
Abstract
Coronaviruses most often infect the respiratory or intestinal tract. Transmissible gastroenteritis virus (TGEV), a group 1 coronavirus, infects the porcine small intestine. Piglets up to the age of 3 weeks die from diarrhea caused by the viral gastroenteritis unless they are protected by antibodies. In addition to the cellular receptor, porcine aminopeptidase N, the TGEV spike protein binds to sialic acid residues. We have shown that the sialic acid binding activity mediates the binding of TGEV to a mucin-like glycoprotein present in porcine brush border membranes. This was shown by performing a virus overlay binding assay with proteins obtained from brush border membranes by lectin precipitation. Because of the reactivity with specific lectins we assume that the recognized glycoprotein has the characteristics of a mucin.
You have full access to this open access chapter, Download protocol PDF
1 Introduction
Transmissible gastroenteritis virus (TGEV) infects susceptible cells by binding to the cellular receptor porcine aminopeptidase N (pAPN) (1). It has been shown that the spike (S) protein of TGEV has, in addition to its pAPN binding site, a sialic acid binding activity, which is located at the N-terminal region of the protein (2). We have shown that binding to sialoglycoproteins in cell culture and in porcine brush border membrane preparations may be a pathogenicity factor affecting the enteropathogenicity of TGEV (3,4). Virus mutants that were not able to bind to sialic acids did not infect piglets (5,6). Thus, binding to sialoglycoproteins or mucins in the gastrointestinal tract may help the virus to reach the cellular receptor pAPN. The sialoglycoproteins serve as first attachment factors.
To further characterize the sugar moieties of the sialoglycoproteins that are bound by TGEV, we isolated brush border membranes from piglets and analyzed them by performing lectin precipitation. Lectin precipitates were further used for a virus overlay binding assay. The lectins used in this study were wheat germ agglutinin (WGA), which specifically binds N-acetylglucosamine and sialic acids; peanut agglutinin (PNA), which specifically recognizes galactose-β(1-3)-N-acetylgalactosamine, a disaccharide present in O-glycosylated proteins; and jacalin, which binds to the same sugars as PNA. The advantage of jacalin is that in contrast to PNA it binds to galactose-β(1-3)-N-acetylgalactosamine without prior neuraminidase treatment. In this way the specific lectin precipitation can be well combined with the virus binding to sialic acids. A high-molecular-mass protein that is recognized by the sialic acid binding activity of TGEV was also readily recognized by WGA and jacalin, suggesting that this sialoglycoprotein is highly O-glycosylated. Therefore we designated it mucin-like glycoprotein (MGP).
2 Materials
2.1 Brush Border Membrane Preparation
-
1.
Preparation buffer A: 100 mM mannit, 10 mM HEPES, Tris pH 7.4/4 °C.
-
2.
Preparation buffer B: 100 mM mannit, 10 mM HEPES, 0.1 mM MgSO4X 7 H2O, Tris pH7.4/4 °C.
-
3.
Vesicle buffer: 100 mM mannit, 100 mM KCl, 10 mM HEPES, Tris pH 7.4/4 °C.
2.2 Virus Purification
-
1.
Dulbecco`s modified Eagle medium (DMEM) with 4500 mg/liter glucose (Gibco/ BRL) supplemented with 10% fetal calf serum (Gibco/BRL).
-
2.
PBS Dulbecco`s without calcium and magnesium (Gibco/BRL).
-
3.
Sucrose (Roth) freshly dissolved in PBS in concentrations of 20, 50, and 60% w/w.
-
4.
Neuraminidase from Vibrio cholerae (1 U/ml, DADE Behring), store at 4 °C (see Note 1).
2.3 Lectin Precipitation
-
1.
Neuraminidase from Vibrio cholerae (1 U/ml, DADE Behring). Store at 4 °C.
-
2.
Sodium acetate buffer solution (50 mM), pH 5.5 with sodium chloride (154 mM) and calcium chloride (9 mM) (see Note 2). Store at 4 °C.
-
3.
Protease inhibitors: leupeptin (Roche), Pefa Block®SC (Roche).
-
4.
TBS pH 7.5: Tris 0.05 M, sodium chloride 0.15 M. Adjust pH with HCl.
-
5.
NP40 lysis buffer: Sodium deoxycholate 0.5 %,TBS pH 7.5, Nonidet® P40 (Roche). Add 1 mM MgCl2, CaCl2, and MnCl2.
-
6.
Wheat germ agglutinin agarose (Sigma), peanut agglutinin agarose (Vector laboratories), jacalin agarose (Sigma). Store at 4 °C.
-
7.
SDS sample buffer (2X): 100 mM Tris-HCl, pH 6.8, 4% (w/v) sodium dodecyl sulfate (SDS), 20% glycerol, 0.02% bromophenol blue. Store in aliquots at –20 °C.
2.4 SDS-Gel Electrophoresis (SDS-PAGE)
-
1.
Separating buffer: 1.5 M Tris-HCl, pH 8.8. Store at 4 °C.
-
2.
Stacking buffer: 1 M Tris-HCl, pH 6.8. Store at 4 °C.
-
3.
30% acrylamide/bis solution. Neurotoxic! Store at 4 °C.
-
4.
10% (w/v) SDS prepared in water. Store at room temperature.
-
5.
10% (w/v) ammonium persulfate (APS) prepared in water. Freeze in aliquots at –20 °C. Aliquot in use must be stored at 4 °C.
-
6.
N,N,N′,N′-tetramethyl-ethylendiamine (TEMED, Bio-Rad). Toxic! Store at 4 °C (see Note 3).
-
7.
10X running buffer: 10 g SDS, 30 g Tris, 144 g glycin filled up to 1 liter with water. Store at room temperature (see Note 4).
-
8.
Prestained molecular weight markers: Rainbow marker (Amersham) or PageRuler™ Prestained Protein Ladder (Fermentas). Store at –20 °C.
2.5 Western Blotting
-
1.
Buffer anode I: 1 M Tris 300 ml, ethanol 200 ml, H2O 500 ml. Adjust to pH 9 with HCl (see Note 5).
-
2.
Buffer anode II: 1 M Tris 25 ml, ethanol 200 ml, H2O 770 ml. Adjust to pH 7.4 with HCl.
-
3.
Buffer cathode: aminocapronic acid 5.25 mg, 1 M Tris 25 ml, ethanol 200 ml, H2O 770 ml. Adjust to pH 9 with HCl.
-
4.
Nitrocellulose membrane (Schleicher & Schuell/Whatman).
-
5.
Filter paper (Schleicher & Schuell/Whatman, 2043A, ref.no.: 10381185).
-
6.
Blocking buffer: 0.5 % blocking reagent (Roche, article number: 1096176) in PBS-0.05% T. Stir at 80 °C for 30 min. After solution store at 4 °C.
2.6 Virus Overlay Binding Assay
-
1.
Phospate buffered saline with/without Tween (PBS, PBS-0.1% T, PBS-0.05% T): Prepare a 10X stock of PBS with NaCl 80 g, KCl 2 g, Na2HPO4 11.5 g, KH2PO4 2 g. The pH has to be 7.5. Dilute 100 ml with 900 ml water for use. Add 0.1 % or 0.05 % Tween, depending on the buffer needed, and stir well (see Note 6).
-
2.
Virus and antibody dilution buffer: PBS-0.05% T.
-
3.
Purified virus (TGEV-NA).
-
4.
First antibody: anti-TGEV-S protein monoclonal antibody 6A.C3 from L. Enjuanes (7).
-
5.
Second antibody: Anti-mouse Ig, biotinylated species-specific whole antibody from sheep (Amersham/GE Healthcare).
-
6.
Streptavidin biotinylated horseradish peroxidase complex (Amersham/GE Healthcare).
-
7.
Enhanced chemoluminescent substrates: BM Chemoluminescence Blotting Substrate (Roche) or Super Signal® (Pierce).
3 Methods
3.1 Brush Border Membrane Preparation
The brush border membrane preparation is performed according to the protocol of Schröder et al. (8).
-
1.
The mucosa of fresh jejunum from sacrificed suckling piglets is abraded from the serosa and frozen in liquid nitrogen. The samples are stored at –80 °C until use.
-
2.
All preparation steps are performed on ice at 4 °C.
-
3.
The mucosa is weighed and 150 ml of preparation buffer A is added.
-
4.
After thawing on ice the mucosa is mixed in a regular household mixer on level 4 twice for 1 min each. After each mixing step, the suspension is filled in a tumbler and left on ice for 2 min. Then the foam is sucked off.
-
5.
The volume of the homogenate is determined in a measuring cylinder.
-
6.
The basolateral brush border membranes are precipitated by adding 1 M MgCl2 in an amount of 1/100 of the homogenate volume. The MgCl2 solution is added drop by drop under continuous stirring on ice followed by further stirring on ice for 15 min.
-
7.
The complete homogenate is then centrifuged for 18 min, 3687 ×g at 4 °C (see Note 7). After centrifugation the supernatant is poured into fresh centrifuge beakers.
-
8.
In a second centrifugation step for 40 min, 16,000 ×g at 4 °C, the apical brush border membrane fraction is pelleted (see Note 8). The pellet is floated in 35 ml of preparation buffer B; i.e., the buffer is rinsed over the pellet until the sediment is detached from the bottom. The floating pieces of the pellet are then homogenized ten times using a potter.
-
9.
To further purify the preparation, the MgCl2 precipitation is repeated followed by a centrifugation for 10 min, 3293 ×g at 4 °C (see Note 9).
-
10.
The supernatant is then centrifuged for 30 min, 26,430 ×g at 4 °C (see Note 10). The resulting pellet is resuspended in 35 ml vesicle buffer and homogenized ten times with the potter.
-
11.
After a final centrifugation step for 40 min, 31,300 ×g at 4 °C, the brush border membranes are homogenized in 1 to 3 ml vesicle buffer by using a 1-ml syringe and a 0.45 ×23-gauge needle (see Note 11).
-
12.
The protein content of the brush border membrane preparation is estimated by using the Bradford assay (BIORAD). To make all protein binding sites available, surface-active saponine (1%) is added in a ratio of 1:2 to all samples. After incubation for 20 min at room temperature the Bradford staining reagent is added.
3.2 Virus Purification
-
1.
ST (swine testicular) cell monolayers in 145-mm cell culture dishes are used for TGEV infection at a multiplicity of infection of 1. For infection, the cells are washed three times with sterile PBS (with Ca2+ and Mg2+) and inoculated with a volume of 300 μl virus suspension per dish. With this small virus volume the plates are incubated at 37 °C for 1 h with careful shaking (see Note 12). Then the inoculum is removed and 20 ml DMEM is added to each plate followed by an incubation at 37 °C for about 24 h (see Note 13).
-
2.
As soon as the CPE is observed, the cell culture supernatant is harvested and centrifuged at 1900 ×g for 10 min, 4 °C, to remove cell debris. The resulting supernatant is subjected to ultracentrifugation at 120,000 ×g for 1 h, 4 °C, to pellet the virus particles (see Note 14). The resulting pellets are resuspended in PBS (total volume 400 to 500 μl).
-
3.
To make all sialic acid binding sites on the S protein available, a neuraminidase treatment of the concentrated virus particles is performed. Neuraminidase from Vibrio cholerae (1 U/ml, DADE Behring) is used in a final concentration of 50 mU/ml. The sample is then incubated at 37 °C for 30 min and cooled on ice.
-
4.
For purification of the virions, a continuous sucrose gradient (20–50% w/w in PBS) with a 60% sucrose cushion is prepared. The sucrose solutions are stirred at 50 °–60 °C and stored at room temperature until use. For the gradient centrifugation, SW 50.1 tubes, ultraclear (Beckman), are used (see Note 15). Per tube, 0.5 ml of the 60% sucrose cushion is applied overlaid by a gradient of 20 and 50% sucrose (using a gradient mixer and a pump). 2.5 ml of the 20% and 2 ml of the 50% solution are put into the gradient mixer. The gradient has a height corresponding to a 4-ml volume. The virus suspension is then carefully put on the top of the gradient using a 100-μl pipette. For purification, the gradient is ultracentrifuged at 150,000 ×g for 3 h, 4 °C (see Note 16).
-
5.
After centrifugation, the visible virus band is carefully harvested using a 1-ml syringe and a 0.9 ×40-gauge needle (see Note 17). After dilution with PBS to a total volume of 4.5 ml, the virions are pelleted by ultracentrifugation (SW 50.1) at 150,000 ×g for 1 h, 4 °C (see Note 16). The viral particles are resuspended in a small volume of PBS (depending on the pellet size in 100 to 300 μl). The sample is stored in aliquots at –20 °C. The purity of the preparation is tested in an SDS-PAGE with Coomassie staining of the proteins in the gel. The protein concentration is determined photometrically at 280 nm (see Note 18).
3.3 Lectin Precipitation
-
1.
Brush border membrane vesicles (1 mg/ml) are incubated at a ratio of 1:2 with neuraminidase from Vibrio cholerae (1 U/ml, DADE Behring) for 1 h at 37 °C (see Note 1). Control samples are treated with sodium acetate buffer in the same manner (in all samples: addition of protease inhibitors: leupeptin and pefa-block).
-
2.
Each sample is diluted in 1 ml NP40 lysis buffer containing 1 mM MgCl2, 1 mM CaCl2, and 1 mM MnCl2 (without protease inhibitors). For membrane lysis, the samples are incubated on ice for 15 min. Membrane debris is centrifuged at 16,000 ×g, 30 min, 4 °C (see Note 19).
-
3.
330 μl of each supernatant is put on 50 μl lectin agarose (wheat germ agglutinin, jacalin, peanut agglutinin) and the tubes are shaken overnight at 4 °C.
-
4.
The next day the agarose samples are washed three times with NP40 lysis buffer (after centrifugation at 16,000 ×g, 3 min, 4 °C). Bound glycoproteins are eluted with 50 μl twofold SDS sample buffer for 10 min at 96 °C. After a final centrifugation step at 16,000 ×g, 5 min, 4 °C, the supernatants containing the lectin-specific glycoproteins are used for SDS-gel electrophoresis.
3.4 SDS-Gel Electrophoresis (SDS-PAGE)
-
1.
These instructions are intended for the use of minigels (70 ×80 ×0.75 mm). Glass plates are cleaned with a rinsable detergent and rinsed with distilled water. After drying they are rinsed with 70% ethanol.
-
2.
Prepare an 8% gel, 0.75 mm thick by mixing 4.6 ml distilled water with 2.5 ml Tris-HCl (1.5 M, pH 8.8), 2.7 ml acrylamide/bis solution (30%), 100 μl SDS (10% in water), 100 μl ammonium persulfate solution (10% in water), and 10 μl TEMED. Pour the gel (∼4 ml), leaving about 1 cm of space for the stacking gel and overlay with isopropanol. The gel should polymerize within 30 min.
-
3.
Pour off the isopropanol. Prepare the stacking gel by mixing 3.4 ml distilled water with 0.63 ml Tris-HCl (1 M, pH 6.8), 0.83 ml acrylamide/bis solution (30%), 50 μl SDS (10% in water), 50 μl ammonium persulfate solution (10% in water), and 10 μl TEMED. Pour about 0.5–1 ml of this mixture over the polymerized separa- ting gel and insert the comb. The stacking gel should polymerize within 30 min.
-
4.
Prepare the running buffer by diluting 100 ml of the 10X running buffer with 900 ml of water. Use a measuring cylinder. Invert the bottle to mix.
-
5.
Once the stacking gel has solidified, assemble the gel running unit. Carefully remove the comb and wash the wells with running buffer by using, e.g., a Hamilton Microlitre™ syringe.
-
6.
Add the running buffer to the lower and upper chambers of the gel unit and load 20 μl of each sample in a well. Load one well with 2.5–5 μl prestained molecular weight marker filled up to 20 μl with 2X SDS sample buffer.
-
7.
Connect the gel unit to a power supply. Run the gel at 80 V until the dye front of the sample buffer is a sharp line and leaves the stacking gel. It is then possible to run the gel at 150 V until the dye front runs off the gel. The whole running time of the gel is about 1.5 h.
3.5 Western Blotting
-
1.
The samples separated by SDS-PAGE are transferred to a nitrocellulose membrane electrophoretically. For this purpose a semidry blotting system with two graphite electrodes is used. Six sheets of filter paper have to be soaked in anode I buffer, three filters in anode II buffer, and nine sheets of paper have to be soaked in the cathode buffer. The nitrocellulose membrane is wetted with pure water before use. Prior to use, all sheets and the membrane are cut to a size of 6 ×8 cm (just a bit larger than the size of the separating gel).
-
2.
The power supply is switched off and the gel unit is disassembled. The stacking gel is cut and discarded. One edge of the separating gel is cut to have a better orientation for blotting.
-
3.
The anode I papers are taken with a pinzette and excess buffer and air bubbles are eliminated from the papers by stripping with clean gloves. The papers are then placed onto the anode and the anode II papers placed onto the anode I papers like a sandwich.
-
4.
The nitrocellulose membrane is carefully taken out of the pure water and put on the papers. A little water is put on the upper side of the membrane, the better to get the gel from the glass slides by creating capillary forces. The free side of the gel is laid on the membrane, and the remaining glass slide on the other side is lifted carefully. A spacer is used to loosen the gel from the slide. After removing possible air bubbles, the cathode papers are put on the gel as described above for the anode papers.
-
5.
With a glass pipette residual air bubbles are stripped out of the sandwich and the cathode is put on the top. About 2 kg are put on the blotting apparatus (e.g., two bottles with 1 liter each) and the apparatus is connected to the power supply. The running conditions are 0.8 mA/cm2. For one membrane with 6 ×8 cm use 38 mA and let the apparatus run for 1 h.
-
6.
Once the transfer is completed, switch off the power supply and carefully lift the upper electrode. Normally some of the papers stick to this upper side. Search for the position of the gel and control if the prestained marker is visible on the nitrocellulose membrane. Discard the gel and mark the bands of the marker with a ballpoint pen because they sometimes get lost during later incubation steps.
-
7.
The nitrocellulose is than incubated in 20 ml blocking buffer overnight at 4 °C on a rocking platform (see Note 20).
3.6 Virus Overlay Binding Assay
-
1.
For the virus overlay binding assay the blocking buffer is discarded and after a short rinse with PBS-0.1% T, the membrane is washed with PBS-0.1% T three times for 10 min each on a rocking platform at room temperature (see Note 21).
-
2.
The membrane is incubated for 1 h at 4 °C with purified TGEV (around 8 μg protein/blot) diluted in 500 μl of PBS-0.05% T covered by Parafilm (see Note22).
-
3.
After another washing period (three times for 10 min each, at room temperature with PBS-0.1% T) the first antibody (mab antiS 6A.C3 from L. Enjuanes) is diluted 1:200 in PBS-0.05% T. Again 500 μl of the dilution are dropped onto the membrane covered by Parafilm (see Note 23). Incubation was at 4 °C for 1 h.
-
4.
The membrane is then again washed three times for 10 min each at room temperature with PBS-0.1% T.
-
5.
The secondary antibody (anti-mouse Ig, biotinylated species-specific whole antibody from sheep, Amersham/GE Healthcare) is prepared 1:1000 in 10 ml PBS-0.05% T and the membrane is incubated in this solution for 1 h at 4 °C under shaking.
-
6.
After the next washing period (three times for 10 min each, at room temperature with PBS-0.1% T), the membrane is incubated with streptavidin biotinylated horseradish peroxidase complex (Amersham/GE Healthcare) diluted 1:5000 in 10 ml PBS-0.05% T for 1 h at 4 °C on a rocking platform.
-
7.
The peroxidase complex dilution is discarded followed by three washings for 10 min each with PBS-0.1% T at room temperature.
-
8.
The bound peroxidase is detected by chemoluminescence using the BM Chemoluminescence Blotting Substrate (Roche) or Super Signal® (Pierce) for the detection of weaker signals (see Note 24). The chemoluminescence signal can be visualized with an X-ray film or by using the Gel Documentation System ChemiDoc™ from BioRad (Fig. 1).
Fig. 1. 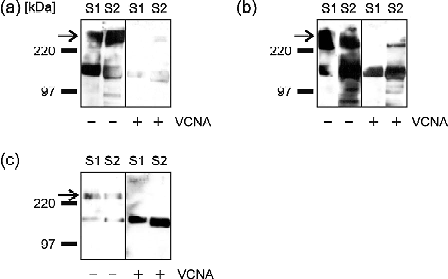
Binding of TGEV to lectin-precipitated brush border membrane proteins from two different suckling piglets (S1, S2). Brush border membranes were treated either with neuraminidase from Vibrio cholerae (+VCNA) or mock-treated (–VCNA) and then precipitated with: (a) wheat germ agglutinin (WGA), (b) jacalin, or (c) peanut agglutinin (PNA) agarose. A virus overlay binding assay was performed after blotting of the brush border membrane proteins. Two main bands were recognized by the virus after lectin precipitation. The lower band of about 150 kDa presents the cellular receptor porcine aminopeptidase N (pAPN). The PNA-precipitation (c, right side) shows clearly that binding to pAPN is not sialic-acid-dependent. TGEV binding to the high-molecular-mass band (arrow) is clearly sialic-acid-dependent, as after VCNA treatment binding is eliminated or at least strongly reduced. The strong precipitation of MGP by WGA gives a hint that it is highly sialylated or has a strong content of N-acetylglucosamine. Jacalin binds to galactose-β(1-3)-N-acetylgalactosamine, a disaccharide present in O-glycosylated proteins. The strong MGP band (b) demonstrates that this protein is highly O-glycosylated. Therefore we designated it mucin-like glycoprotein (MGP). For the same reason it is precipitated by PNA (c), but PNA precipitation is not as good without neuraminidase treatment. After neuraminidase treatment (+VCNA) the virus is not able to bind to MGP because of the missing sialic acids.

4 Notes
-
1.
Neuraminidase from Vibrio cholerae is no longer available from DADE Behring. Alternatively, one can use, e.g., type V neuraminidase from Clostridium perfringens (Sigma).
-
2.
The pH of sodium acetate buffer solution has to be adjusted with acetic acid.
-
3.
A small aliquot of the TEMED bottle is stored at 4 °C for current use. In this way the original bottle is not opened too frequently, retaining the quality of the TEMED.
-
4.
Never adjust the pH of the running buffer, e.g. with HCl. This will disturb the running conditions during electrophoresis. The buffer has to be used without pH adjustment.
-
5.
It is better for the environment to use ethanol for buffers instead of methanol, which has to be discarded separately under specific conditions.
-
6.
The pH of the PBS solution has to be at 7.5 without any adjustment. It just has to be checked.
-
7.
For example, 3687 ×g corresponds to 6000 rpm in a GSA rotor.
-
8.
16,000 ×g corresponds to 12,500 rpm in a GSA rotor.
-
9.
3293 ×g corresponds to 6000 rpm in an SS34 rotor.
-
10.
26,430 ×g corresponds to 17,000 rpm in an SS34 rotor.
-
11.
31,300 ×g corresponds to 18,500 rpm in an SS34 rotor. For the homogenization of the pellet, first carefully rinse the buffer over the pellet to loosen parts of it. Homogenization has to be performed on ice because it takes some time until the whole pellet is suspended.
-
12.
A small virus volume is important to get an efficient infection. In this way the particles have the best contact with the cell surface.
-
13.
For virus production at least five or, better, ten dishes with a diameter of 145 mm (Greiner) have to be used.
-
14.
120,000 ×g corresponds to 28,000 rpm in an SW32 Beckman rotor. The centrifuge tubes have to be filled with 35 ml supernatant each. Do not use a smaller volume because then the tubes will be damaged by the centrifugation forces. Use ultraclear SW28 centrifuge tubes to see the white virus pellets better. Yellow pellets could be a hint that cellular proteins are still present in the supernatant. For this reason the first centrifugation step for getting rid of the cell debris is very important.
-
15.
It is important to use ultraclear tubes. In this way the “white“ band formed by purified virus particles can be seen at the respective density after centrifugation.
-
16.
150,000 ×g corresponds to 35,000 rpm in an SW55 Beckman rotor.
-
17.
The “white“ virus particle band can best be seen with the help of scattering light produced by an external light source. The needle has to be carefully directed vertically into the tube from the top up to the height of the band. The harvested volume should not exceed 1 ml per tube.
-
18.
Different dilutions of BSA or transferrin are used as a standard. For the first measurement, the virus preparation is diluted 1:100. Sometimes higher concentrations have to be measured to get an extinction reading in a representative range. With the standard staining methods that are used by most of the assays for the determination of protein concentration (e.g., BCA assay) it is not possible to detect the TGEV proteins, so the photometric measurement at 280 nm is used.
-
19.
16,000 ×g corresponds to 14,000 rpm in an Eppendorf microcentrifuge.
-
20.
For blocking, different buffers as well as nonfat dry milk were tried. A lot of them, especially the nonfat dry milk, lead to high backgrounds in the virus overlay binding assay. With blocking reagent from Roche, high backgrounds owing to the wrong blocking substance can be avoided.
-
21.
Vigorous washing of the membrane is necessary as in a virus overlay binding assay high backgrounds could be a problem.
-
22.
For reduction of a possible background, virus and antibodies are diluted in PBS-0.05% T instead of just PBS.
-
23.
By using Parafilm one needs just 500 μl virus or antibody dilution for one membrane. In this way the virus preparation lasts for a longer time.
-
24.
Usually Super Signal® (Pierce) is used, as the signals after the virus overlay assay may be quite weak. If the signals are stronger, it is better to use BM Chemoluminescence Blotting Substrate (Roche) as the background is lower with this substrate.
References
Delmas, B., Gelfi, J., L' Haridon, R., Vogel, L. K., Sjostrom, H., Noren, O., and Laude, H. (1992) Nature 357, 417–420.
Schultze, B., Krempl, C., Ballesteros, M. L., Shaw, L., Schauer, R., Enjuanes, L., and Herrler, G. (1996) J. Virol. 70, 5634–5637.
Schwegmann-Wessels, C., Zimmer, G., Schroder, B., Breves, G., and Herrler, G. (2003) J. Virol. 77, 11846–11848.
Schwegmann-Wessels, C., Zimmer, G., Laude, H., Enjuanes, L., and Herrler, G. (2002) J. Virol. 76, 6037–6043.
Bernard, S., and Laude, H. (1995) J. Gen. Virol. 76(Pt 9), 2235–2241.
Krempl, C., Schultze, B., Laude, H., and Herrler, G. (1997) J. Virol. 71, 3285–3287.
Gebauer, F., Posthumus, W. P., Correa, I., Sune, C., Smerdou, C., Sanchez, C. M., Lenstra, J. A., Meloen, R. H., and Enjuanes, L. (1991) Virology 183, 225–238.
Schröder, B., Hattenhauer, O., and Breves, G. (1998) Endocrinology 139, 1500–1507.
Acknowledgments
The authors would like to thank Professor Gerhard Breves and Professor Bernd Schröder for the brush border membrane preparation protocol and Marion Burmester for technical assistance. This work was supported by Deutsche Forschungsgemeinschaft (SFB 280).
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2008 Humana Press, a part of Springer Science+Business Media, LLC
About this protocol
Cite this protocol
Schwegmann-Wessels, C., Herrler, G. (2008). Identification of Sugar Residues Involved in the Binding of TGEV to Porcine Brush Border Membranes. In: Cavanagh, D. (eds) SARS- and Other Coronaviruses. Methods in Molecular Biology, vol 454. Humana Press, Totowa, NJ. https://doi.org/10.1007/978-1-59745-181-9_22
Download citation
DOI: https://doi.org/10.1007/978-1-59745-181-9_22
Published:
Publisher Name: Humana Press, Totowa, NJ
Print ISBN: 978-1-58829-867-6
Online ISBN: 978-1-59745-181-9
eBook Packages: Springer Protocols