 M acetic acid) in 60 min at a constant flow rate of 200 nl/min (see Note 25 ).
3.8 Mass Spectrometry Data Analysis
-
1.
Finnigan *.raw files are converted to *.dta files using BioWorks software, version 3.1 SR1 (Thermo Electron Corporation).
-
2.
For this process the program is set to track the scan limits automatically and calculate for peptides with a mass from 300 to 5,000 amu, automatically detecting the charge state and MS level (MS or MS/MS). The threshold was set to 100 counts.
-
3.
Subsequently, Mascot generic files were generated through in-house developed software.
-
4.
These files were used to search the IPI_Human 3.36 database [17] on an in-house Mascot server [4] allowing up to 2 missed cleavages, a peptide mass tolerance of 50 ppm and a fragment mass tolerance of 0.8 Da (see Note 26 ).
-
5.
Peptide modifications allowed in the searches were carbamidomethyl modification of cysteine (fixed) and oxidation of methionine, tryptophan, and histidine (variable) (see Note 27 ).
-
6.
Proteins matching the criteria for at least two reliable peptides (rank 1; unique; individual score higher than 29 [1 % false positive rate]), and with a protein score higher than 64 were considered as positive identified proteins.
-
7.
Raw data files and Mascot html results pages were loaded into the MSQuant program [5] adapted for SILAC-based quantitative analysis.
-
8.
All quantified peptides are verified by manual inspection of the spectra used for quantification. To identify statistically significant (and removing) different protein abundances between samples (p < 0.05), data from 3 independent experiments were loaded into the StatQuant program for statistical analysis [18] (see Note 28 ).
4 Notes
-
1.
For 200 ml LB broth, dissolve 2 g tryptone, 1 g yeast extract, 2 g sodium chloride in 200 ml deionized water. For agar plates, add 3 g agar to the mixture. Autoclave for 20 min and allow the solution to cool to 55 °C. Store the liquid LB broth at room temperature or 4 ºC. For agar plates, pour approximately 15 ml into petri dishes and let solidify at room temperature. Store the plates at 4 ºC.
-
2.
For 29 % w/w sucrose solution dissolve 65.08 g sucrose in 200 ml of 10 mM Tris, pH 7.4; for the 35 % w/w: 80.60 g sucrose in 200 ml of 10 mM Tris, pH 7.4 and for the 62 % w/w sucrose: 161 g sucrose in 200 ml of 10 mM Tris, pH 7.4. Prepare the solutions in advance and dissolve with rotation ON at 4 °C. Sucrose solutions can be stored at −80 °C.
-
3.
Medium is removed from the cells and cells are washed by rinsing them carefully with PBS, 2 ml of trypsin is added to the cells and the flask is incubated in the incubator at 37 °C/5 % CO2 for a few minutes. Cells are removed from the surface by carefully tapping the flask. Add medium (10 ml) and transfer the cells into a 15 ml tube (Corning). Cells are centrifuged at 500 × g for 5 min. Resuspend the cell pellet in 1 ml medium and the cell density is determined by cell countering under the microscope using a cell counter. A specific number of cells (see Note 4 ) is transfered to new flasks or plates in medium and incubated in the incubator at 37 °C/5 % CO2.
-
4.
Different volumes of DMEM and number of HeLa cells are used, depending on the type of plate, indicated at the specific steps of the protocol.
-
5.
For overnight cultures, add 1 Salmonella colony (from a freshly grown agar plate) into 4 ml LB broth in a 15 ml tube (Corning) and incubate for 16–18 h at 37 °C with continuous shaking. Add 1 ml of the overnight culture to 32 ml of LB broth in a 50 ml tube (Corning) and incubate for another 3.5 h at 37 °C with continuous shaking to reach the exponential phase, as described before [13].
-
6.
HeLa cells are infected with Salmonella at a multiplicity of infection (MOI) of 100. This means that 100 times more bacteria then HeLa cells are added to the cells. For this, the bacteria from the 33 ml culture are centrifuged for 15 min at 1,000 × g after which the pellet is resuspended in 1 ml DMEM supplemented with 10 % FCS (no pen/strep!). By performing a growth curve of Salmonella, we determined that after 3.5 h the concentration of bacteria in the 33 ml culture 3.5 h is 8.3 × 108 bacteria/ml. The growth curve of Salmonella should be repeated for the strain that is used in the experiments. Since the number of HeLa cells is known (specified in the specific steps of the protocol), the volume of bacteria that corresponds with an MOI of 100 can be calculated. HeLa cells are carefully rinsed with PBS and incubated in DMEM, supplemented with 10 % FCS (no pen/strep) and the right amount of bacteria. Cells are incubated for 15 min at 37 °C. During these 15 min, the Salmonella bacteria attach to the HeLa cells.
-
7.
After the attachment of the bacteria to the cells, the medium is removed from the cells and the cells are washed with PBS (adding and removing of 15 ml of PBS) to get rid of cell debris and any loose bacteria. Cells and bacteria are then incubated in medium containing 100 μg/ml gentamycin. The stock solution of gentamycin has a concentration of 10 mg/ml. Therefore, this solution should be diluted 1,000 times in DMEM/10 % FCS.
-
8.
The “heavy” amino acids l-arginine-13C6 15N4 hydrochloride and l-lysine-13C6 15N2 hydrochloride, or the “light” amino acids l-arginine-12C6 14N4 hydrochloride and l-lysine-12C6 14N2 are added to specialized DMEM lacking l-arginine and l-lysine, at a final concentration of 84 mg/L and 146 mg/L for arginine and lysine, respectively. Instead of using commercially available normal “light” DMEM, we choose to add the light amino acids to specialized DMEM lacking l-arginine and l-lysine. In this way, we prepared the heavy and light media in exactly the same manner.
-
9.
10 % Dialyzed FCS and 100 IU of penicillin/ml and 100 μg of streptomycin/ml are added to specialized DMEM. Dialyzed FCS is used because dialysis depletes FCS of small molecules such as amino acids, hormones and cytokines while avoiding the precipitation of serum proteins. In this way, “normal” l-arginine and l-lysine remains absent in specialized DMEM.
-
10.
To check the incorporation of the “heavy” amino acids into the cells, MALDI-TOF-TOF analysis is performed as described [8]. For this analysis, cells passaged in light and heavy medium are trypsinized, centrifuged for 10 min at 500 × g and pellets are resuspended in 1 ml lysis buffer (20 mM Tris–HCl, pH 7.6, 150 mM NaCl, 1 % nonidet P-40, 0.5 % NaDOC, 0.1 % SDS, 2 μg/ml aprotinin, 2 μg/ml leupeptin, 1 μg/ml pepstatin, 1 mM PMSF). After an incubation on ice for 10 min, the nuclei are spun down by centrifugation for 5 min at 600 × g at 4 ºC, and the supernatant is used for MALDI-TOF-TOF analysis.
-
11.
For the SILAC experiments 4 T175 flasks with 1 × 107 cells are infected with Salmonella at a MOI of 100. Therefore, 100 × 107 bacteria are needed per flask, which is 400 × 107 bacteria in total. Since the 1:33 culture contains 8.3 × 108 Salmonella/ml, 400 × 107 bacteria corresponds with 4.8 ml of bacterial culture. This volume of bacteria is taken and centrifuged for 5 min at 1,000 × g after which the pellet is resuspended in 40 ml of DMEM/10 % FCS. 10 ml is added per flask and infection is carried out as described in Subheading 3.2. The other 4 flasks are mock-infected. In that case, the same procedure is followed but instead of Salmonella bacteria, DMEM is used. The SILAC experiments must be repeated at least three times; 2 experiments in which the “heavy” cells are infected with Salmonella and 1 experiment in which the “light” cells are infected.
-
12.
After Salmonella infection, remove the medium from the cells, and wash the cells carefully with PBS. Add 5 ml homogenization buffer per flask. Using cell scrapers harvest the cells from the flasks and combine them in a 1:1 ratio in a 50 ml tube. Centrifuge the cells at 500 × g for 10 min (4 ºC). In order to wash the cells, resuspend the pellet in cold Homogenization Buffer (HB) and transfer them to a 15 ml tube (10 min, 500 × g, 4 ºC). Measure the volume of the pellet and add four times HB with protease inhibitors, incubate on ice.
-
13.
Carefully remove the HB as the pellet is loose after this step.
-
14.
It is important to estimate as good as possible the volume of the pellet and to add not more than 5 volumes (of the pellet estimated volume) of HB to ensure sufficiency and reproducibility of the homogenization.
-
15.
Check the homogenization efficiency at the microscope using a vital stain such as Trypan Blue after every 10–20 strokes. In general 50–60 strokes are needed to ensure disruption of about 90 % of the cells. However, the number of strokes are dependent of the pressure given by the operator. Be careful not to homogenize the cells excessively as too harsh conditions may affect the integrity of intracellular membranes and contaminate your preparation.
-
16.
If the sucrose concentration is out of the range 36.5–37.5 (w/w), adjust by adding either 10 mM Tris–HCl buffer (pH 7.4) or sucrose solution (2 M). Use the following formulas to calculate the required volume:
$$ \frac{V_{\mathrm{original}}\times \left({C}_{\mathrm{wanted}}-{C}_{\mathrm{original}}\right)}{\left({C}_{\mathrm{stock}\;\mathrm{solution}}-{C}_{\mathrm{wanted}}\right)}={V}_{\mathrm{to}\kern0.24em \mathrm{add}} $$When the sucrose concentration is higher than 37.5:
When sucrose concentration of the homogenate is lower than 36.5:
$$ \frac{V_{\mathrm{original}}\times \left({C}_{\mathrm{wanted}}-{C}_{\mathrm{original}}\right)}{\left({C}_{\mathrm{wanted}}\right)}={V}_{\mathrm{to}\kern0.24em \mathrm{add}} $$V original = Volume of homogenate solution after the addition of 62 %(w/w) sucrose.
V to add = Volume to add of either 2 M sucrose solution or 10 mM Tris–HCl buffer.
C original = Sucrose concentration (in M) of homogenate solution after adding 62 % (w/w) sucrose.
C wanted = Desired concentration of sucrose (i.e., 37 % (w/w)).
C stock solution = Stock solution of sucrose (2 M).
-
17.
When preparing the sucrose gradient use a bent pipet tip to slowly overlay the sucrose layers without disturbing the interfaces. The interfaces have to be easily recognized after layering. Only then the floated Golgi-enriched fraction is easy to see and collect.
-
18.
After centrifugation carefully remove the tube from the centrifuge bucket. Use a syringe with a needle of 22G to puncture the tube and to collect the Golgi-enriched band (see Fig. 5).
Fig. 5 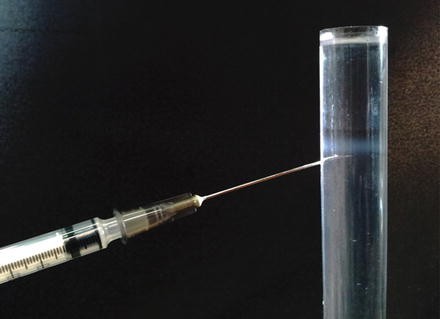
Floated Golgi-enriched fraction. After flotation an opalescent band at the interface between 35 and 29 % sucrose will be visible. Collect the band by using 1 ml syringe with a 22G needle. The collected fraction is about 1 ml and has a protein concentration of about 0.15 μg/μl. The sucrose concentration is about 30 % (w/w)
-
19.
Determine the protein concentration of the collected fraction (usually 0.15 μg/μl), aliquot the Golgi fraction into Eppendorf tubes, snap-freeze the tubes in liquid nitrogen and then store at −80 °C.
-
20.
Protein concentration of the Golgi-enriched fraction is measured using the Bradford protein assay. The volume corresponding to 80 μg of protein is added to an Eppendorf tube, centrifuged at maximal speed for 10 min (4 ºC). Pellet is resuspended in 20 μl of Laemmli sample buffer containing 10 mM DTT and heated for 5 min at 95 °C. Samples are cooled on ice.
-
21.
Adding a drop of sample buffer to un-used wells will help to form straight dye front during the electrophoresis.
-
22.
After Coomassie staining, each gel lane is cut into 24-equally sized pieced, using a razor blade. Cut around the edges of the complete lane and remove this lane from the gel. Next, equally divide the lane into 24 pieces (see Fig. 4). Transfer each piece into a Eppendorf tube.
-
23.
DTT is added to reduce cysteines and to disrupt sulfur bridges. Iodoacetamide is added to prevent the formation of new sulfur bridges. The cycle with AcN and Ambic dehydrates and rehydrates the minced gel slices for washing and absorbance of reagents and trypsin in the gel slices. During this cycle, the solutions have only to be added for a few seconds, after which the solutions can be removed again.
-
24.
In this step, the peptides derived from trypsin-digested proteins are loaded onto the trapping column. This step can be compared to the loading of proteins into the stacking gel when performing a SDS-PAGE gel. No separation takes place at this step.
-
25.
After loading of all peptides onto the column, valves are switched and the gradient from solution A to B starts running. More complex samples, e.g., samples containing peptides from various proteins, require longer run times. Peptides are eluted from the trapping column, with the most hydrophilic peptides coming off first, and the most hydrophobic peptides coming off last.
-
26.
In the identification of peptides, some room for error (incorrect measurements of peptide and peptide fragment masses) is allowed. The more accurate the mass spectrometer can determine peptide and fragment mass, the smaller the error will be, yielding higher confidence in identifications.
-
27.
During preparation of peptide mixtures, some peptide modifications can occur. As such modifications affect peptide (and fragment) mass, and mass changes have to be taken into account to deduce to identity of each peptide and fragment. All cysteine residues in the sample are reduced and subsequently alkylated using iodoacetamide, yielding a mass addition of 57.02146 amu on each cysteine. Oxidation, which can occur but does not necessarily affect all methionine, tryptophan or histidine residues, yields a mass change of 15.99491 amu.
-
28.
Nowadays, many programs for quantitative analysis of mass spectrometry are available. Many of them have the option to calculate differential quantities based on different labeling methods such as SILAC, iTRAQ or label-free quantitation. Also protein identification is integrated, generating a robust quantitative proteomics analysis pipeline.

References
Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A et al (2002) Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics 1(5):376–386
Oda Y, Huang K, Cross FR, Cowburn D, Chait BT (1999) Accurate quantitation of protein expression and site-specific phosphorylation. Proc Natl Acad Sci U S A 96(12):6591–6596
Mannova P, Fang R, Wang H, Deng B, McIntosh MW, Hanash SM et al (2006) Modification of host lipid raft proteome upon hepatitis C virus replication. Mol Cell Proteomics 5(12):2319–2325
Perkins DN, Pappin DJ, Creasy DM, Cottrell JS (1999) Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20(18):3551–3567
Schulze WX, Mann M (2004) A novel proteomic screen for peptide-protein interactions. J Biol Chem 279(11):10756–10764
Shi L, Chowdhury SM, Smallwood HS, Yoon H, Mottaz-Brewer HM, Norbeck AD et al (2009) Proteomic investigation of the time course responses of RAW 264.7 macrophages to infection with Salmonella enterica. Infect Immun 77(8):3227–3233
Shui W, Gilmore SA, Sheu L, Liu J, Keasling JD, Bertozzi CR (2009) Quantitative proteomic profiling of host-pathogen interactions: the macrophage response to Mycobacterium tuberculosis lipids. J Proteome Res 8(1):282–289
van Balkom BW, van Gestel RA, Brouwers JF, Krijgsveld J, Tielens AG, Heck AJ et al (2005) Mass spectrometric analysis of the Schistosoma mansoni tegumental sub-proteome. J Proteome Res 4(3):958–966
Vester D, Rapp E, Gade D, Genzel Y, Reichl U (2009) Quantitative analysis of cellular proteome alterations in human influenza A virus-infected mammalian cell lines. Proteomics 9(12):3316–3327
Vogels MW, van Balkom BW, Heck AJ, de Haan CA, Rottier PJ, Batenburg JJ et al (2011) Quantitative proteomic identification of host factors involved in the Salmonella typhimurium infection cycle. Proteomics 11(23):4477–4491
Vogels MW, van Balkom BW, Kaloyanova DV, Batenburg JJ, Heck AJ, Helms JB et al (2011) Identification of host factors involved in coronavirus replication by quantitative proteomics analysis. Proteomics 11(1):64–80
Steele-Mortimer O (2008) Infection of epithelial cells with Salmonella enterica. Methods Mol Biol 431:201–211
Beuzon CR, Meresse S, Unsworth KE, Ruiz-Albert J, Garvis S, Waterman SR et al (2000) Salmonella maintains the integrity of its intracellular vacuole through the action of SifA. EMBO J 19(13):3235–3249
Balch WE, Dunphy WG, Braell WA, Rothman JE (1984) Reconstitution of the transport of protein between successive compartments of the golgi measured by the coupled incorporation of N-acetylglucosamine. Cell 39(2 Pt 1):405–416
Brugger B, Sandhoff R, Wegehingel S, Gorgas K, Malsam J, Helms JB et al (2000) Evidence for segregation of sphingomyelin and cholesterol during formation of COPI-coated vesicles. J Cell Biol 151(3):507–518
Balch WE, Rothman JE (1985) Characterization of protein transport between successive compartments of the golgi apparatus: asymmetric properties of donor and acceptor activities in a cell-free system. Arch Biochem Biophys 240(1):413–425
Kersey PJ, Duarte J, Williams A, Karavidopoulou Y, Birney E, Apweiler R (2004) The international protein index: an integrated database for proteomics experiments. Proteomics 4(7):1985–1988
van Breukelen B, van den Toorn HW, Drugan MM, Heck AJ (2009) StatQuant: a post-quantification analysis toolbox for improving quantitative mass spectrometry. Bioinformatics 25(11):1472–1473
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer Science+Business Media New York
About this protocol
Cite this protocol
Kaloyanova, D., Vogels, M., van Balkom, B.W.M., Helms, J.B. (2015). Quantitative Proteomic Identification of Host Factors Involved in the Salmonella typhimurium Infection Cycle. In: Schatten, H., Eisenstark, A. (eds) Salmonella. Methods in Molecular Biology, vol 1225. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-1625-2_2
Download citation
DOI: https://doi.org/10.1007/978-1-4939-1625-2_2
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-1624-5
Online ISBN: 978-1-4939-1625-2
eBook Packages: Springer Protocols