
Abstract
Quantification of the number of infectious viruses in a sample is a basic virological technique. In this chapter we provide a detailed description of three techniques to estimate the number of viable infectious avian coronaviruses in a sample. All three techniques are serial dilution assays, better known as titrations.
You have full access to this open access chapter, Download protocol PDF
1 Introduction
Technological advances in particle analysis have made it possible to quantify the number of virus particles in a sample with increasing accuracy. Techniques such as specialized flow cytometry [1], dynamic light scattering [2], quantitative capillary electrophoresis [3], and fluorescence correlation spectroscopy [4] can determine the number of particles in a sample within hours. The choice of technique depends on the sort of virus and the matrix in which it is suspended. All aforementioned techniques differentiate particles on the basis of physical properties such as size or antibody affinity. As a consequence particle analysis cannot differentiate infectious from noninfectious virus particles.
There is only one technique available that can reliably quantify the number of infectious particles in a sample. This technique, developed many decades ago [5], exploits the fact that virus can propagate in biological systems such as embryonated eggs or cell cultures. Propagation of a virus is generally accompanied by changes in cell morphology (referred to as cytopathic effect or CPE), which can be visualized using a microscope, or even by eye. Some viruses do not induce CPE, in which case an antibody based assay (immunofluorescence or ELISA) is needed to determine presence or absence of virus. During a titration assay, tissue cultures or embryonated eggs are incubated with tenfold serial dilutions of a virus containing sample and several days later the cytopathic effect is scored. From these scores, the virus titer is calculated using the methods described by Spearman and Kaerber [6, 7] or Reed and Muench [8]. The virus titer is defined as the reciprocal of the dilution at which 50 % of the inoculated embryos or tissue cultures show CPE. In this chapter we use the method of Spearman and Kaerber to calculate the titer, as this calculation can cope with unequal group sizes. Unequal group sizes frequently arise when eggs are lost to aspecific death of the embryo or bacterial infection.
Coronaviruses in general have a narrow host range and many clinical isolates only replicate in primary cells. Replication of most field isolates of infectious bronchitis coronavirus is restricted to embryonated eggs or tracheal organ culture. Most isolates, however, can be adapted to propagation in primary chicken kidney (CK) cells. Adaptation typically requires several passages and selects for viral subpopulations and can induce mutations [9]. Passaging of IBV in either embryonated eggs or primary cell cultures leads to attenuation of the virus in vivo [10–12]. The most striking example is the IBV Beaudette strain, which has been passaged hundreds of times in eggs and primary chicken kidney cells [13, 14]. Although IBV Beaudette propagates very well on eggs, CK cells and even in Vero cells, the virus is highly attenuated in vivo and vaccination using Beaudette provides little protection against infection with pathogenic strains of IBV [15].
For quantification of IBV field isolates, embryonated chicken eggs are the most suitable substrate. A protocol on titration of IBV on embryonated chicken eggs is provided in the first part of this protocol. IBV strains which have been adapted to grow in cultures of primary chicken cells can be titrated on these cells using either the TCID50 method or plaque titration. Protocols for both methods are provided in this chapter. Plaque-forming unit (PFU) titration yields more accurate and reproducible results then the TCID50 method, it is however more labor intensive. Both methods are presented in this chapter.
2 Materials
2.1 Titration of Avian Infectious Bronchitis Virus in Fertilized Eggs by EID50
-
1.
Fertilized specific pathogen free (SPF) eggs, 9–11-day-old (see Note 1 ).
-
2.
Diluent: 2.5 % w/v tryptose, 1,000 U/ml penicillin, 1,000 μg/ml streptomycin.
-
3.
Disinfectant: 70 % alcohol in water.
-
4.
Egg shell drill or punch.
-
5.
Sterile 1 ml syringes.
-
6.
Needles, preferably 25 G; 16 mm.
-
7.
Hobby glue or melted wax to seal the inoculation site.
-
8.
Egg candling light.
-
9.
Egg incubator with rocker.
2.2 Tissue Culture Infective Dose (TCID50) Titration
-
1.
96-well plates containing 80–100 % confluent CK cells.
-
2.
Titration medium: 1:1 mix of medium M199: Ham’s F-10 nutrient mixture supplemented with 0.5 % fetal calf serum (FCS), 0.1 % w/v tryptose phosphate broth, 0.1 % w/v sodium bicarbonate, 0.1 % w/v HEPES, 100 U/ml penicillin, 100 μg/ml streptomycin.
-
3.
Multistepper pipette (Socorex or equivalent).
-
4.
Inverted microscope.
-
5.
Positive control sample with known titer.
2.3 Plaque-Forming Unit Titration
-
1.
Six-well plates containing 60–90 % confluent CK cells.
-
2.
1× BES cell culture medium: EMEM, 10 % w/v tryptose phosphate broth, 0.2 % w/v bovine serum albumin (BSA), 20 mM N,N bis(2-hydroxethyl)-2-aminoethanesulfonic acid (BES), 0.4 % w/v sodium bicarbonate, 2 mM l-glutamine, 250 U/ml nystatin, 100 U/ml penicillin, 100 μg/ml streptomycin.
-
3.
2× BES cell culture medium: 2× EMEM, 20 % w/v tryptose phosphate broth, 0.4 % w/v BSA, 40 mM BES, 0.8 % w/v sodium bicarbonate, 4 mM l-glutamine, 500 U/ml nystatin, 200 U/ml penicillin, 200 μg/ml streptomycin.
-
4.
Sterile phosphate buffered saline (PBS).
-
5.
2 % w/v agarose in water (autoclaved).
-
6.
10 % w/v formaldehyde in PBS.
-
7.
0.1 % w/v crystal violet in water.
-
8.
Microwave.
-
9.
Water bath.
-
10.
Small spatula.
3 Methods
3.1 Titration of Avian Infectious Bronchitis Virus in Fertilized Eggs by EID50
-
1.
Candle the eggs using the candling light and draw a line on the shell marking the edge of the air sac. Draw an X approximately 5 mm above this line, which marks the inoculation site.
-
2.
Assign ten eggs per dilution and select those dilutions (at least three) that include the 50 % end point of the sample.
-
3.
Prepare tenfold serial dilutions in diluent.
-
4.
Disinfect the eggs by spraying them with disinfectant.
-
5.
After the eggs have dried drill or pierce a hole in the egg shell at the marked inoculation site.
-
6.
Inoculate ten eggs per dilution each with 0.2 ml volume via the allantoic cavity by holding the syringe and needle vertically and by inserting the needle approximately 16 mm into the egg (Fig. 1a, b).
Fig. 1 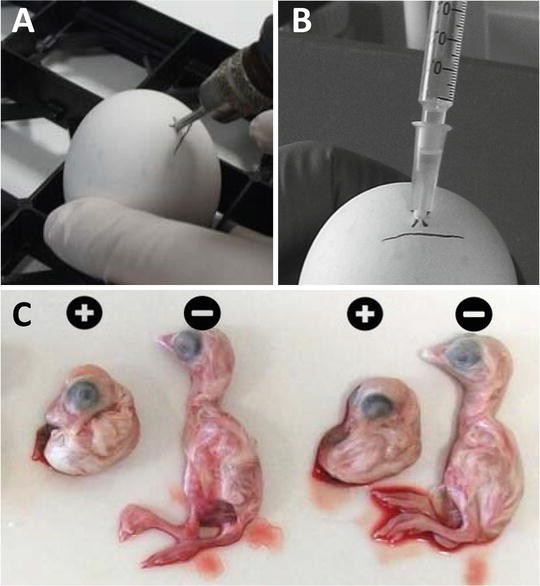
Egg inoculation into the allantoic cavity. (a) Drill or pierce a hole in the egg 5 mm above the edge of the air sac. (b) Inoculate each egg with a 0.2 ml volume by inserting the needle approximately 16 mm into the egg. (c) Typical IBV induced malformations 2 days post infection with strain 4/91. Plus indicates embryos infected with IBV, minus indicates non-infected embryos
-
7.
After inoculation, the hole in each egg is sealed with hobby glue or melted wax.
-
8.
Incubate the eggs in an egg incubator with rocker at 37.8 °C and 60–65 % humidity.
-
9.
Candle the eggs after 24 h of incubation. Embryo mortality occurring up till 24 h post inoculation is considered nonspecific and therefore these eggs are discarded.
-
10.
Incubate the eggs for 6 more days in an egg incubator with rocker at 37.8 °C and 60–65 % humidity. Candle the eggs at the end of the incubation period to identify embryos that have died. Subsequently, macroscopically evaluate all surviving embryos for the presence of lesions characteristic for IBV infection (stunting and curling; Fig. 1c). Embryos that have died and embryos that exhibit lesions characteristic for IBV infection are considered positive (see Note 2 ).
-
11.
Any embryo with IBV specific CPE is regarded positive. Virus titers in the original sample, expressed as 10log EID50/ml are calculated using the method described by Spearman and Kaerber [6, 7], using the following formula:\( Titre=\left({x}_0-\frac{d}{2}+d{\displaystyle \sum}\frac{r}{n}\right) \)
In which:
x 0: logarithm of the inverse value of the lowest dilution at which all embryos are positive.
d: logarithm of the dilution factor (d=1 when using tenfold serial dilutions).
n: number of eggs used per dilution.
r: number of positive eggs at that dilution.
-
12.
Example of calculation of virus titer
Using the result of the titration depicted in Fig. 2, the virus titer is calculated as follows:
Fig. 2 
Schematic result of an egg titration. Each circle represents one egg and crosses indicate aspecific death. Plus signs indicate embryos with IBV specific malformations
$$ \mathrm{Titre}\left({}{}^{10}log{\mathrm{EID}}_{50}/0.2\;\mathrm{ml}\right)=\left(4-\frac{1}{2}+1\times \left(\frac{5}{10}+\frac{1}{9}\right)\right)= $$$$ 5.1{}{}^{10}\kern0.1em \log EI{D}_{50}\kern0.1em /\kern0.1em 0.2 ml={10}^{5.1}\kern0.1em /\kern0.1em 0.2={5.8}^{10} \log EI{D}_{50}\kern0.1em /\kern0.1em ml $$


3.2 Tissue Culture Infective Dose (TCID50) Titration
-
1.
CK cells are seeded in 96-well plates at 7.5 × 104 cells/well, 1 or 2 days before the titration. One plate is needed per sample. At the time of titration, the monolayer should be nearly confluent.
-
2.
Prepare tenfold serial dilutions of the samples in titration medium (see Note 3 ).
-
3.
Empty the medium from the 96-well plate containing CK cells into a waste container and gently tap the plate dry on a stack of tissues.
-
4.
Fill the wells of column 1 and 12 of the 96-well plate with 100 μl/well titration medium. These are the negative control wells (Fig. 3a).
Fig. 3 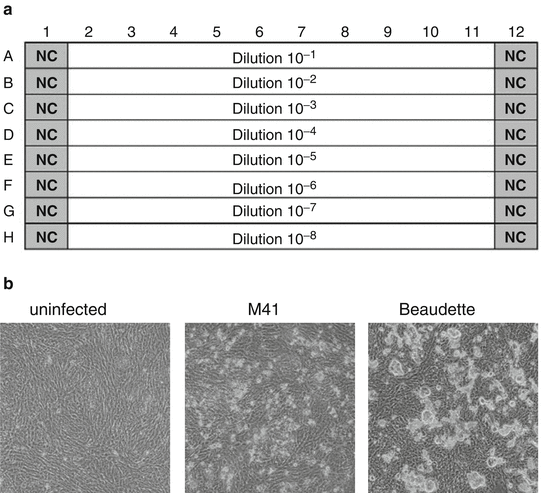
Layout of sample dilutions in the 96-well titration plate. NC negative control (a). Typical CPE of IBV 2 days post infection of CK cells with M41 or Beaudette (b)
-
5.
Starting with the highest dilution, dispense 100 μl/well in row H using the multistepper pipette. Proceed with filling the descending dilutions in rows G till A.
-
6.
Incubate the 96-well plates for 3–4 days at 37 °C and 5 % CO2.
-
7.
After 3–4 days incubation, score all wells for IBV specific CPE using a microscope. Although the CPE may vary per IBV strain, it is generally characterized by clusters of rounded cells on top of the monolayer. At low dilutions the monolayer may be partly destroyed, exemplified by the IBV Beaudette strain (Fig. 3b).
-
8.
Titers are calculated using the method described by Spearman and Kaerber (Subheading 3.1, step 11) and are expressed in 10log (TCID50)/ml.

3.3 Plaque-Forming Unit Titration
3.3.1 Infection of Cells
-
1.
CK cells are seeded into six-well plates 3 days prior to titration. When performing the titration, the monolayer should 60–90 % confluent.
-
2.
Prepare tenfold serial dilutions of virus in 1× BES.
-
3.
Remove media from cells and wash once with sterile PBS.
-
4.
Remove PBS from the cells and add 500 μl of diluted virus to each well. Duplicate wells should be inoculated for each dilution.
-
5.
Incubate cells at 37 °C for 1 h to allow virus attachment.
-
6.
Melt 2 % agar in a microwave and then transfer to a 42 °C water bath. Allow the agar to equilibrate in temperature.
-
7.
Mix the partially cooled agar with 2× BES pre-warmed to 37 °C to generate 1× BES + 1 % agar. Keep at 42 °C until needed to prevent premature setting (see Note 4 ).
-
8.
Remove virus inoculum and overlay cells with 2.5 ml of the 1× BES/agar mix.
-
9.
Leave cells at room temperature for approximately 5 min until agar has solidified.
-
10.
Incubate at 37 °C and 5 % CO2 for 3 days for plaques to develop.
3.3.2 Staining Cells and Determining Titer
-
1.
Overlay agar with 1 ml per well 10 % formaldehyde in PBS.
-
2.
Incubate at room temperature for 1 h.
-
3.
Remove formaldehyde and ensure disposal according to local regulations.
-
4.
Using a small spatula, flick off the agar from the cells (see Note 5 ).
-
5.
Wash cells by shaking the plate upside down in a sink containing water.
-
6.
Add 0.5 ml 0.1 % crystal violet to each well.
-
7.
Incubate at room temperature for 10 min.
-
8.
Remove crystal violet and dispose of according to local regulations.
-
9.
Wash plate by shaking upside down in a sink of water.
-
10.
Pat plate dry and leave upside down at room temperature to fully dry.
-
11.
Plaques should be clearly visible as holes in the monolayer (Fig. 4). Count the number of plaques per well at the dilution with clearly defined, individual (not overlapping) plaques (typically 10–50 plaques/well). Ensure duplicate wells are counted and an average taken.
Fig. 4 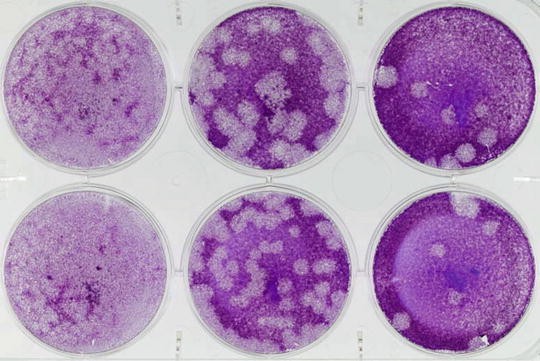
Example plaque assay plate. CK cells were infected with tenfold serial dilutions of IBV in duplicate and incubated at 37 °C for 3 days. Cells were then fixed and stained with 0.1 % crystal violet
-
12.
Determine titer using the following equation:
$$ \mathrm{Titre}\left(\mathrm{P}\mathrm{F}\mathrm{U}/\mathrm{ml}\right)=\frac{\mathrm{averagenumberofplaques}}{\mathrm{dilutionfactor}\times \mathrm{inoculumvolume}\left(0.5\mathrm{ml}\right)} $$ -
13.
For most accurate results, the plaque assay should be repeated three times and the average titer determined.

4 Notes
-
1.
SPF eggs should be used for titration of IBV, as non-SPF eggs may contain IBV specific antibodies that can interfere with the replication of IBV.
-
2.
Non-egg adapted IBV isolates may induce very little IBV specific aberrations of the embryos. When titrating such viruses, the eggs are incubated for an additional 2–3 days post inoculation. Candle the eggs at the end of the incubation period to identify embryos that have died. Subsequently, collect allantoic fluid of each of the surviving embryos and test them in a monoclonal based antigen capture ELISA as described in refs. [16, 17]. Dead embryos and those of which the allantoic fluid contains IBV as established by ELISA are considered positive.
-
3.
IBV titers usually do not exceed 108.8 TCID50/ml. Therefore 10−8 should be adequate as highest dilution. When the virus titer of the sample is known, select a number of tenfold dilutions (at least three) that include the 50 % end-point dilution. If the titer is unknown, select a broader range of tenfold dilutions that most likely include the 50 % end-point dilution. For a TCID50 titration typically all the wells of the 96-well plate are used
-
4.
Alternative methods also exist for mixing media and agar. If there is concern regarding the overlay setting too quickly or risk of contamination from the water bath, hot agar can be mixed directly with cold media (4 °C). Once the mixture feels warm to the touch, rather than hot, it can be added to cells.
-
5.
The simplest method for removing agar from the cells is to hold the plate upside down with the lid removed. The small spatula is inserted between the agar and the wall of the well. Once the base of the well is reached, a small amount of pressure is applied to remove the agar, being careful not to scrape off the cells. The whole agar plug should then fall out easily.
References
Ferris MM, Stepp PC, Ranno KA et al (2011) Evaluation of the Virus Counter® for rapid baculovirus quantitation. J Virol Methods 171:111–116
Driskell JD, Jones CA, Tompkins SM et al (2011) One-step assay for detecting influenza virus using dynamic light scattering and gold nanoparticles. Analyst 136:3083–3090
Mironov GG, Chechik AV, Ozer R et al (2011) Viral quantitative capillary electrophoresis for counting intact viruses. Anal Chem 83:5431–5435
Schwille P, Bieschke J, Oehlenschläger F (1997) Kinetic investigations by fluorescence correlation spectroscopy: the analytical and diagnostic potential of diffusion studies (evolutionary biotechnology—from theory to experiment). Biophys Chem 66:211–228
Dulbecco R (1952) Production of plaques in monolayer tissue cultures by single particles of an animal virus. Proc Natl Acad Sci U S A 38:747–752
Spearman C (1908) The method of ‘right and wrong cases’ (‘constant stimuli’) without Gauss’s formulae. Br J Psychol 2:227–242
Kaerber G (1931) Beitrag zur kollektiven behandlung pharmakologischer reihenversuche. Arch Exp Pathol Pharmacol 162:480–487
Reed L, Muench H (1938) A simple method of estimating fifty percent endpoints. Am J Hyg 27:493–497
Gillette KG (1973) Plaque formation by infectious bronchitis virus in chicken embryo kidney cell cultures. Avian Dis 17:369–378
Gelb J Jr, Cloud SS (1983) Effect of serial embryo passage of an Arkansas-type avian infectious bronchitis virus isolate on clinical response, virus recovery, and immunity. Avian Dis 27:679–687
Huang YP, Wang CH (2006) Development of attenuated vaccines from Taiwanese infectious bronchitis virus strains. Vaccine 24:785–791
Jackwood MW, Hilt DA, Brown TP (2003) Attenuation, safety, and efficacy of an infectious bronchitis virus GA98 serotype vaccine. Avian Dis 47:627–632
Yachida S, Aoyama S, Takahashi N et al (1979) Growth kinetics of embryo- and organ-culture adapted Beaudette strain of infectious bronchitis virus in embryonated chicken eggs. Avian Dis 23:127–131
Cunningham CH, Spring MP, Nazerian K (1972) Replication of avian infectious bronchitis virus in African green monkey kidney cell line VERO. J Gen Virol 16:423–427
Hodgson T, Casais R, Dove B et al (2004) Recombinant infectious bronchitis coronavirus Beaudette with the spike protein gene of the pathogenic M41 strain remains attenuated but induces protective immunity. J Virol 78:13804–13811
Ignjatovic EJ, Ashton F (1996) Detection and differentiation of avian infectious bronchitis viruses using a monoclonal antibody-based. Avian Pathol 25:721–736
Naqi SA, Karaca K, Bauman B (1993) A monoclonal antibody-based antigen capture enzyme-linked immunosorbent assay for identification of infectious bronchitis virus serotypes. Avian Pathol 22:555–564
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer Science+Business Media New York
About this protocol
Cite this protocol
Kint, J., Maier, H.J., Jagt, E. (2015). Quantification of Infectious Bronchitis Coronavirus by Titration In Vitro and In Ovo. In: Maier, H., Bickerton, E., Britton, P. (eds) Coronaviruses. Methods in Molecular Biology, vol 1282. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-2438-7_9
Download citation
DOI: https://doi.org/10.1007/978-1-4939-2438-7_9
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-2437-0
Online ISBN: 978-1-4939-2438-7
eBook Packages: Springer Protocols