
Abstract
Neutralizing antibodies are a critical component in the protection or recovery from viral infections. In the absence of available vaccines or antiviral drugs for many important human viral pathogens, the identification and characterization of new human monoclonal antibodies (hmAbs) that are able to neutralize viruses offers the possibility for effective pre- and/or post-exposure therapeutic modalities. Such hmAbs may also help in our understanding of the virus entry process, the mechanisms of virus neutralization, and in the eventual development of specific entry inhibitors, vaccines, and research tools. The majority of the more recently developed antiviral hmAbs have come from the use of antibody phage-display technologies using both naïve and immune libraries. Many of these agents are also enveloped viruses possessing important neutralizing determinants within their membrane-anchored envelope glycoproteins, and the use of recombinant, soluble versions of these viral glycoproteins is often critical in the isolation and development of antiviral hmAbs. This chapter will detail several methods that have been successfully employed to produce, purify, and characterize soluble and secreted versions of several viral envelope glycoproteins which have been successfully used as antigens to capture and isolate human phage-displayed monoclonal antibodies.
You have full access to this open access chapter, Download protocol PDF
1 Introduction
For the vast majority of viral pathogens there is a paucity of drug-based therapies. Rather, it has been the development of vaccines that has been the mainstay of prevention and intervention strategies for combating human and animal viral diseases. There are presently 15 viral vaccines approved for human use, excluding various subtypes, and the majority of these are live-attenuated formulations (reviewed in (1)). In general terms, these live-attenuated viral vaccines are highly effective because they elicit a balanced immune response in the recipient host, stimulating both cell-mediated and humoral immunity. However, for many viruses, especially those that have associated highly pathogenic characteristics as with Biological Safety Level-4 (BSL-4) restricted agents or retroviruses such as human immunodeficiency virus type 1 (HIV-1), the use of live-attenuated vaccines is not feasible.
A number of studies have demonstrated the importance of neutralizing antibodies in the protection or recovery from viral infections (2, 3). Indeed, as obligate intracellular parasites, viruses pose significant challenges for the development of effective antiviral therapeutics. Neutralizing polyclonal antibodies have a long history of being effective against some viruses and more recently, monoclonal antibodies (mAbs) have also shown success. The humanized mAb Synagis (palivizumab), which is currently the only mAb against a viral disease approved by the US Food and Drug Administration (FDA), has been widely used as a prophylactic measure against respiratory syncytial virus (RSV) infections in neonates and immune-compromised individuals and is more cost-effective and efficacious than the original polyclonal product (4). Most recently, the anti-RSV palivizumab has been improved, and motavizumab has been shown to potently inhibit viral replication in the upper respiratory tract in a cotton rat model (5). Virus-neutralizing antibodies can also be administered passively to acutely infected individuals and be highly efficacious. The mechanism of passively administered antibody therapy can be viewed as that of an antiviral drug: suppressing infection and permitting the host to mount an effective immune response (6). Today, passively administered antibody is routinely used as an effective antiviral therapy or prophylactic for hepatitis B, varicella-zoster, rabies virus, measles virus, and others (reviewed in (2)). In most cases their use is a first-line therapy as a post-exposure measure or in circumstances where vaccination is not possible. However, serum polyclonal antibody preparations have associated problems related to toxicity and potential allergic reactions, as well as lot-to-lot variation and uncertain dosing regimes (7).
The major advances in furthering the development of specific mAbs has been through the use of bacterial phage-display platforms with combinatorial antibody libraries (8, 9). Further, these phage libraries can be prepared to encode human antibodies as Fabs which contain the light-chain and the first two domains of the heavy-chain or single-chain variable domain fragments (scFvs) containing the variable domains of the light and heavy chains, and this technology has been complemented by innovative affinity maturation strategies to improve antibody binding (reviewed in (10)). These techniques in human phage-display antibody platforms have facilitated the rapid identification and isolation of specific human mAbs, eliminating the immunization, hybridoma development, and humanization processes. In the absence of available vaccines or antiviral drugs, the identification and characterization of new human monoclonal antibodies (hmAbs) that are able to neutralize viruses offers the possibility for effective pre- and post-exposure therapeutic modalities. Such antibodies may also help in our understanding of the virus entry process and its underlying mechanisms, the viral neutralization mechanisms, and in the eventual development of specific entry inhibitors, vaccines, and research tools. There have been many recent examples of the development and isolation of hmAbs using phage-display methodologies reactive against important human viral pathogens including HIV-1 (11–16), the paramyxoviruses, Hendra virus (HeV) and Nipah virus (NiV) (17), and the human SARS coronavirus (18).
Many of these viral pathogens are also enveloped viruses, and it is almost without exception that all neutralizing antibodies to enveloped viruses are directed against the virus’ envelope glycoproteins, and traditionally the antibody response has been the immunologic measure of vaccine efficacy (19). All known viral envelope glycoproteins are homo- or heterooligomers in their mature and functional forms (20) and multimeric proteins, like these, generally interact over large areas which often translate into important structural differences between monomeric subunits and the mature oligomer. This feature can also impart significant differences in antigenic structure which has been shown for a number of proteins such as the trimeric influenza HA glycoprotein (21) and HIV-1 gp120/gp41 (22). In addition, some viruses pose significant additional challenges such as antigenic variation of their structural proteins that are important neutralization determinants. Perhaps the best-characterized example of this particular problem is with primary HIV-1 isolates that exist across the many varied HIV-1 subtypes (23). In efforts to circumvent this issue, further improvements and enhancements in the techniques of phage-displayed antibody library panning have been developed in order to better select for broadly reactive mAbs or for mAbs reactive to particular subunits of a multi-subunit viral glycoprotein such as sequential antigen panning (SAP) and competitive antigen panning (CAP) methodologies (14, 15).
It is often critical that the antigens used for the panning and isolation of hmAbs from phage libraries be produced and/or purified using methods whereby they retain a near-native structure and conformation, such as an oligomeric configuration. A useful approach to develop viral membrane glycoproteins suitable for panning phage libraries or as antigens for eliciting antibody responses that recognize their native form is to engineer soluble and secreted versions of the molecules. Often, this approach yields a quaternary structure similar to their native counterparts, and for animal viruses eukaryotic expression systems are typically employed such as recombinant bacculovirus or vaccinia virus, or transient or stable expression in cell culture (22, 24–36). This chapter will detail several methods that have been successfully employed to produce, purify, and characterize soluble and secreted versions of several viral envelope glycoproteins which have been successfully used as antigens to capture and isolate human phage-displayed mAbs.
2 Materials
2.1 Cell Culture
-
1.
Cell lines: BS-C-1 (ATCC #CCL 26), HeLa cells (ATCC #CCL 2), 293T was provided by Dr. G. Quinnan (Uniformed Services University, MD).Media: Dulbecco’s modified Eagle’s medium (Quality Biologicals, Gaithersburg, MD) supplemented with 2 mM L-glutamine and either 10% cosmic calf serum (CCS) (DMEM-10) (HyClone, Logan, UT) or 2.5% CCS (DMEM-2.5); Eagle’s minimal essential medium (Quality Biologicals) supplemented with 2 mM L-glutamine and either 10% CCS serum (EMEM-10) or 2.5% CCS (EMEM-2.5); 0.25% trypsin/0.02% EDTA (Quality Biologicals); sterile 1× PBS for cell culture (Quality Biologicals); OptiMEM I Reduced Serum Medium (1X), liquid with L-glutamine (Invitrogen Corp.).
-
2.
Plasticware: 25 cm2, 75 cm2, and 150 cm2 tissue culture flasks (Falcon, BD laboratories); polypropylene 50 ml conical centrifuge tubes (Falcon, BD laboratories); 6-, 24-, and 96-well flat-bottom tissue culture plates (Falcon, BD laboratories); 850 cm2and 1700 cm2 ribbed roller bottles (Corning Inc); 250 conical-bottom centrifuge tube (Corning Inc.); sterilizing filter units, low protein binding membrane, 0.22 µm PES (Corning Inc.).
-
3.
Hygromycin B, 50 mg/ml (Invitrogen Corp.)
2.2 Recombinant Vaccinia Viruses and Expression Plasmids
-
1.
Recombinant vaccinia viruses: vAC4 (vaccinia virus encoding HIV-1 gp140R2) (37, 38); vKB-16 (vaccinia virus encoding HeV sG) (24); vKB-22 (vaccinia virus encoding NiV sG) (39).
-
2.
Expression plasmid constructs are prepared using cytomegalovirus (CMV) enhancer/promoter-driven expression vector phCMV1 (Gelantis), and pcDNA3.1/Hygro(+) (Invitrogen Corp.).
-
3.
HRP-conjugated Rabbit anti-S-tag antibody (Benthyl Laboratories, Inc.)
-
4.
HRP substrate: Supersignal West Pico Chemiluminescent Substrate (Pierce)
-
5.
S-protein agarose (EMD Biosciences, Inc.)
2.3 DNA Transfection
-
1.
Expression plasmid containing gene of interest.
-
2.
0.1% Gelatin.
-
3.
DMEM serum free, DMEM-10.
-
4.
Fugene® 6 transfection reagent (Roche).
2.4 Immuno- and Affinity Precipitations
-
1.
Lysis buffer: 1% Triton® X-100, 0.1 M Tris pH 8.0, 0.1 M NaCl.
-
2.
S-protein Agarose, 50% slurry (EMD Biosciences, Inc.).
-
3.
Complete protease inhibitor (Roche).
-
4.
Antibody that reacts with the protein of interest.
2.5 Affinity Purification
Filter sterilization is recommended for all buffers.
2.5.1 HIV-1 gp140
-
1.
Lentil lectin SepharoseTM 4B (GE Healthcare).
-
2.
XK26 column (GE Healthcare).
-
3.
Centriprep® molecular weight cut off centrifugal filter units, YM 50 (Millipore Corp).
-
4.
Acrodisc® Syringe Filter 0.2 µm HT Tuffryn low protein-binding membrane (Millipore Corp.).
-
5.
Equilibration buffer: PBS, 0.5% Triton® X-100, 0.02 M Tris-HCl, pH 7.5.
-
6.
Wash buffer I: PBS, 0.5% Triton® X-100, 0.3 M NaCl, 0.02 M Tris-HCl, pH 7.5.
-
7.
Wash buffer II: PBS, 0.02 M Tris-HCl, pH 7.5.
-
8.
Elution buffer: PBS, 0.02 M Tris-HCl, pH 7.5, 0.5 M methyl α-D-mannopyranoside.
-
9.
High-pH buffer: 0.5 M NaCl, 0.02 M Tris-HCl, pH 8.5.
-
10.
Low-pH buffer: 0.5 M NaCl, 0.02 M Tris-HCl, pH 5.5.
-
11.
Regeneration buffer: 0.02 M Tris-HCl, pH 7.5, 0.001 M MnCl2, 0.001 M CaCl2.
-
12.
Protein buffer: PBS, pH 7.0.
2.5.2 Henipavirus Soluble G (sG)
-
1.
S-protein Agarose (EMD Biosciences, Inc.).
-
2.
XK26 column (GE Healthcare).
-
3.
Centriprep® molecular weight cut-off centrifugal filter units, YM 30 (Millipore Corp.).
-
4.
Acrodisc® Syringe Filter 0.2 µm HT Tuffryn low protein-binding membrane (Millipore Corp.).
-
5.
Wash buffer I: PBS, 0.1% Triton® X-100, 0.3 M NaCl.
-
6.
Wash buffer II: PBS, 0.1% Triton® X-100.
-
7.
20% Ethanol.
-
8.
Elution buffer: 0.2 M citrate, pH 2.
-
9.
Neutralization buffer: 1 M Tris-HCl pH 8.0 or 1 M HEPES buffer pH 9.0 (see Note 1).
-
10.
Protein buffer: PBS, pH 7.0.
2.5.3 Henipavirus Soluble F (sF)
-
1.
S-protein Agarose (EMD Biosciences, Inc).
-
2.
XK26 column (GE Healthcare).
-
3.
Centriprep® molecular weight cut-off centrifugal filter units, YM 30 (Millipore Corp.).
-
4.
Acrodisc® Syringe Filter 0.2 µm HT Tuffryn low protein-binding membrane (Millipore Corp.).
-
5.
Wash buffer I: PBS, 0.5% Triton® X-100, 0.5 M NaCl, 0.1 M L-Arginine, 0.02 M Tris-HCl, pH 7.5.
-
6.
Wash buffer II: PBS, 0.1% Triton® X-100, 0.1 M L-Arginine.
-
7.
20% Ethanol.
-
8.
Elution buffer: 0.2 M citrate, 0.2 M L-Arginine, pH 2.
-
9.
Neutralization buffer: 1 M HEPES buffer pH 9.0 (see Note 2).
-
10.
Protein buffer: PBS, 0.01% Triton® X-100, 0.1 M L-Arginine.
2.6 Polyacrylamide Gel Electrophoresis and Western Blotting
2.6.1 Blue Native PAGE (Invitrogen)
-
1.
NativePAGE™ Novex® 3–12% Bis-Tris Gels 1.0 mm, 15 well.
-
2.
NativePAGE™ Running Buffer (20X).
-
3.
NativePAGE™ Cathode Buffer Additive (20X).
-
4.
NativePAGE™ 5% G-250 Sample Additive.
-
5.
NativePAGE™ Sample Buffer (4X).
-
6.
NativeMark™ Unstained Protein Standard.
-
7.
Fixing solution (40% methanol, 10% acetic acid).
-
8.
Destain solution (8% acetic acid).
-
9.
XCell SureLock™ Mini-Cell (Invitrogen Corp.).
2.6.2 Western Blotting and Immunodetection
-
1.
PVDF (polyvinylidene difluoride) membrane (Bio-Rad Laboratories, Inc.) for native gel analysis.
-
2.
NuPAGE® Transfer Buffer (20X).
-
3.
Blocking buffer: 5% skim milk blocker (Bio-Rad Laboratories, Inc.), 1× PBS, 0.2% Tween 20.
-
4.
Wash buffer: 1× PBS, 0.2% Tween 20.
-
5.
Rabbit anti-S-tag antibody, HRP conjugated (Benthyl Laboratories, Inc.).
-
6.
Supersignal West Pico Chemiluminescent Substrate (Pierce).
-
7.
XCell II™ Blot Module (Invitrogen Corp.).
2.7 Size-Exclusion Chromatography
All buffers should be sterile-filtered and degassed.
-
1.
Preparative column: HiLoad 16/60 Superdex 200 prep grade XK 16 gel filtration column (GE Healthcare).
-
2.
Analytic column: Superdex 200 10/300 GL gel filtration column (GE Healthcare).
-
3.
Molecular weights (29 kDa −669 kDa) calibration kit (Sigma).
-
4.
Centriprep® molecular weight cut-off centrifugal filter units (Millipore Corp.).
-
5.
Phosphate buffer Saline (PBS).
-
6.
20% Ethanol.
-
7.
PBS, 0.01% Triton® X-100.
-
8.
A fast protein liquid chromatography (FPLC) system or similar ones such as a P-500 pump (GE Healthcare), UA-6 UV detector (ISCO), and fraction collector (ISCO).
2.8 Sucrose Gradient Ultracentrifugation
-
1.
5%, 20% Sucrose in 0.1 M Tris-HCl pH 7.5, 0.1 M NaCl.
-
2.
Fraction collector (ISCO).
-
3.
Polyallomer 14-mm by 95-mm tubes (Beckman Coulter, Inc.).
-
4.
Gradient master (Biocomp).
-
5.
SW40 rotor (Beckman Coulter, Inc.).
-
6.
Fraction recovery system (Beckman Coulter, Inc.).
3 Methods
The methods described below outline the steps for recombinant viral glycoprotein production using vaccinia virus for HIV-1 gp140 and HeV and NiV soluble G glycoprotein (sG) and affinity purification of the glycoproteins. In addition, recombinant viral glycoprotein production and affinity purification from stable HeV and NiV soluble F glycoprotein (sF)-secreting 293T cell lines will be detailed. Additional purification and oligomeric analyses methods are also detailed, including size-exclusion gel filtration, sucrose gradient ultracentrifugation analysis, and Blue Native polyacrylamide gel electrophoresis (BN-PAGE) analysis.
The design and construction of recombinant vaccinia viruses encoding HIV-1 gp140 and HeV and NiV sG as well as the characterization of the recombinant, soluble, and secreted viral glycoproteins have been detailed elsewhere(24, 40). In this section for the purposes of additional illustration there will be an emphasis on the production and purification of soluble and secreted forms of the HeV and NiV F glycoprotein, including the analysis of oligomeric characteristics as well as the generation of stable cell lines secreting recombinant sF.
3.1 Vaccinia Virus Expressed Soluble Recombinant Viral Glycoproteins
3.1.1 Cell Culture and Protein Expression
3.1.1.1 Preparation of Cells in Roller Bottles
-
1.
Prepare 150-cm2 tissue culture flasks of BS-C-1 cells. One confluent flask of cells should be transferred into one 850-cm2 roller bottle (see Note 3).
-
2.
Aspirate media from each flask, wash the cells once with sterile PBS, and add 3 ml of trypsin-EDTA per flask. When the cells begin to detach, dislodge the cells and add 15 ml of EMEM-10 (if employing HeLa cells, replace EMEM with DMEM) to each flask, resuspend the cells by pipetting up and down.
-
3.
Transfer the cells to a large 250-ml conical bottom tube and centrifuge the cells at 500 g for 10 min to pellet the cells.
-
4.
Resuspend the cell pellet in EMEM-10 to a volume of five times the number of T-150 flasks that were harvested (e.g., cells from two T-150 flasks should be resuspended in 10 ml of EMEM-10) (see Note 4).
-
5.
Add 200 ml of EMEM-10 and add 5 ml of the cell/media suspension to each roller bottle.
-
6.
Flow 10% CO2, 90% air into each roller bottle for 30 s at approximately 150 kPa and tighten the caps of the roller bottles (see Note 5).
-
7.
Incubate the roller bottles in 37°C at 0.5 rpm for 5 days or until the cells are completely confluent to form a compact monolayer.
3.1.1.2 Infection of Cells with Purified Vaccinia Virus
-
1.
Preparation of virus inoculum
-
a.
Thaw and vortex the purified recombinant vaccinia virus to be used for protein expression.
-
b.
The virus inoculum is prepared in 5 ml of OptiMEM for each roller bottle to be infected at an MOI of 5 pfu/cell. A confluent roller bottle is estimated to have approximately 150 × 106 BSC-1 cells or 100 × 106 HeLa- cells (7.5 × 108 pfu for BSC-1 cells or 5 × 108 pfu for HeLa- cells per roller bottle).
-
a.
-
2.
Aspirate EMEM-10 and wash each roller bottle with 30 ml of pre-warmed sterile PBS three times. Avoid keeping the cells in PBS and without media for extended time.
-
3.
Add 5 ml of virus inoculum (MOI = 5) to each bottle and gently roll to spread the inoculum over the cell monolayer.
-
4.
Immediately add an additional 15 ml of OptiMEM to each roller bottle and gently roll to spread the inoculum over the cell monolayer.
-
5.
Return the roller bottles to the 37°C incubator and allow the infection to proceed for 3 h at 0.5 rpm.
-
6.
Following the 3-h incubation, add an additional 120 ml of OptiMEM and flow each roller bottle with CO2 as described in Section 3.1.1.1, Step 6.
-
7.
Incubate the cells in 37°C incubator at 0.5 rpm for 40–50 h before harvesting the supernatant.
3.1.1.3 Harvesting Supernatant from Roller Bottles
-
1.
Empty the media from each roller bottle into 250-ml conical-bottom centrifuge tubes and centrifuge at 2000 g for 20 min.
-
2.
Collect the supernatant and add protease inhibitor according to manufacturer’s recommendation and Triton® X-100 to a final concentration of 0.5%.
-
3.
Filter the supernatant through a 0.22-?m PES (low protein-binding) filter unit (see Note 6).
-
4.
The supernatant can be stored at 4°C and ready for affinity purification. We recommend to process the supernatant as soonest as possible.
3.1.2 Affinity Purification
This is the first step of purification. Depending on the protein’s nature or the tag engineered to the protein, different approaches can be carried out for affinity purification. Here, we describe the use of (1) lentil lectin purification of the highly glycosylated HIV-1 gp140 and (2) S-protein agarose purification for the N-terminally S-peptide-tagged HeV/ NiV sG.
3.1.2.1 Gp140 Purification Through Lentil Lectin Sepharose Affinity Column
All the buffers should be degassed and sterile-filtered.
-
1.
Load 25-ml bed volume of Lentil Lectin SepharoseTM 4B into a XK 26 column and allow the beads to settle.
-
2.
Add 20% ethanol 3–5 cm above the sepharose surface. Push the column inlet adaptor downwards until tightly contacting with the ethanol leaving 2–3 cm of ethanol from the sepharose surface to the column inlet. Avoid formation of air bubbles. Tighten the connection via the O-ring using the knob on the top of the column inlet adaptor.
-
3.
Wash the beads with 3–5 bed volumes of equilibration buffer. Use a peristaltic pump to apply buffer through the input tubing.
-
4.
Apply the prepared supernatant from Section 3.1.1.3 to the column. Use the peristaltic pump to maintain a constant flow rate of 3–4 ml/min. Save the flow-through until the purified protein has been obtained. A small portion (1 ml) can be used for immunoprecipitation (Section 3.2.2.2).
-
5.
Wash the column with 300 ml of wash buffer I follow by 100 ml of wash buffer II to remove Triton.
-
6.
To elute the protein, apply elution buffer to the column, stop the flow after the first 10 ml of flow-through and incubate for 15 min. Collect 25 ml of the elution in a sterile 50-ml tube, stop the flow, and incubate for 15 min. Repeat elution and incubation for 3–5 times to collect several 25-ml elutions.
-
7.
Combine the eluate and concentrate it to less than 2 ml followed by buffer exchange into protein buffer (PBS) using Centriprep® molecular weight cut-off centrifugal filter units.
-
8.
Filter the purified protein through a 0.2-µm low protein-binding syringe filter membrane and store at 4°C. Pre-wet the membrane with protein buffer before filtering.
-
9.
To regenerate the column, apply 50 ml of high-pH buffer followed by 50 ml of low-pH buffer through the column. Repeat this step two times.
-
10.
After the last low-pH wash, apply 200-ml regeneration buffer through the column followed by 200 ml PBS.
-
11.
Finally, apply 200 ml 20% EtOH through the column and store the beads in 20% EtOH at 4°C. The beads can be reused for up to five times.
3.1.2.2 HeV/NiV sG Purification Through S-Protein Agarose Affinity Column
-
1.
Load 15 ml bed volume of S-protein agarose into a XK 26 column. Always maintain at least 2–3 cm of buffer above the surface of the agarose.
-
2.
Allow the column inlet adaptor to be 3–5 cm above the buffer level allowing input buffer to enter the column dropwise. Use a peristaltic pump to apply buffer through the inlet tubing. Solution passing through the column outlet depends on gravity.
-
3.
Wash the column with 6× bed volumes of PBS followed by 6× bed volumes of wash buffer II (refer to Section 2.5.2 for buffers).
-
4.
Apply the prepared supernatant from Section 3.1.1.3 to the column. Maintain a flow rate of 3–4 ml/min by adjusting the peristaltic pump and a stopcock connected at the outlet tubing of the column.
-
5.
Wash the column with 3X bed volumes of wash buffer I followed by 6× bed volumes of wash buffer II and subsequently 6× bed volumes of PBS.
-
6.
To elute the bound protein, stop the pump and allow the PBS to drain until it reaches the surface of the beads. Add 15 ml of elution buffer. Incubate the elution buffer with the beads for 10 min after collecting the first 8 ml of flow-through (this should still be the PBS).
-
7.
Collect 15 ml of the elution into a 50-ml sterile conical centrifuge tube containing 10 ml of neutralization buffer. Check the pH with pH strips and add more neutralization buffer if needed to obtain neutral pH.
-
8.
Repeat the elution and incubation 2–3 times.
-
9.
Wash the column with 6X bed volume of PBS follow by 6X bed volume of 20% ethanol. Store the beads in ethanol. The beads can be reused for up to five times.
-
10.
Combine all neutralized elution with the first 8 ml flow-through from Step 6.
-
11.
Concentrate the elution to less than 2 ml and buffer-exchange the elution into protein buffer (PBS) using Centriprep® molecular weight cut-off centrifugal filter units.
-
12.
Filter the purified protein through a 0.2-µm low protein-binding filter membrane and store at 4°C. Pre-wet the membrane with the same protein buffer before filtering.
3.2 Stable Cell-Line Expressing Soluble Recombinant HeV/NiV sF
3.2.1 Plasmid Construction Considerations
Several considerations may be required while constructing the expression plasmid: for example, Kozak translation initiation sequence and an ATG start codon for proper initiation of translation, promoter/enhancer and termination sequences, the expression plasmid employed, the type of antibiotic selection, the cell line employed, codon optimization of the viral RNA sequence, mutation and modifications on the amino acid sequence to enhance expression and secretion, the tag required in facilitating protein purification, etc. The sF construct described here has been optimized based on all the above concerns. Figure 2.1 show the diagram of the construct of our sF GCN.

Hendra virus and Nipah virus sF GCN expression plasmid. The Sac I flanking fragment of the CMV promoter from phCMV-1 vector was inserted into the Sac I site upstream to the multiple cloning site of pcDNA 3.1 Hygro (+). The transmembrane and cytoplasmic tail domain of F was replaced by the trimeric coil-coiled motif GCN (MKQIEDKIEEILSKIYHIENEIARIKKLIGE) and the 15-amino-acid S-peptide (KETAAAKFERQHMDS) was appended to the C-terminal of the construct. The sF sequence was codon-optimized and inserted into the promoter-modified pcDNA 3.1 Hygro (+) at the multiple cloning site via Xho I and Apa I sites. The Kozak translation initiation sequence and an ATG start codon for proper initiation of translation is shown with the start codon in uppercase.
The vector employed was a promoter-modified pcDNA 3.1 Hygro (+). The enhanced CMV promoter was imported from phCMV 1 vector that allows high expression level of the human and mouse codon-optimized sF. The Hygromycin selection marker allows the selection of transfected 293T cell which is resistant to the commonly used Geneticin antibiotic. Replacing the C-terminal transmembrane and cytoplasmic tail domain of F is the trimeric GCN4 motif (41, 42) that allows stabilization of the protein trimeric structure (26, 43) and hence enhances the expression and secretion of the protein. This is followed by the 15-amino-acid S-peptide that facilitates purification and immunodetection of the sF by S-protein agarose and anti-S-peptide antibody. A factor Xa cleavage site (IEGR) is also engineered upstream to the S-peptide tag allowing removal of the tag by enzymatic digestion. Standard cloning procedures were employed to construct the plasmid.
3.2.2 Transient Expression
Before generating a stable cell line, several optimizations may be required to obtain the best expression level. This can be investigated in transient expression using various cell lines to obtain the best result. We describe here the transient transfection of the sF construct in 293T cells using lipid-based transfection procedure. The expression level can be analyzed using immuno- or affinity precipitations from the cell lysate and supernatant followed by SDS-PAGE and Western blotting to access the secretion level. Figure 2.2 shows a representative result of transient transfection of various sF constructs to assess the secretion level by Western blotting. The addition of GCN tail and codon optimization by Geneart, Inc. greatly enhanced expression and secretion of the sF.

Transient expression of various sF constructs. 293T cells were transfected with various sF constructs in the promoter-modified pcDNA 3.1 Hygro (+) vector. Cell lysate and supernatant were precipitated with S-protein agarose. Western blot detection was performed using HRP-conjugated rabbit anti-S-peptide antibody. WT, wild-type sequence; CO, codon-optimized sequence; Lys, lysate; Sup, supernatant.
3.2.2.1 Transfection
-
1.
A day prior to transfection, seed 0.4 × 106/well in 3-ml DMEM-10 of 293T cell onto a six-well tissue culture plate pre-coated with 0.1% gelatin (see Note 7). Allow the cells to adhere and grow at 37°C, 5–8% CO2 overnight.
-
2.
Cells should be 70–90% confluent the next day. Prepare transfection mixture as follows: Add 6 µl of Fugene transfection reagent to 0.5 ml of serum-free DMEM. Gently mix the solution. Add 2 µg of plasmid DNA to the mixture and vortex briefly. Incubate the transfection mixture at room temperature for 30 min.
-
3.
Transfect one well with the vector DNA without the gene of interest as negative control.
-
4.
Aspirate the medium from the six-well plate and replace with 0.5 ml of fresh DMEM-10 per well. Add the transfection mixture to the cell at the side of the well without disturbing the monolayer. Gently rock the plate to mix.
-
5.
Incubate the cells at 37°C, 5–8% CO2 overnight or at least 3 h before adding an additional 0.5 ml of DMEM-10.
-
6.
Continue to incubate the cells at 37°C, 5–8% CO2 for 36–48 h to allow protein expression and secretion to take place.
-
7.
Proceed to Section 3.2.2.2 to harvest cells and supernatant for immuno- or affinity precipitations to assess the secretion level.
3.2.2.2 Cell Harvest and Affinity Precipitation
-
1.
Collect the supernatant from the six-well plate into a 1.5-ml centrifuge tube. Centrifuge at 18,000 g for 1 min to pellet cells.
-
2.
Transfer the supernatant to a fresh tube and add 30 µl of 50% slurry S-agarose. Rotate at room temperature for 1 h.
-
3.
Add one tablet of complete mini protease inhibitor to 10 ml of lysis buffer. Add 0.7 ml of this lysis buffer/well to the cells remaining on the plate. Allow lysis on ice for 5–10 min.
-
4.
Transfer the lysate to a 1.5-ml centrifuge tube and rotate at 4°C for 5–10 min to allow complete lysis.
-
5.
Centrifuge the lysate at 18,000 g for at least 5 min to pellet cell debris.
-
6.
Transfer the cleared lysate to a fresh tube and add 30 µl of 50% slurry S-agarose beads. Rotate at room temperature for 1 h.
-
7.
After the affinity binding, centrifuge the samples at 3000 g for 1 min to pellet the agarose beads. Aspirate the unbound lysate or supernatant without disturbing the agarose beads.
-
8.
Wash the agarose beads by adding 1 ml of lysis buffer and mix by inverting the tube several times.
-
9.
Repeat steps 7 and 8 at least twice.
-
10.
After aspirating the lysis buffer at the last washing step, add 50 µl of 2× SDS-PAGE sample loading buffer with 50 µl of 2-Mercaptoethanol per 1 ml of sample buffer to the agarose.
-
11.
Vortex and boil the samples for 5 min. Centrifuge the samples at 3000 g for 1 min. Use 25 µl for standard SDS-PAGE followed by Western blotting procedures.
-
12.
For immunodetection, use 1/20,000 dilution of HRP-conjugated Rabbit anti-S-tag antibody in blocking buffer for 1 h. We use the Supersignal West Pico Chemiluminescent Substrate as the HRP substrate. See Fig. 2.2 for Western blot result.
3.2.3 Cell Line Establishment
Once the best expression construct has been optimized with the best cell line, a stable cell line can be generated. In order to select a stable cell clone expressing the soluble glycoprotein, the minimum concentration of the appropriate selection antibiotic required to kill the non-transfected host cell line needs to be determined. Because natural resistance varies among cell lines, we recommend testing a range of antibiotic concentrations on 25% confluent cells. Choose the concentration that prevents growth within 2–3 days and kills all cells within 5–7 days. The procedures described here have been optimized for 293T cells.
3.2.3.1 Selection of Transfected Cell
-
1.
Repeat transfection and immunodetection as described in Sections 3.2.2.1 and 3.2.2.2. Make duplicate wells for each transfection, one for expression assessment and the other for cell line generation.
-
2.
At 36–48 h post-transfection, harvest the cell and supernatant of one well and take only the supernatant of the other well. Proceed to Section 3.2.2.2 to ensure expression and secretion.
-
3.
To the cells remaining on the plate for cell line generation, add 2 ml of PBS to wash the cells without disturbing the monolayer.
-
4.
Remove the PBS and add 0.5 ml of trypsin-EDTA. Allow trypsinization for 3–5 s before aspirating the trypsin.
-
5.
Use 1-ml fresh DMEM-10 medium to resuspend the cells and transfer the cells into a 25-cm2 tissue culture flask.
-
6.
Add 4 ml of DMEM-10 to the flask and allow cells to adhere at 37°C, 5–8% CO2 for several hours or overnight.
-
7.
Once cells have adhered, replace the medium with fresh DMEM-10 supplemented with 150 µg/ml Hygromycin B.
-
8.
Prepare another flask with the same selection medium for non-transfected cells to monitor the action of the antibiotic.
-
9.
Change and replace the selection medium when necessary by observing the color change of the medium. Changing the medium will also remove dead cells.
-
10.
Non-expressing cells should die within several days and expressing colonies will start to form. For 293T cells, this may take 7–10 days depending on the transfection efficiency. When all dead cells have been removed and colonies are growing well, the cells should be transferred into a 75-cm2 tissue culture flask to form monolayer.
-
11.
Aspirate medium from the 25-cm2 flask and add 2 ml of PBS. Gently rock the flask before aspirating the PBS.
-
12.
Then, add 1 ml of trypsin-EDTA and leave for 5 s. Aspirate the trypsin and use 2 ml of fresh DMEM-10 medium to resuspend the cells and transfer the cells into a 75-cm2 tissue culture flask.
-
13.
Add 18 ml of DMEM-10 supplemented with 150 µg/ml Hygromycin B to the flask. Allow the cells to grow and form monolayer in 37°C, 5–8% CO2.
-
14.
Once cells have reached confluence, split them at 1 in 5 dilution in selection medium. Repeat this for another two passages. Keep 1 ml of supernatant for affinity precipitation to monitor the secretion level.
-
15.
When cells reach confluence at the third passage, proceed to Section 3.3.3.2.
3.2.3.2 Single-Cell Isolation
-
1.
Aspirate the medium from the 75-cm2 flask and add 8-ml PBS. Gently rock the flask before aspirating the PBS.
-
2.
Then, add 4 ml of trypsin-EDTA and leave for 5 s. Aspirate the trypsin and use 10 ml of fresh DMEM-10 medium to resuspend the cells and transfer the cells into a 50-ml conical centrifuge tube.
-
3.
Pellet the cells at 800 g for 10 min and resuspend the pellet in 20-ml DMEM-10.
-
4.
Count the cells using trypan blue to exclude dead cells. Dilute 30 µl of the cell suspension to 8 cells/ml in 20-ml DMEM-10 medium. Freeze the remaining cells in liquid nitrogen.
-
5.
Prepare two 96-well flat-bottom tissue culture plates. Fill 200 µl/well of PBS to the outer 36 wells and 100 µl/well of DMEM-10 supplemented with 300 µg/ml Hygromycin B to the inner 60 wells.
-
6.
Add 100 µl/well of the diluted cells from step 4 to the 100-µl selection medium in the inner 60 wells of the prepared 96-well plates from step 5. Each well now should have less than one cell.
-
7.
Allow colonies to form in 37°C, 5–8% CO2 for 10–14 days. Observe each well under microscope to identify single-colony wells after 10 days.
-
8.
When the color of the medium starts to change, transfer all single colonies individually by pipetting up and down into 24-well plates with 1-ml selection medium added to the each well.
-
9.
Allow monolayer to form in 37°C, 5–8% CO2 for 4–6 days. When cells reach 70%–90% confluence, take 0.9 ml of supernatant from each well for immunoprecipitation and Western blotting (Section 3.2.2.2) to assess for the strongest secreting clone. Replace the selection medium in the 24-well plate. Once the best clone has been determined, it can then be expanded into a larger tissue culture flask.
-
10.
Aspirate the medium from the well of the selected clone, add 1 ml PBS. Add 0.3 ml trypsin-EDTA after removing the PBS and allow trypsinization for 3 s. Remove trypsin and resuspend cells in 1 ml DMEM-10.
-
11.
Transfer the resuspended cells into a 75-cm2 tissue culture flask added with 19 ml of selection medium. Allow the cells to grow to confluence in 37°C, 5–8% CO2; this may take up to 1 week.
-
12.
Repeat the cloning (Steps 1–11) at least once to ensure stable expression and to obtain the best possible secreting clone.
-
13.
Once a desirable clone has been obtained, expand the cell to larger flasks for freezing to keep as stock. Proceed to Section 3.2.4 for protein expression.
3.2.4 Cell Culture for Expression
Once a stable cell line has been established, it can then be grown into roller bottles for larger-scale protein production. The following protocol describes the large-scale production of secreted sF from 293T cells in roller bottles. For protein expression, addition of selection antibiotic is not necessary. Because of the weak adherent nature of 293T derivative cells, gelatin treatment of the roller bottle surface is necessary to avoid the formation of cell clumps which will decrease the protein secretion level.
-
1.
Add 50 ml of 0.1% sterile gelatin solution into each 1700-cm2 roller bottle. Rotate at 1 rpm at 37°C overnight or at least 1 h.
-
2.
Remove gelatin solution and wash once with 50 ml sterile PBS and once with DMEM medium.
-
3.
Prepare 150-cm2 flasks of sF 293T cell. One confluent flask of cells should be transferred into one 1700-cm2 roller bottle.
-
4.
Proceed to steps 2–7 in Section 3.1.1.1.
-
5.
When the cells have reached near-confluence in the roller bottles, collect the DMEM-10 medium. Depending on the expression level, protein can be harvested from the serum medium (see Note 8).
-
6.
Wash the cells with 50 ml PBS gently without disturbing the monolayer and remove the PBS.
-
7.
Add 120 ml of OptiMEM and pump with CO2 to each roller bottle as described in Section 3.1.1.1, Step 6.
-
8.
Allow protein secretion for 4–5 days or until the cell starts to detach. Harvest the supernatant as described in Section 3.1.1.3.
3.2.5 S-Affinity Purification
This step is essentially similar to Section 3.1.2.2 except replacing all buffers to the list in Section 2.5.3 and omitting the PBS wash in step 5. The protein concentration and yield can then be estimated. We use the Bradford method (44) to estimate the protein concentration at 595-nm OD. At this point, a small portion of the purified protein can be analyzed on SDS-PAGE follow by Coomassie staining (Fig. 2.3 ). Proceed to Section 3.3 for molecular weight purification.

SDS-PAGE followed by Coomassie stain analysis of S-protein agarose purified HeV and NiV sF GCN. Volumes of 0.5 µl and 1.0 µl of protein purified from DMEM supplemented by 10% calf serum (D-10) and reduced serum medium (OptiMEM) was analyzed on a NuPAGE® 4–12% Bis Tris Gel under denaturing and reduced condition.
3.3 Molecular Weight Purification by Gel Filtration
Following S-affinity purification, the concentrated purified protein can be analyzed on SDS-PAGE followed by Coomassie staining to assess the purity and yield. In most cases, there will be presence of small amount of contaminating proteins especially when purification is done from serum medium. Hence, further purification is needed.
Gel filtration chromatography separates molecules on the basis of size. It is often reserved for the final step of purification to achieve higher purity. This method can also be used to estimate the molecular masses of globular proteins in their native condition base on their migration through a chromatographic matrix packed in a column. Pre-packed columns with various matrix of different fractionation range are available commercially from GE healthcare.
The approximate molecular mass of a certain protein can be estimated from a calibrated curve obtained from a series of standard proteins with known molecular masses. Standard calibration protein kits can be obtained commercially from, for example, GE healthcare, Sigma, etc., with well-described calibrating protocols. Once the molecular weight of the different oligomeric species has been determined, a preparative chromatography can be carried out to isolate the desired oligomeric species.
Here, we describe the use of a calibrated Superdex 200 10/300 GL gel filtration column to estimate the approximate molecular mass of the different oligomeric species of the soluble viral glycoprotein using example of HeV and NiV sF GCN. We also describe the use of HiLoad 16/60 Superdex 200 prep grade gel filtration column XK 16 to separate the different oligomeric species of affinity-purified sF.
3.3.1 Analytic Chromatography
Before analyzing the soluble viral glycoprotein, calibrate the column with a series of protein standards. The Superdex 200 10/300 GL gel filtration column employed here has been calibrated with the molecular weights (29–669 kDa) calibration kit. As depicted in Fig. 2.4 , the calibration curve generated was used to estimate the approximate size of the HeV sF GCN. Well-described calibrating protocols are included in commercially available gel filtration molecular weights calibration kit. Below is a simple procedure used to determine the approximate size of the sF.
-
1.
The inlet tubing of the column should be connected to the appropriate buffer through the P-500 pump. Set the back pressure limit to 1.5 MPa.
-
2.
Connect the column outlet tubing to the optical unit through a flow cell of the UV detector.
-
3.
Wash the column with at least 3× column volume (3× 24 ml) with sterile deionized water with a flow rate of 24 ml/h.
-
4.
Equilibrate the column with the appropriate protein buffer from Section 2.5 with at least 3× of the column volume with the same flow rate.
-
5.
Set the sensitivity to 1.0 ABS for 3–5 mg of sF, sG, and gp140 and 0.2 ABS for 70 µl of the recommended concentration of the protein standards. Do not exceed 5 mg of protein and 500 µl of sample volume. Apply each protein standards as a separate run.
-
6.
Set the baseline for the UV detector and set the chart speed to 30 cm/h. Proceed to next step for sample application once a stable baseline is observed.
-
7.
Stop the pump and disconnect the column’s inlet tubing from the pump. Apply affinity-purified protein or protein standard to the column by using a 1-ml syringe and reconnect the input tubing to the pump. Use the same sample volume for all protein standards and the unknown sample.
-
8.
Start the flow and set the chart speed to 30 cm/h. Use a flow rate of 24 ml/h.
-
9.
Start fraction collector after the first 3 ml has been eluted. Collect fractions at 60 s intervals for at least 40 fractions.
-
10.
Stop collection when the last peak has been observed.
-
11.
Wash the column with at least 2× column volume of sterile deionized water followed by 3× column volume of 20% ethanol. Store column in 20% ethanol.
-
12.
Obtain the elution volume (V e) by measuring the volume of the eluent from the point of sample application to the center of the elution peak. Determine the void volume (V o), which is the V e of blue dextran (2 mg/ml).
-
13.
Calculate the K av values for each protein by using the formula K av = (V e – V o)/(V c – V o), where V c is the total column volume.
-
14.
Generate a calibration curve by plotting the calculated K av for each protein standard against the log of the known molecular weight.
-
15.
Calculate the estimated molecular weight of the soluble viral glycoprotein from the calibrated curve.
-
16.
Take 2–10 µl from each fraction of all peaks from the soluble glycoprotein for native gel analysis (Section 3.5) to observe the oligomeric species. Figure 2.5 shows a representative result of the elution profile HeV sF GCN and the Coomassie blue and Western blot of a native gel analysis of the fractions.

Size-exclusion chromatography analysis of HeV sF GCN. A panel of high-molecular-weight standards was separated on a Superdex 200 size 10/300 GL exclusion column, and a calibration curve was generated. Samples of S-affinity-purified HeV sF GCN were separated on the calibrated Superdex 200 column and fractionated. The Kav values of the major peaks were calculated, and the apparent molecular weight estimates were determined using the calibration curve.

Gel filtration analysis of S-affinity-purified HeV sF GCN. An amount of 4 mg of protein was analyzed on a Superdex 200 10/300 GL gel filtration column. Fractions of 400 µl were collected. (A) Gel filtration elution profile of the protein. The approximate molecular weight of the major peak was calculated from the calibrated curve to be 327 kDa. Some large aggregates of the protein elute near the void volume (Vo). (B) A volume of 10 µl of each of the fractions from gel filtration was analyzed on BN-PAGE stained with Coomassie. (C) A volume of 2 µl of each of the fractions from gel filtration was analyzed on BN-PAGE followed by Western blotting. The bands were detected using rabbit anti-S-peptide HRP-conjugated antibody.
3.3.2 Preparative Chromatography
-
1.
Repeat steps 1–11 of Section 3.3.1 using the of HiLoad 16/60 Superdex 200 prep grade gel filtration column. The back pressure limit should be set to 0.3 MPa. The column flow rate should be 60 ml/h. Set the sensitivity to 1.0 ABS for 8–10 mg of sF, sG, and gp140. Use 3 ml syringe for larger sample volume. The column volume is approximately 120 ml. The chart speed can be set to 15 cm/h.
-
2.
Proceed to step 16 of Section 3.3.1. Figure 2.6 shows a representative result of the elution profile HeV sF GCN and the Coomassie blue of a native gel analysis of selected peaks.
-
3.
Pool the selected peak fractions and concentrate it to 1 ml using Centriprep® molecular weight cut-off centrifugal filter units.
-
4.
Observe 2–10 µg of the purified protein again on a native gel. Figure 2.6C shows the purified HeV sF GCN trimer on a native gel.

Purification of trimeric HeV sF GCN using preparative gel filtration. An amount of 8.5 mg of S-affinity-purified HeV sF GCN were separated on a HiLoad 16/60 Superdex 200 prep grade gel filtration column. Fractions of 1 ml were collected. (A) Gel filtration elution profile of the protein. (B) Native gel analysis of the selected peak fractions. A volume of 10 µl of each selected fraction were analyzed on BN-PAGE and observed by Coomassie stain. Fractions 26–28 were pooled and concentrated to obtain pure trimer. (C) An amount of 5 µg of purified trimer were then analyzed on BN-PAGE.
3.4 Sucrose Gradient Ultracentrifugation
Native oligomeric forms of the purified soluble glycoprotein can also be analyzed by sucrose gradient centrifugation. As shown in Fig. 2.7 , two species (large aggregate and trimer) of HeV sF GCN were observed in the sucrose gradient. This is consistent with the gel filtration profile as shown in Fig. 2.5 .
-
1.
Underlay 6 ml of 5% sucrose with 6 ml of 20% sucrose in polyallomer 14 mm × 95 mm tubes.
-
2.
Generate a linear sucrose gradient with a gradient master at an angle of 81.5° for 1 min 55 s at a speed of 15 rpm.
-
3.
Overlay 200 µg of soluble viral glycoprotein on top of the gradients.
-
4.
Centrifuge at 40,000 rpm (approximately 284,000 g) for 20 h at 4°C using an SW40 rotor.
-
5.
Collect fractions of approximately 800 µl from the bottom of the gradient using a Beckman fraction recovery system and automated fraction collector.
-
6.
Take 15 µl from each fraction for native gel analysis to observe the oligomeric species. Figure 2.7 shows the Western blot of a native gel analysis of the fractions from 200 µg HeV sF GCN.
Fig. 2.7. 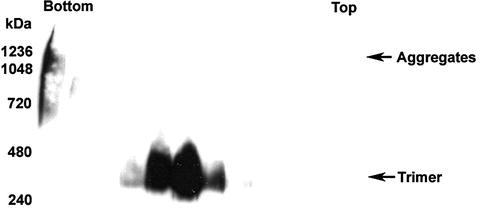
Sucrose gradient analysis of oligomeric forms of HeV sF GCN. An amount of 200 µg of S-affinity-purified protein was layered onto continuous (5–20%) sucrose gradients and fractioned. A volume of 15 µl of each fraction was analyzed on BN-PAGE followed by Western blotting. The bands were detected using rabbit anti-S-peptide HRP-conjugated antibody. The bottom and top of the gradient fractions are indicated.

3.5 Native Gel Analysis by BN-PAGE System
Native gel analysis allows the visualization of the soluble glycoprotein on polyacrylamide gel electrophoresis in its native form. This allows the estimation of the size of the native oligomeric species. It is most helpful in analyzing the fractions separated from gel filtration and sucrose gradient.
The BN gel system is based on the originally described system by Schagger and von Jagow (45). Many modifications have been made based on the original method, for example, in observing the native forms of HIV-1 Env (30, 46, 47). We find that the commercially available BN-PAGE system from Invitrogen Corp. is most suitable for our soluble glycoprotein analysis. It requires little or no optimization and is included with well-described step-by-step protocol. In this BN-PAGE system, the Coomassie® G-250 binds to proteins and confers a negative charge while maintaining the proteins in their native state. Below are the procedures we use to analyze our soluble glycoproteins using the BN-PAGE system followed by Western blotting.
3.5.1 Electrophoresis
-
1.
Prepare running buffers as follows:
-
a.
Dilute 20X NativePAGE™ running buffer to 1X solution in deionized H2O.
-
b.
Dilute 20X NativePAGE™ Cathode Buffer Additive to 1X solution for Coomassie detection and to 0.1X solution for Western blotting in 1X NativePAGE™ running buffer.
-
a.
-
2.
Prepare samples as follows
For 20 µl, add 5 µl of 4X NativePAGE™ sample buffer and 0.5 µl of NativePAGE™ 5% G-250 to 14.5 µl of protein sample.
-
3.
Set up the electrophoresis system using the NativePAGE™ Novex® 3–12% Bis-Tris Gels 1.0 mm, 15 well and the XCell SureLock™ Mini-Cell as described by the manufacturer.
-
4.
Rinse the wells with 1X running buffer and load the samples before adding the cathode buffer.
-
5.
Load 5 µl of NativeMark™ Unstained Protein Standard for Coomassie stain and 7 µl added with 1 µl of 5% G-250 for Western blotting.
-
6.
Perform the electrophoresis at 150 V for 2 h at room temperature.
3.5.2 Coomassie Stain
The G-250 in the cathode buffer stains the proteins during electrophoresis. Therefore, further staining is not required. However, this depends on the sensitivity required. More sensitive staining protocol is available by the manufacturer.
-
1.
After electrophoresis, place gel in container with fixing solution enough to cover the gel. Microwave the gel to near-boiling and cool for 1 min on an orbital shaker.
-
2.
Repeat Step 1 for 2–3 times and allow the gel to cool at room temperature for 15 min on an orbital shaker with gentle shaking.
-
3.
Replace the fix solution with destain solution and microwave to near-boiling for 2–3 times as described in Steps 1 and 2.
-
4.
Shake the gel on an orbital shaker at room temperature until the desired background is obtained. See Figures 2.5B, 2.6B, and 2.6C for examples of Coomassie-stained gels.
3.5.3 Western Blotting
-
1.
Use only 0.1X cathode buffer in 1X NativePAGE™ running buffer for Western blot.
-
2.
Prepare 1X transfer buffer from 20X NuPAGE® Transfer Buffer in 20% methanol.
-
3.
Place PVDF membrane in methanol for several seconds and rinse with deionized water.
-
4.
Equilibrate PVDF membrane and filter paper in 1X transfer buffer for at least 5 min.
-
5.
After electrophoresis, place gel in transfer buffer with 0.1% SDS. Let this incubate while setting up the transfer apparatus using the XCell II™ Blot Module as described by the manufacturer.
-
6.
Transfer at 30 V constant for 1 h at room temperature.
-
7.
After transfer, incubate the membrane in 20 ml 8% acetic acid for 15 min. Rinse with deionized water and air dry the membrane
-
8.
Re-wet the membrane with methanol, and then rinse it with deionized water.
-
9.
Place membrane in blocking buffer and incubate at room temperature for at least 1 h or 4°C for overnight.
-
10.
Proceed to immunodetection. To detect S-peptide-tagged sF or sG, use 1/20,000 dilution of Rabbit anti-S-tag antibody, HRP-conjugated in blocking buffer.
-
11.
Wash membrane for 5 min in wash buffer and repeat wash for three times. Add Supersignal West Pico Chemiluminescent Substrate and incubate for 5 min at room temperature. Proceed to film exposure. See Figures 2.5C and 2.7 for results.
-
12.
After developing the film, place the membrane in Coomassie blue stain to stain the protein ladder. Wash excess stain with methanol briefly and mark the ladder bands on the membrane.
4 Notes
-
1.
Tris is an inhibitor for DTSSP cross-linker. If DTSSP will be used with the protein in downstream analysis, HEPES can be used as an alternate neutralization buffer.
-
2.
Arginine prevents protein aggregation and is used in protein refolding. It also helps in elution of proteins and antibody. Addition of arginine is optional, but we find that it is necessary for sF purification.
-
3.
Cells should be very dense. Estimation of the following days of incubation for cell growth is based on each T-150 flask containing greater than 3.0 × 107 cells. To obtain the maximum yield of pure protein, the cells must be very dense when seeded into roller bottles and when infected with purified recombinant vaccinia virus.
-
4.
Ensure cell clumps are broken up by repeatedly pipetting the suspension up and down. An even cell suspension is necessary for an even monolayer in the roller bottles. Formation of cell clumps in the roller bottles will decrease the protein yield.
-
5.
A 10-ml pipette without a cotton stopper is connected to a tube that connects to a 10% CO2, 90% air tank with a pressure controller and indicator; a 0.2-µm filter membrane should be connected to the tubing.
-
6.
Pre-wet the filter unit membrane with PBS, 0.1% Triton® X-100 or the appropriate washing buffer before filtering the supernatant. This can be done by filtering 50–100 mlL of wash buffer prior to sample filtering. We find that this procedure will decrease protein lost during filtration. This step is essential for purification of sF.
-
7.
Gelatin treatment is optional. However for 293T and derivatives cells, gelatin-treated surface allows the cells to adhere better during the medium-changing procedures of transfection and hence yields better transfection results.
Coat 2 ml per well of sterile 0.1% Gelatin onto a six-well tissue culture plate. Incubate the plate at 37°C for 1 h or 4°C overnight. Rinse twice with sterile PBS before using.
-
8.
Harvest the DMEM medium and follow procedures in Section 3.1.1.3. Use this medium for S-column purification as described in Section 3.2.5. There will still be contaminating serum protein at this stage. Apply the neutralized elution to another round of S-column purification in smaller scale. Scale down all buffers for 2 ml bed volume of S-Agarose. Use 10 ml Poly-Prep Chromatography Columns (Bio-Rad Laboratories, Inc). The flow rate is entirely depending on gravity. Depending on the expected protein yield, several columns may be required. It is estimated that 1 ml bed volume of S-Agarose is able to capture at least 1 mg sF. We use three columns for sF GCN and more than 6 mg can be recovered from supernatant of ten roller bottles.
References
Graham, B. S. and Crowe, J., J.E. (2007) Immunization against viral diseases. In Fields Virology (Knipe, D. M. and Howley, P. M., eds.), pp. 489–538, Lippincott Williams & Wilkins, Philadelphia.
Casadevall, A., Dadachova, E., and Pirofski, L. A. (2004) Passive antibody therapy for infectious diseases. Nat. Rev. Microbiol. 2, 695–703.
Dimitrov, D. S. (2004) Virus entry: molecular mechanisms and biomedical applications. Nat. Rev. Microbiol. 2, 109–122.
Zeitlin, L., Cone, R. A., Moench, T. R. and Whaley, K. J. (2000) Preventing infectious disease with passive immunization. Microbes Infect. 2, 701–708.
Wu, H., Pfarr, D. S., Johnson, S., Brewah, Y. A., Woods, R. M., Patel, N. K., White, W. I., Young, J. F., and Kiener, P. A. (2007) Development of Motavizumab, an ultra-potent antibody for the prevention of respiratory syncytial virus infection in the upper and lower respiratory tract. J. Mol. Biol. 368, 652–665.
Burton, D. R. (2002) Antibodies, viruses and vaccines. Nat. Rev. Immunol. 2, 706–713.
Casadevall, A. (1999) Passive antibody therapies: progress and continuing challenges. Clin. Immunol. 93, 5–15.
Rader, C. and Barbas, C. F., 3rd (1997) Phage display of combinatorial antibody libraries. Curr. Opin. Biotechnol. 8, 503–508.
Hayden, M. S., Gilliland, L. K., and Ledbetter, J. A. (1997) Antibody engineering. Curr. Opin. Immunol. 9, 201–212.
Hudson, P. J. and Souriau, C. (2001) Recombinant antibodies for cancer diagnosis and therapy. Expert Opin. Biol. Ther. 1, 845–855.
Choudhry, V., Zhang, M. Y., Sidorov, I. A., Louis, J. M., Harris, I., Dimitrov, A. S., Bouma, P., Cham, F., Choudhary, A., Rybak, S. M., Fouts, T., Montefiori, D. C., Broder, C. C., Quinnan, G. V., Jr., and Dimitrov, D. S. (2007) Cross-reactive HIV-1 neutralizing monoclonal antibodies selected by screening of an immune human phage library against an envelope glycoprotein (gp140) isolated from a patient (R2) with broadly HIV-1 neutralizing antibodies. Virology 363, 79–90.
Zhang, M. Y., Xiao, X., Sidorov, I. A., Choudhry, V., Cham, F., Zhang, P. F., Bouma, P., Zwick, M., Choudhary, A., Montefiori, D. C., Broder, C. C., Burton, D. R., Quinnan, G. V., Jr., and Dimitrov, D. S. (2004) Identification and characterization of a new cross-reactive human immunodeficiency virus type 1-neutralizing human monoclonal antibody. J. Virol. 78, 9233–9242.
Zhang, M. Y., Shu, Y., Sidorov, I., and Dimitrov, D. S. (2004) Identification of a novel CD4i human monoclonal antibody Fab that neutralizes HIV-1 primary isolates from different clades. Antiviral Res. 61, 161–164.
Zhang, M. Y., Shu, Y., Phogat, S., Xiao, X., Cham, F., Bouma, P., Choudhary, A., Feng, Y. R., Sanz, I., Rybak, S., Broder, C. C., Quinnan, G. V., Evans, T., and Dimitrov, D. S. (2003) Broadly cross-reactive HIV neutralizing human monoclonal antibody Fab selected by sequential antigen panning of a phage display library. J. Immunol. Methods 283, 17–25.
Zhang, M. Y., Choudhry, V., Sidorov, I. A., Tenev, V., Vu, B. K., Choudhary, A., Lu, H., Stiegler, G. M., Katinger, H. W., Jiang, S., Broder, C. C. and Dimitrov, D. S. (2006) Selection of a novel gp41-specific HIV-1 neutralizing human antibody by competitive antigen panning. J. Immunol. Methods 317, 21–30.
Moulard, M., Phogat, S. K., Shu, Y., Labrijn, A. F., Xiao, X., Binley, J. M., Zhang, M. Y., Sidorov, I. A., Broder, C. C., Robinson, J., Parren, P. W., Burton, D. R., and Dimitrov, D. S. (2002) Broadly cross-reactive HIV-1-neutralizing human monoclonal Fab selected for binding to gp120-CD4-CCR5 complexes. Proc. Natl. Acad. Sci. USA 99, 6913–6918.
Zhu, Z., Dimitrov, A. S., Bossart, K. N., Crameri, G., Bishop, K. A., Choudhry, V., Mungall, B. A., Feng, Y. R., Choudhary, A., Zhang, M. Y., Feng, Y., Wang, L. F., Xiao, X., Eaton, B. T., Broder, C. C. and Dimitrov, D. S. (2006) Potent neutralization of Hendra and Nipah viruses by human monoclonal antibodies. J. Virol. 80, 891–899.
Zhu, Z., Chakraborti, S., He, Y., Roberts, A., Sheahan, T., Xiao, X., Hensley, L. E., Prabakaran, P., Rockx, B., Sidorov, I. A., Corti, D., Vogel, L., Feng, Y., Kim, J. O., Wang, L. F., Baric, R., Lanzavecchia, A., Curtis, K. M., Nabel, G. J., Subbarao, K., Jiang, S., and Dimitrov, D. S. (2007) Potent cross-reactive neutralization of SARS coronavirus isolates by human monoclonal antibodies. Proc. Natl. Acad. Sci. USA 104, 12123–12128.
Quinnan, G. V. (1997) Immunization against viral diseases. In Antiviral Agents and Human Viral Disease (Galasso, G., Whitley, R. and Merigan, T. C., eds.), pp. 791–834, Raven Press, New York.
Doms, R. W., Lamb, R., Rose, J. K. and Helenius, A. (1993) Folding and assembly of viral membrane proteins. Virology 193, 545–562.
Wiley, D. C. and Skehel, J. J. (1987) The structure and function of the hemagglutinin membrane glycoprotein of influenza virus. Ann. Rev. Biochem. 56, 365–394.
Broder, C. C., Earl, P. L., Long, D., Abedon, S. T., Moss, B., and Doms, R. W. (1994) Antigenic implications of human immunodeficiency virus type 1 envelope quaternary structure: oligomer-specific and -sensitive monoclonal antibodies. Proc. Natl. Acad. Sci. USA 91, 11699–11703.
Garber, D. A., Silvestri, G., and Feinberg, M. B. (2004) Prospects for an AIDS vaccine: three big questions, no easy answers. Lancet Infect. Dis. 4, 397–413.
Bossart, K. N., Crameri, G., Dimitrov, A. S., Mungall, B. A., Feng, Y. R., Patch, J. R., Choudhary, A., Wang, L. F., Eaton, B. T. and Broder, C. C. (2005) Receptor binding, fusion inhibition, and induction of cross-reactive neutralizing antibodies by a soluble G glycoprotein of hendra virus. J. Virol. 79, 6690–6702.
Earl, P. L., Broder, C. C., Long, D., Lee, S. A., Peterson, J., Chakrabarti, S., Doms, R. W., and Moss, B. (1994) Native oligomeric human immunodeficiency virus type 1 envelope glycoprotein elicits diverse monoclonal antibody reactivities. J. Virol. 68, 3015–3026.
Yin, H. S., Wen, X., Paterson, R. G., Lamb, R. A., and Jardetzky, T. S. (2006) Structure of the parainfluenza virus 5 F protein in its metastable, prefusion conformation. Nature 439, 38–44.
Yin, H. S., Paterson, R. G., Wen, X., Lamb, R. A., and Jardetzky, T. S. (2005) Structure of the uncleaved ectodomain of the paramyxovirus (hPIV3) fusion protein. Proc. Natl. Acad. Sci. USA.
Yang, X., Farzan, M., Wyatt, R., and Sodroski, J. (2000) Characterization of stable, soluble trimers containing complete ectodomains of human immunodeficiency virus type 1 envelope glycoproteins. J. Virol. 74, 5716–5725.
Stamatatos, L., Lim, M. and Cheng-Mayer, C. (2000) Generation and structural analysis of soluble oligomeric gp140 envelope proteins derived from neutralization-resistant and neutralization-susceptible primary HIV type 1 isolates [In Process Citation]. AIDS Res. Hum. Retroviruses 16, 981–994.
Schulke, N., Vesanen, M. S., Sanders, R. W., Zhu, P., Lu, M., Anselma, D. J., Villa, A. R., Parren, P. W., Binley, J. M., Roux, K. H., Maddon, P. J., Moore, J. P. and Olson, W. C. (2002) Oligomeric and conformational properties of a proteolytically mature, disulfide-stabilized human immunodeficiency virus type 1 gp140 envelope glycoprotein. J. Virol. 76, 7760–7776.
Malvoisin, E. and Wild, F. (1994) Characterization of a secreted form of measles virus haemagglutinin expressed from a vaccinia virus recombinant. J. Gen. Virol. 75, 3603–3609.
Heinz, F. X., Mandl, C. W., Holzmann, H., Kunz, C., Harris, B. A., Rey, F., and Harrison, S. C. (1991) The flavivirus envelope protein E: isolation of a soluble form from tick-borne encephalitis virus and its crystallization. J. Virol. 65, 5579–5583.
Mirza, A. M., Sheehan, J. P., Hardy, L. W., Glickman, R. L., and Iorio, R. M. (1993) Structure and function of a membrane anchor-less form of the hemagglutinin-neuraminidase glycoprotein of Newcastle disease virus. J. Biol. Chem. 268, 21425–21431.
Seto, N. O. and Gillam, S. (1994) Expression and characterization of a soluble rubella virus E1 envelope protein. J. Med. Virol. 44, 192–199.
Wang, Z. M., Tong, L. L., Grant, D. and Cihlar, T. (2001) Expression and characterization of soluble human parainfluenza virus type 1 hemagglutinin-neuraminidase glycoprotein. J. Virol. Methods 98, 53–61.
Gaudin, Y., Moreira, S., Benejean, J., Blondel, D., Flamand, A., and Tuffereau, C. (1999) Soluble ectodomain of rabies virus glycoprotein expressed in eukaryotic cells folds in a monomeric conformation that is antigenically distinct from the native state of the complete, membrane-anchored glycoprotein. J. Gen. Virol. 80, 1647–1656.
Zhang, P. F., Cham, F., Dong, M., Choudhary, A., Bouma, P., Zhang, Z., Shao, Y., Feng, Y. R., Wang, L., Mathy, N., Voss, G., Broder, C. C., and Quinnan, G. V., Jr. (2007) Extensively cross-reactive anti-HIV-1 neutralizing antibodies induced by gp140 immunization. Proc. Natl. Acad. Sci. USA May 31, [Epub ahead of print].
Quinnan, G. V., Jr., Yu, X. F., Lewis, M. G., Zhang, P. F., Sutter, G., Silvera, P., Dong, M., Choudhary, A., Sarkis, P. T., Bouma, P., Zhang, Z., Montefiori, D. C., Vancott, T. C., and Broder, C. C. (2005) Protection of rhesus monkeys against infection with minimally pathogenic simian-human immunodeficiency virus: correlations with neutralizing antibodies and cytotoxic T cells. J. Virol. 79, 3358–3369.
Mungall, B. A., Middleton, D., Crameri, G., Bingham, J., Halpin, K., Russell, G., Green, D., McEachern, J., Pritchard, L. I., Eaton, B. T., Wang, L. F., Bossart, K. N., and Broder, C. C. (2006) Feline model of acute Nipah virus infection and protection with a soluble glycoprotein-based subunit vaccine. J. Virol. 80, 12293–12302.
Broder, C. C. and Earl, P. L. (1999) Recombinant vaccinia viruses. Design, generation, and isolation. Mol. Biotechnol. 13, 223–245.
Harbury, P. B., Zhang, T., Kim, P. S. and Alber, T. (1993) A switch between two-, three-, and four-stranded coiled coils in GCN4 leucine zipper mutants. Science 262, 1401–1407.
Harbury, P. B., Kim, P. S. and Alber, T. (1994) Crystal structure of an isoleucine-zipper trimer. Nature 371, 80–83.
Yang, X., Lee, J., Mahony, E. M., Kwong, P. D., Wyatt, R., and Sodroski, J. (2002) Highly stable trimers formed by human immunodeficiency virus type 1 envelope glycoproteins fused with the trimeric motif of T4 bacteriophage fibritin. J. Virol. 76, 4634–4642.
Bradford, M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254.
Schagger, H. and von Jagow, G. (1991) Blue native electrophoresis for isolation of membrane protein complexes in enzymatically active form. Anal. Biochem. 199, 220–231.
Beddows, S., Schulke, N., Kirschner, M., Barnes, K., Franti, M., Michael, E., Ketas, T., Sanders, R. W., Maddon, P. J., Olson, W. C., and Moore, J. P. (2005) Evaluating the immunogenicity of a disulfide-stabilized, cleaved, trimeric form of the envelope glycoprotein complex of human immunodeficiency virus type 1. J. Virol. 79, 8812–8827.
Binley, J. M., Sanders, R. W., Master, A., Cayanan, C. S., Wiley, C. L., Schiffner, L., Travis, B., Kuhmann, S., Burton, D. R., Hu, S. L., Olson, W. C., and Moore, J. P. (2002) Enhancing the proteolytic maturation of human immunodeficiency virus type 1 envelope glycoproteins. J. Virol. 76, 2606–2616.
Acknowledgment
This work was supported in part by Middle Atlantic Regional Center of Excellence (MARCE) for Biodefense and Emerging Infectious Disease Research, NIH AI057168 and AI054715 grants to C.C.B.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2009 Humana Press, a part of Springer Science+Business Media, LLC
About this protocol
Cite this protocol
Chan, YP., Yan, L., Feng, YR., Broder, C.C. (2009). Preparation of Recombinant Viral Glycoproteins for Novel and Therapeutic Antibody Discovery. In: Dimitrov, A. (eds) Therapeutic Antibodies. Methods in Molecular Biology™, vol 525. Humana Press. https://doi.org/10.1007/978-1-59745-554-1_2
Download citation
DOI: https://doi.org/10.1007/978-1-59745-554-1_2
Published:
Publisher Name: Humana Press
Print ISBN: 978-1-934115-92-3
Online ISBN: 978-1-59745-554-1
eBook Packages: Springer Protocols