
Abstract
As obligate intracellular parasites, viruses need to cross the plasma membrane and deliver their genome inside the cell. This step is initiated by the recognition of receptors present on the host cell surface. Receptors can be major determinants of tropism, host range, and pathogenesis. Identifying virus receptors can give clues to these aspects and can lead to the design of intervention strategies. Interfering with receptor recognition is an attractive antiviral therapy, since it occurs before the viral genome has reached the relative safe haven within the cell. This chapter describes the use of an immunoprecipitation approach with Fc-tagged viral spike proteins followed by mass spectrometry to identify and characterize the receptor for the Middle East respiratory syndrome coronavirus. This technique can be adapted to identify other viral receptors.
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others


Key words
1 Introduction
The first step of the infection cycle of a virus is characterized by the interaction between the viral particle and the cell surface receptor. This interaction is followed by a series of events that lead to the delivery of the viral genome inside the cytoplasm. Viruses can use diverse types of molecules to bind and enter cells. The presence of a receptor is the principal determinant of cell, tissue and organ tropism, host range, and virulence. Therefore, identifying a receptor can give clues on pathogenesis, mode of transmission, zoonotic transmission potential and can lead to the design of targeted intervention strategies.
For the last three decades the identification of virus receptors has been a major goal in virology. A group of viruses of which many receptors are known are coronaviruses (CoVs). Coronaviruses infect a wide range of avian and mammalian hosts and they are known for their ability to cross the species barrier [1]. This is exemplified by the 2003 severe acute respiratory syndrome (SARS) pandemic that was caused by the SARS-CoV [2]. In 2012, a novel zoonotic CoV was identified from a patient from Saudi Arabia that presented with a severe pneumonia [3]. This virus belongs to the same genus as SARS-CoV and was named Middle East respiratory syndrome coronavirus (MERS-CoV).
For CoVs, the viral Spike (S) protein primarily determines host and cell tropism. It is a type I membrane glycoprotein that is assembled in trimers in the viral envelope. The S protein can be functionally divided into two distinct subunits, S1 and S2. The S1 subunit binds to a cell surface receptor, whereas S2 facilitates fusion with cellular membranes.
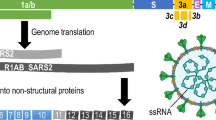
Although virus receptors can be identified using several methods [4–8], we identified the MERS-CoV receptor using Fc-tagged S1 proteins in an immunoprecipitation assay followed by mass spectrometry [9]. This assay is basically similar to the method described by Li et al., for the identification of the SARS-CoV receptor [10]. In this assay, the S1 subunit of MERS-CoV is ligated into a fusion vector to generate an S1-Fc fusion protein, for expression in HEK-293T cells and purification using protein A-sepharose beads. Incubation of the S1-Fc proteins with whole cell lysate of virus-susceptible cells allows the precipitation of the virus receptor with the tagged S1. This complex can then be pulled down from the lysate using protein A-sepharose beads. Subsequently, mass spectrometry is employed to identify candidate protein receptors (Fig. 1). These candidates must be evaluated functionally, which is done using flow-cytometric binding assays, infection blocking experiments using antibodies against the candidate receptor, and finally by attempting to infect non-susceptible cells that have been transfected with the candidate receptor. This method has been successfully employed for the rapid identification of the SARS-CoV and MERS-CoV receptor [9, 10] and is suitable for identification of protein receptors with reasonable affinity. Glycan receptors cannot be identified using the described method; treatment of susceptible cells with glycosidases prior to infection can give an insight into the type of viral receptor. Success of the protein receptor pull-down using the S1-Fc as bait depends on the affinity of S1-receptor interaction. A FACS-based S1-Fc cell-binding assay provides good insight in the strength of this interaction. The FACS-based S1-Fc assay is also instrumental to identify cell lines with high levels of receptor expression that can be used as a source for receptor affinity-isolation. Alternatively, in the absence of a suitable cell line, homogenates of tissue targeted by the virus can also be used for immunoprecipitation of the receptor.

Schematic drawing of the identification of the MERS-CoV receptor. The S1 subunit of the MERS-CoV S protein is expressed as an IgG Fc-tagged protein in HEK-293T cells and purified using protein A-sepharose beads. Incubating the S1-Fc protein with whole cell lysate of susceptible cells allows the precipitation of the virus protein receptor with the tagged S1. This complex can be pulled down from the lysate using protein A-sepharose beads. Subsequently, mass spectrometry is employed to identify candidate receptors
2 Materials
2.1 RNA Isolation, cDNA Synthesis, PCR, and Cloning
-
1.
The virus used in this protocol is used as an example. A stock of MERS-CoV-EMC isolate was prepared at 107 TCID50/ml and stored at −70 °C.
-
2.
RNA isolation: viral RNA isolation kit or tissue RNA isolation kit (Qiagen) or equivalent.
-
3.
Reverse transcriptase, e.g., SuperScript II or equivalent.
-
4.
100 mM dithiothreitol (DTT).
-
5.
RNase inhibitor.
-
6.
10 mM random primers.
-
7.
Pfu Ultra II Fusion HS DNA polymerase or equivalent.
-
8.
10 mM dNTPs.
-
9.
10 mM gene specific forward primer, e.g., MERS-CoV S1 forward primer cgaattcaccATGATACACTCAGTGTTTCTAC: the nucleotides in upper case represent MERS-CoV and those in lower case a suitable restriction endonuclease site for cloning purposes; the example here contains an EcoRI site.
-
10.
10 mM gene specific reverse primer, e.g., MERS-CoV S1 reverse primer cggatccGGTGTGAGAGTACTAGGTGTC: the nucleotides in upper case represent MERS-CoV and those in lower case a suitable restriction endonuclease site for cloning purposes; the example here contains a BamHI site.
-
11.
PCR cleanup kit.
-
12.
DNA gel extraction kit.
-
13.
Competent E. coli, e.g., Top10 competent cells.
-
14.
Cloning vectors: pCAGGS (Addgene), pFUSE-hIgG1-Fc2 (InvivoGen), and pCDNA3.1(+) (Life Technologies) or similar.
-
15.
Restriction enzymes, e.g., EcoRI and BamHI.
-
16.
SOC medium: 2 % w/v tryptone, 0.5 % w/v yeast extract, 10 mM NaCl, 2.5 mM KCl, 10 mM MgSO4, 20 mM glucose. Adjust to pH 7.5 using NaOH.
-
17.
T4 DNA Ligase.
-
18.
Maxi prep DNA kit (Qiagen) or equivalent.
-
19.
LB medium: 1 % w/v tryptone, 0.5 % w/v yeast extract, 1 % w/v NaCl. Adjust pH to 7.0 with 5 N NaOH. Sterilize by autoclaving.
-
20.
LB Amp medium: LB medium, 100 μg/ml ampicillin added after sterilization. Store at 4 °C.
-
21.
LB Amp plates: LB medium, 1.5 % agar added prior to sterilization. Sterilize by autoclaving and allow to cool to 45 °C. Add ampicillin to a final concentration of 100 μg/ml. Dispense into 90 mm petri dishes and store at 4 °C.
2.2 Expression of S1-Fc Fusion Proteins
-
1.
HEK-293T cells. HEK-293T growth medium: DMEM, 10 % fetal calf serum (FCS), 100 U/ml penicillin, 100 mg/ml streptomycin, 2 mM glutamine, 1 % nonessential amino acids, and 1 mM sodium pyruvate.
-
2.
T175 cell culture flasks.
-
3.
293T cell expression medium (as described in ref. 11) 293SF II medium (life Technologies), 1 % GlutaMAX, 0.3 % primatone, 0.2 % glucose, 0.37 % NaHCO3 and 1.5 % DMSO.
-
4.
Serum-free DMEM.
-
5.
1 mg/ml Polyethylenimine (PEI) stock solution: Add 50 mg PEI to 50 ml of endotoxin-free dH2O, place in a 75 °C water bath and vortex every 10 min until completely dissolved, cool to room temperature, neutralize to pH 7.0, filter-sterilize using a 0.22 μm filter, aliquot and store at −20 °C.
-
6.
Protein-A sepharose CL-4B (GE Healthcare) or equivalent.
-
7.
1 M Tris–HCl pH 8.0.
-
8.
Protein elution buffer: 0.5 M acetic acid pH 3. Add 29 ml of acetic acid to a beaker, make up to 1 l by adding 971 ml dH2O, adjust to pH 3 using NaOH.
-
9.
3 M Tris–HCl pH 8.8.
2.3 Immunoprecipitaion
-
1.
Huh-7 cells
-
2.
Huh-7 growth medium: RPMI1640, 10 % FCS, 100 U/ml penicillin, and 100 mg/ml streptomycin.
-
3.
100 mm cell culture dishes.
-
4.
Phosphate-buffered saline (PBS).
-
5.
Protein-A sepharose CL-4B (GE Health care) or equivalent.
-
6.
Protein elution buffer: 0.5 M acetic acid pH 3 as in Subheading 2.2.
-
7.
Lysis buffer: 3.3 mg/ml n-decyl-β-d-maltopyranoside (DDM), protease inhibitor cocktail complete Mini (Roche).
-
8.
Rubber policemen or plastic cell scrapers.
2.4 Mass Spectrometry
-
1.
Tris-Glycine SDS sample buffer 2×: 1.25 ml 1 M Tris–HCl (pH 6.8), 4 ml 10 % (w/v) SDS, 2 ml glycerol, 1 ml 0.1 % (w/v) bromophenol Blue, 1 ml 2 M DTT, make up to 10 ml with distilled water and store at room temperature.
-
2.
10 % pre-cast Tris-Glycine SDS-PAGE gels (Bio-Rad) or equivalent.
-
3.
Tris-Glycine SDS Running buffer: 250 mM Tris base, 1,920 mM glycine, 1.0 % SDS, pH 8.3.
-
4.
Absolute methanol.
-
5.
Coomassie blue staining solution: 100 mg of Coomassie brilliant Blue R250, 10 ml acetic acid, 50 ml methanol, 40 ml dH2O.
-
6.
Destaining solution: 10 ml acetic acid, 50 ml methanol, and 40 ml dH2O.
-
7.
100 mM ammonium bicarbonate (NH4HCO3).
-
8.
Acetonitrile.
-
9.
50 mM NH4HCO3.
-
10.
20 mM dithiothreitol (DTT).
-
11.
200 mM Tris(carboxyethyl)phosphine (TCEP).
-
12.
55 mM iodoacetamide (IAA).
-
13.
Trypsin, mass spectrometry grade.
-
14.
Trypsin stock solution: dissolve 100 μg of trypsin in 1 ml of 1 mM HCl, aliquot into 10 μl samples, store at −80 °C.
-
15.
0.5 % formic acid in 30 % acetonitrile.
-
16.
Razor blade and tweezers.
-
17.
Filter paper.
-
18.
Mickle gel slicer (Brinkman) or equivalent.
-
19.
Ultrasonic water bath.
-
20.
SpeedVac.
-
21.
EASY-nLC coupled to a Q Exactive mass spectrometer (both Thermo Scientific).
-
22.
ReproSil C18 reversed-phase column (Dr. Maisch GmbH).
2.5 Validation of Receptor Identification
-
1.
Cos-7 cells.
-
2.
Cos-7 cell growth medium: DMEM, 10 % FCS, 100 U/ml penicillin, and 100 mg/ml streptomycin.
-
3.
Trypsin–EDTA: 0.25 % w/v trypsin, 0.02 % w/v EDTA in PBS.
-
4.
Hemocytometer or cell counting chamber.
-
5.
Anti-DPP4 or antibody against other protein of interest.
-
6.
Flow cytometer.
-
7.
4 % formaldehyde.
-
8.
10 % normal goat serum or serum corresponding species from which secondary antibody is raised.
-
9.
Anti-SARS nsp4 or antibody against other viral protein.
-
10.
Goat anti-rabbit FITC or other suitable secondary antibody.
-
11.
Fluorescence microscope.
3 Methods
3.1 RNA Isolation
The virus used in this method, MERS-CoV EMC, was described previously [12] and is used as an example.
-
1.
Isolate viral RNA from 140 μl of virus stock at 107 TCID50/ml using the viral RNA isolation kit, following manufacturer’s instructions. The tissue RNA isolation kit was used to isolate RNA from 2 × 107 Huh-7 cells, following manufacturer’s instructions (see Note 1 ).
3.2 cDNA Synthesis
To convert RNA into cDNA we use SuperScript II reverse transcriptase but other reverse transcriptase enzymes can also be used.
-
1.
For a 20 μl reaction mix, 10 μl RNA, 1 μl 10 mM dNTPs, 1 μl 10 mM random primers, and 1.5 μl dH2O.
-
2.
Incubate at 65 °C for 10 min and then at 4 °C for 2 min (see Note 2 ).
-
3.
Place on ice.
-
4.
Make reverse transcriptase (RT) mix (for a 20 μl total reaction volume) as follows: 4 μl 5× SuperScript II reaction buffer, 1 μl 100 mM DTT, 0.5 μl RNase inhibitor (20 U/μl), 1 μl 10 mM random primers (Promega), and 1 μl SuperScript II reverse transcriptase (200 U/μl).
-
5.
Add 6.5 μl of RT mix and incubate as follows: 25 °C for 5 min, 50 °C for 45 min, 70 °C for 20 min.
-
6.
Store at 4 °C.
3.3 PCR Amplification of the S1 Region
We strongly recommend the use of Pfu Ultra II Fusion HS DNA polymerase, although other enzymes may be used. The PCR instructions in this protocol are optimized for the use of this polymerase.
-
1.
Prepare the PCR mix (48 μl reaction volume) as follows: 5.0 μl 10× Pfu Ultra II Fusion HS DNA polymerase buffer, 2.5 μl 10 mM dNTPs, 1.5 μl 10 mM gene specific forward primer, 1.5 μl 10 mM gene specific reverse primer, 1.0 μl Pfu Ultra II Fusion HS DNA polymerase, and 36.5 μl dH2O.
-
2.
Add 2.0 μl of the cDNA reaction mix from Subheading 3.2, step 6 to the 48 μl PCR mix and mix by pipetting the solution up and down several times.
-
3.
Incubate the PCR cDNA mix as follows: 94 °C for 3 min; then incubate for 39 cycles at: 94 °C for 20 s, 58 °C for 30 s, 72 °C for 2 min, with a final extension at 72 °C for 10 min.
-
4.
Store PCR reaction at 4 °C (see Note 3 ).
-
5.
Analyze the PCR products by standard agarose gel electrophoresis.
-
6.
If a single PCR product of the expected size is detected, remove polymerase, dNTPs, and primers using a standard PCR cleanup kit.
-
7.
If multiple products are detected, separate PCR products by gel electrophoresis, remove an agarose slice containing the required product and use a gel extraction kit to isolate the DNA from the agarose slice.
-
8.
Elute the required PCR product from the cleanup or gel purification kit column in dH2O.
-
9.
Analyze the PCR product using an appropriate restriction enzyme or enzymes followed by standard agarose gel electrophoresis to confirm that the PCR product is as expected.
-
10.
Quantify the nucleic acid concentration of the PCR product using NanoDrop 1000 or similar spectrophotometer.
-
11.
Store the PCR product at −20 °C.
3.4 Cloning and Expression of the S1-Fc Fusion Protein
-
1.
The gene fragment encoding the Fc part of human IgG1 is PCR amplified from pFUSE-hIgG1-Fc2 with flanking restriction sites using the forward cgaattcagatctTGAGCCCAAATCTTGTGAC and reverse primer cggatccTCATT TACCCGGAGACAGG. Subsequently, the EcoRI/BamHI digested PCR fragment can be cloned into the EcoRI-BglII digested pCAGGS vector creating the pCAGGS-Fc vector.
-
2.
Digest the S1 PCR product and the pCAGGS-Fc vector by adding the following into separate 1.5 ml tubes: 2 μl of the appropriate 10× restriction buffer, 1 μg of DNA (PCR product or vector), H2O up to 20 μl, 20 U of the appropriate restriction enzyme (EcoRI and BamHI in the example in Subheading 2.1).
-
3.
Mix gently by pipetting and incubate at 37 °C for 1 h.
-
4.
Electrophorese the restriction digests of the vector and S1 PCR product in a agarose gel, identify the products and cut out the gel slices containing the digested products.
-
5.
Purify the DNA products from the agarose slices using a gel extraction kit, elute the DNA into H2O and quantify the nucleic acid concentrations using a NanoDrop.
-
6.
Ligate the digested S1 PCR product into the pCAGGS-Fc vector by adding the following to a 1.5 ml tube: 2 μl 10× T4 ligation buffer, DNA of digested vector and S1 PCR product in a 1:3 molar ratio, H2O up to 19 μl, 1 μl T4 DNA ligase, mix gently by pipetting (see Note 4 ).
-
7.
Incubate the ligation mixture at 16 °C for 1 h.
-
8.
Transform competent E. coli cells by adding 2–5 μl of ligation mixture to 50 μl competent cells.
-
9.
Incubate on ice for 30 min.
-
10.
Heat-shock cells for 30 s at 42 °C in a thermocycler or water bath.
-
11.
Add 250 μl of SOC medium.
-
12.
Incubate at 37 °C for 1 h while shaking.
-
13.
Plate 100 μl on prewarmed LB Amp plates.
-
14.
Incubate at 37 °C overnight.
-
15.
Next day, pick colonies using a sterile toothpick for colony PCR screening and storage.
3.5 Colony PCR
-
1.
Transfer a small amount of a colony into 25 μl of LB medium.
-
2.
Make a PCR mix as follows: 2 μl 10× PCR polymerase buffer, 1 μl 10 mM dNTPs, 0.6 μl 10 mM forward primer, 0.6 μl 10 mM reverse primer, 1 μl of the colony mix, 1 μl PCR polymerase, 13.8 μl dH2O.
-
3.
Incubate the PCR mix for 30 cycles at 94 °C for 20 s, 58 °C for 30 s, 72 °C for 2 min, with a final extension at 72 °C for 10 min.
-
4.
Analyze the PCR products by standard agarose gel electrophoresis.
-
5.
Inoculate PCR positive clones in 2 ml LB Amp medium and to grow at 37 °C for ~8 h while shaking.
-
6.
From this 2 ml culture, inoculate 500 μl into a 500 ml of LB Amp medium.
-
7.
Allow the bacteria to grow ~8 h at 37 °C while shaking.
-
8.
Next day, extract plasmid DNA from the bacteria using a maxi prep DNA kit, according to manufacturer’s instructions.
-
9.
Perform a restriction digest and analyze the products by standard agarose gel electrophoresis to confirm the plasmid DNA is correct.
-
10.
Determine DNA concentration using a NanoDrop or spectrophotometer and prepare a DNA stock of 1 μg/μl.
3.6 Large-Scale Expression and Purification of S1-Fc Fusion Proteins
-
1.
Seed HEK-293T cells in 20T175 flasks in 40 ml of 293T cell growth medium and incubate at 37 °C with 5 % CO2 for approximately 24 h until 60–70 % confluent.
-
2.
Prepare a working stock of 1 mg/μl PEI. This can be kept at 4 °C.
-
3.
Two hours prior to transfection, remove medium from 293T cells and replace with 30 ml of fresh prewarmed 293T cell growth medium.
-
4.
For each T175 flask, prepare the DNA transfection solution as follows: add 18 μl of 1 μg/μl pCAGGS-MERS-CoV-S1-Fc plasmid DNA (Subheading 3.5, step 10) to 3 ml of serum-free DMEM and mix by pipetting.
-
5.
Add 54 μl of 1 mg/μl PEI to the transfection solution and mix.
-
6.
Incubate at room temperature for 30 min.
-
7.
Add the DNA/PEI complex dropwise to the T175 flask and gently swirl to mix.
-
8.
Incubate cells 4–12 h (determine experimentally).
-
9.
Aspirate the medium from the transfected cells and replace with 40 ml of HEK-293T expression medium, incubate at 37 °C with 5 % CO2 for 6 days.
-
10.
Prepare 50 % (w/v) protein-A sepharose beads: Add 0.25 g of protein-A sepharose CL-4B to a tube, add 10 ml PBS to form a slurry, centrifuge for 2 min at 2,000 × g, remove supernatant, and repeat two more times. Pellet the beads for 2 min at 2,000 × g and resuspend in 1.4 ml PBS per tube (50 % w/v), the final volume will be ~2.8 ml.
-
11.
Collect the expression medium from the transfected HEK-293T cells into 50 ml tubes and centrifuge at 2,850 × g for 10 min to remove cell debris.
-
12.
Transfer medium to new 50 ml tubes and centrifuge again at 2,850 × g for 15 min.
-
13.
Transfer cleared medium to new 50 ml tubes and keep it on ice; take a 100 μl aliquot and store at −20 °C.
-
14.
Add 0.5 ml of washed protein-A sepharose beads (50 % w/v) and 800 μl of 1 M Tris–HCl pH 8.0 to each 40 ml supernatant to neutralize the pH and incubate overnight, rotating at 4 °C (see Note 5 ).
-
15.
Collect the protein-A sepharose beads by centrifugation at 2,000 × g for 15 min (see Note 6 ).
-
16.
Pool all the protein-A sepharose beads together in a 50 ml tube and wash three times with 10 ml PBS.
-
17.
After the final centrifugation, resuspend the protein-A sepharose beads in 1 ml of 0.5 M acetic acid pH 3 elution buffer and incubate for 1 min at room temperature.
-
18.
Centrifuge the protein-A sepharose beads at 14,000 × g for 10 min and transfer the supernatant to a 1.5 ml tube.
-
19.
Repeat steps 17 and 18 twice more to elute any remaining S1-Fc protein from the protein-A sepharose beads.
-
20.
To remove any remaining protein-A sepharose beads in the supernatant repeat step 18 once and transfer supernatant to a fresh tube.
-
21.
To neutralize the pH of the eluted S1-Fc protein, add 200 μl of 3 M Tris–HCl pH 8.8 (final pH 7.5).
-
22.
Quantify the protein concentration using a NanoDrop at 280 nm.
-
23.
To analyze the size of the eluted S1-Fc protein, run 2 μg of the protein in a standard 10 % SDS-PAGE gel.
-
24.
Aliquot the S1-Fc protein and store at −80 °C.
3.7 Immunoprecipitation
3.7.1 Preparation of Cell Lysate
-
1.
Seed 5 × 107 Huh-7 cells in 100 mm dishes with growth medium and incubate at 37 °C for 24 h to allow the cells to become confluent.
-
2.
Wash the adherent cells twice with ice-cold PBS and allow the PBS to drain off.
-
3.
Add 1 ml of DDM lysis buffer onto the cells and gently rock the dish to cover the entire cell sheet.
-
4.
Scrape adherent cells off the dish with either a rubber policeman or a plastic cell scraper and transfer the cell suspension into a fresh centrifuge tube. Gently rock the suspension on either a rocker or an orbital shaker at 4 °C for 15 min to lyse the cells.
-
5.
Centrifuge the lysate at 14,000 × g in a precooled microcentrifuge for 1 min.
-
6.
Immediately transfer the supernatant to a fresh centrifuge tube and discard the pellet.
3.7.2 Preclearing of the Huh-7cell Lysate
-
1.
Prepare a 50 % (w/v) protein-A sepharose bead slurry as in Subheading 3.6, step 10.
-
2.
Add 100 μl of the protein A-sepharose bead slurry to every 1.5 ml of cell lysate and incubate at 4 °C for 10 min on a rocker or orbital shaker (see Note 7 ).
-
3.
Remove the beads by centrifugation at 14,000 × g at 4 °C for 1 min and carefully transfer supernatant to a fresh tube.
3.7.3 Immunoprecipitation
-
1.
Add 2.5 μg of the purified S1-Fc fusion protein (Subheading 3.6, step 24) to 1.5 ml of the Huh-7 precleared lysate and incubate for 1 h at room temperature on a rocker or an orbital shaker.
-
2.
Use 1.5 ml of the Huh-7 precleared lysate, without the purified S1-Fc fusion protein, as a negative control and incubate as described in step 1.
-
3.
Capture any immunocomplexes between the S1-Fc fusion protein and the precleared Huh-7 cell lysate by adding 150 μl of the protein A-sepharose 50 % bead slurry to 1.5 ml of the lysates in Subheading 3.7, step 2, gently mix overnight at 4 °C on either a rocker or an orbital shaker.
-
4.
Collect the protein A-sepharose beads by pulse centrifugation (i.e., 5 s in the microcentrifuge at 14,000 × g). Discard the supernatant and wash the protein A-sepharose beads twice with DDM lysis buffer and once in PBS alone. Discard the supernatants.
-
5.
Resuspend the protein A-sepharose beads in 200 μl of PBS and store at 4 °C.
3.8 Mass Spectrometry Analysis by Nanoflow LC-MS/MS
3.8.1 SDS-PAGE
-
1.
Prepare 100 mM NH4HCO3–acetonitrile wash solution as follows: 1:1 (v:v) of 100 mM NH4HCO3 and acetonitrile.
-
2.
Pellet 100 μl of the protein A-sepharose beads (Subheading 3.7.3, step 5) by pulse centrifugation and discard the supernatant.
-
3.
Resuspend the protein A-sepharose beads in 30 μl 2× Tris-Glycine SDS sample buffer, mix gently and incubate at 100 °C for 10 min (see Note 8 ).
-
4.
Centrifuge at 14,000 × g for 1 min
-
5.
Load 15 μl of supernatant on a 10 % pre-cast Tris-Glycine SDS-PAGE gel, electrophorese the sample for 40 min at 100 V; Alternatively, the supernatant can be transferred to a fresh 1.5 ml tube and stored frozen at −20 °C for later use, frozen supernatants should be reboiled for 5 min directly prior to loading on a gel.
-
6.
Transfer SDS-PAGE gel to a clean cell culture dish or other plastic container and cover with Coomassie blue staining solution. Incubate at room temperature for 45 min while shaking.
-
7.
Destain the gel with destaining solution until bands can clearly be seen and leave the gel in dH2O in a clean cell culture dish.
-
8.
Cut out the lane of interest using a clean razor blade and tweezers and put the complete lane onto two dH2O-wetted filter papers (1.5 × 10 cm).
-
9.
Clean the razor blade of the Mickle gel slicer with methanol and then dH2O.
-
10.
Place the filter paper with the gel lane on top onto the sled of the Mickle gel slicer and cut the gel lane into 1 mm slices.
-
11.
Depending on the complexity of the protein mixture in the gel lane, transfer two or three adjacent slices to 1.5 ml tubes that contain 600 μl of NH4HCO3–acetonitrile wash solution, so that you divide the complete gel lane over 20–30 sample tubes.
3.8.2 Destaining and Washing of Gel Pieces
-
1.
Destain the gel slices by shaking at 4 °C overnight in the 100 mM NH4HCO3–acetonitrile wash solution (see Note 9 ).
-
2.
Aspirate off the wash solution with a gel loading tip, replace with 0.5 ml of fresh NH4HCO3–acetonitrile wash solution, and shake for 1 h at 4 °C.
-
3.
Wash with 200 μl dH2O once and with 200 μl NH4HCO3–acetonitrile wash solution twice for 15 min.
-
4.
Shrink the gel pieces using 200 μl 100 % acetonitrile and flick the tube until the gel pieces turn white.
-
5.
Incubate for 5 min at room temperature.
-
6.
Aspirate off acetonitrile and air-dry the gel slices for 5 min.
3.8.3 Reduction and Alkylation of Proteins (See Note 10 )
-
1.
Freshly prepare gel swelling solution as follows: 5 ml 100 mM NH4HCO3, 5 ml freshly prepared 20 mM DTT.
-
2.
Freshly prepare alkylation solution as follows: Dissolve 102 mg of Iodoacetamide (IAA) in 5 ml of dH2O and then add 5 ml of 100 mM NH4HCO3.
-
3.
Swell each gel slice in 200 μl gel swelling solution.
-
4.
Incubate for 1 h at 37 °C.
-
5.
Remove the gel swelling solution and add 200 μl alkylation solution to each gel slice.
-
6.
Incubate for 1 h at room temperature in the dark.
-
7.
Wash the gel slices twice with 200 μl of the NH4HCO3–acetonitrile wash solution for 15 min.
-
8.
Shrink the gel pieces in 200 μl 100 % acetonitrile.
3.8.4 In-Gel Digestion
-
1.
Just before use, prepare a 10 μg/ml trypsin working solution by diluting the 100 μg/ml trypsin stock solution with 50 mM NH4HCO3.
-
2.
Add 10 μl of the 10 μg/ml trypsin working solution to every 1 mm gel slice so that each slice is fully immersed in the trypsin working solution.
-
3.
Incubate the gel slices for 30 min at room temperature.
-
4.
Check that the gel pieces are still fully covered by the trypsin solution, if not, add some more trypsin working solution.
-
5.
Incubate overnight at 37 °C; shaking not necessary.
3.8.5 Extraction of Peptides from the Gel
-
1.
Centrifuge the gel slices for 10 s and remove trypsin.
-
2.
Add 50 μl of the 0.5 % formic acid solution in 30 % acetonitrile peptide extraction solution, sonicate in an ultrasonic bath for 2 min at room temperature, then incubate for 30 min at room temperature.
-
3.
Transfer the supernatant to a clean 1.5 ml tube or into a 96-well plate.
-
4.
Repeat steps 3 and 4 twice and combine the supernatants.
-
5.
Dry the combined supernatants in a SpeedVac.
3.8.6 Mass Spectrometry
-
1.
Dissolve the peptides in 30 μl of 0.1 % formic acid.
-
2.
Analyze the samples by nanoflow LC-MS/MS on an EASY-nLC coupled to a Q Exactive mass spectrometer, operating in positive mode and equipped with a nanospray source.
-
3.
Trap peptide mixtures on a ReproSil C18 reversed-phase column (column dimensions 1.5 cm × 100 μm, packed in-house) at a flow rate of 8 μl/min−1.
-
4.
Separate peptides on a ReproSil C18 reversed-phase column (column dimensions 15 cm × 50 μm, packed in-house) using a linear acetonitrile gradient from 0 to 80 % (A = 0.1 % formic acid; B = 80 % (v/v) acetonitrile, 0.1 % formic acid) in 70 or 120 min and at a constant flow rate of 200 nl/min−1.
-
5.
Spray the column eluent directly into the electrospray ionization (ESI) source of the mass spectrometer.
-
6.
Acquire mass spectra in continuum mode; fragment the peptides in data-dependent mode by higher-energy collisional dissociation (HCD).
-
7.
For data analysis, create peak lists automatically from raw data files using a software suite such as Mascot Distiller software (Matrix Science), Proteome Discoverer (Thermo), or MaxQuant. Use a database search engine such as Mascot or Andromeda (MaxQuant) for searching peak lists against a Uniprot fasta database that contains all human protein sequences. For control purposes, merge the human protein database with a fasta database containing all for example MERS virus protein sequences. Perform the database search analysis on either an in-house server or a multi-core desktop PC.
-
8.
Human Dipeptidyl peptidase 4 (DPP4 or CD26) tryptic peptides are expected to be detected specifically in MERS-CoV-S1-Fc IP samples, in relation to the examples given.
3.9 Cloning and Expression of Human DPP4 or Appropriate Receptor
3.9.1 RNA Isolation and PCR Amplification of Human DPP4
-
1.
Isolate total RNA from Huh-7 cells and make cDNA as described in Subheadings 3.1 and 3.2.
-
2.
Amplify complete human DPP4 using Pfu Ultra II fusion HS DNA polymerase using the PCR protocol described in Subheading 3.3 using specific primers for human DPP4 or the gene sequence of the protein of interest.
3.9.2 Cloning and Expression of Human DPP4
-
1.
Clone the complete DPP4 gene or gene of interest into the pcDNA 3.1 (+) expression vector into the BamH1 and NotI site (pcDNA-hDPP4 plasmid).
-
2.
Check the construct by sequence analysis.
Prepare cells not susceptible to virus infection (e.g., Cos-7 cells) and transfect them with the DPP4-expression plasmid. Transfect pcDNA-hDPP4 using PEI (plasmid PEI ratio 1:3)
-
3.
At 24 h post transfection the cells can be tested for the cell surface expression of DPP4 using FACS analysis and susceptibility of infection by infecting those cells with MERS-CoV-EMC.
3.9.3 Identification Receptor Expressing Cells by Flow Cytometry Analysis
-
1.
After 24 h of transfection (Subheading 3.9.2, step 2), wash the cells once with PBS and add 1 ml of Trypsin EDTA.
-
2.
Incubate at 37 °C for approximately 5 min (until complete disassociation of the cells).
-
3.
Add 1 ml of PBS and mix it by pipetting up and down.
-
4.
Transfer the cell suspension in to a new tube and add additional 5 ml of PBS.
-
5.
Centrifuge at 400 × g for 5 min.
-
6.
Resuspend the cells in 2 ml PBS and count the cells using the counting chamber.
-
7.
Place 5 × 105 cells in a 96 well “v” bottom plate and add S1-Fc or 5 μg/ml antibody against protein under investigation such as goat anti-DPP4 polyclonal antibodies in this example, or without any protein as a control, in volume of 50 μl.
-
8.
Incubate on ice for 30 min.
-
9.
Wash the cells three times with PBS containing 0.5 % BSA.
-
10.
Add 50 μl of FITC-labelled goat anti-human IgG or FITC-labelled rabbit anti goat serum (5 μg/ml) or any other labelled secondary antibody depending on the origin of the antibody in step 7.
-
11.
Incubate on ice for 30 min and wash the cells three times with PBS.
-
12.
Resuspend the cells in 190 μl of PBS.
-
13.
Analyze the cells for any fluorescence by flow cytometry (Fig. 2).
Fig. 2 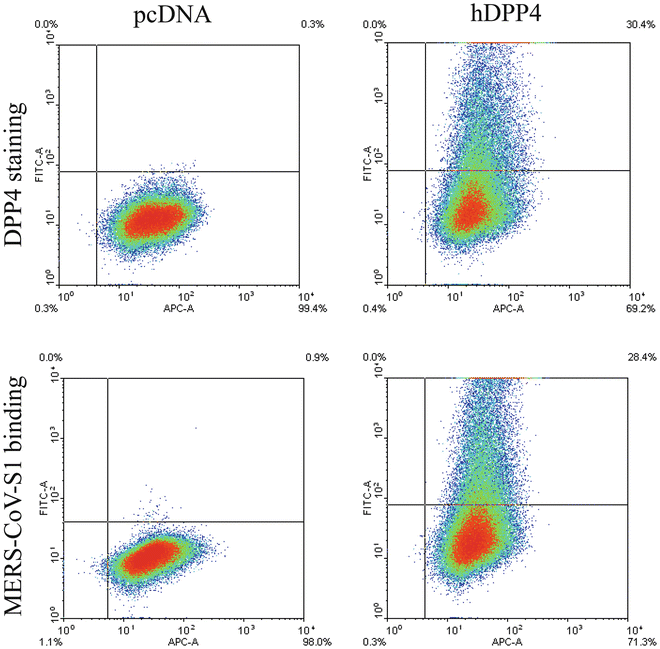
Human DPP4 expression and MERS-CoV-S1 binding to cells transfected with human DPP4 plasmid or empty plasmid as analyzed by FACS analysis. DPP4 staining (upper panel ) and MERS-CoV-S1 binding (lower panel ) are shown

3.9.4 Virus Infection Assay
-
1.
Transfect pcDNA-DPP4 plasmid or plasmid containing the gene under investigation in Cos-7 and empty plasmid as control.
-
2.
After 24 h of transfection, wash the cells with Cos-7 growth medium and incubate the cells with virus under investigation, e.g., MERS-CoV-EMC for 1 h.
-
3.
Wash the cells two times with Cos-7 growth medium containing 1 % FCS to remove any unbound virus and after final wash add 3 ml of fresh medium.
-
4.
Incubate the cells at 37 °C with 5 % CO2 for 24 h.
-
5.
Fix the cells with 4 % formaldehyde solution for 10 min.
-
6.
Wash the cells three times with PBS.
-
7.
Add 500 μl of 70 % ethanol and keep the plate at 4 °C until immunofluorescent staining.
-
8.
Wash the cells three times with PBS.
-
9.
Add 200 μl of 10 % normal goat serum or serum corresponding to the species from which the secondary antibody in step 14 is derived.
-
10.
Incubate the cells at 37 °C for 30 min.
-
11.
Remove the 10 % normal goat serum and add 200 μl of any antibody to a specific virus protein, for example rabbit-anti-SARS-CoV nsp4 (5 μg/ml) is cross-reactive for MERS-CoV-EMC.
-
12.
Incubate the cells at 37 °C for 1 h.
-
13.
Wash the cells three times with PBS.
-
14.
Add 200 μl of goat anti-rabbit serum conjugated with FITC (5 μg/ml).
-
15.
Incubate the cells at 37 °C for 1 h.
-
16.
Wash the cells three times with PBS and analyze using a fluorescent microscope (Fig. 3).
Fig. 3 
Characterization of human DPP4 as a functional receptor for MERS-CoV. Human DPP4 plasmid or a control plasmid was transfected in non-susceptible Cos-7 cells and after 24 h the cells were infected with MERS-CoV-EMC. Subsequently, the cells were fixed after 24 h of infection and stained for MERS-CoV. (a) Empty plasmid transfected cells. (b) Human DPP4 plasmid transfected cells

4 Notes
-
1.
When using another RNA isolation kit, please refer to the recommendations of the respective manufacturer.
-
2.
The amount of RNA that can be used for this reaction volume is 100 pg to 1 μg RNA. If more RNA is used, (e.g., 2 μg), add the appropriate amount of reagents needed.
-
3.
When using another polymerase than Pfu, please refer to the recommendations of the respective manufacturer.
-
4.
Add the ligase last.
-
5.
It is recommended to cut the tip of the pipette-tip off when working with sepharose beads to avoid disruption of the beads.
-
6.
Before aspiration of the medium, take a 100 μl aliquot and store at −20 °C.
-
7.
Preclearing the lysate will reduce nonspecific binding of proteins to the agarose or sepharose when it is used later on in the assay.
-
8.
Be sure that all equipment that you use for running gels (trays, boxes, dishes, tips, etc.) is clean and try to keep equipment that you use for mass spec gels apart from other electrophoresis equipment in your lab. The more the keratin contaminants, the less the protein identifications in the end.
-
9.
A few hours is sufficient.
-
10.
These steps can be skipped if you have alkylated the proteins before running the gel.
References
Parrish CR, Holmes EC, Morens DM et al (2008) Cross-species virus transmission and the emergence of new epidemic diseases. Microbiol Mol Biol Rev 72:457–470
Drosten C, Günther S, Preiser W et al (2003) Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med 348:1967–1976
Zaki AM, van Boheemen S, Bestebroer TM et al (2012) Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med 367:1814–1820
Dalgleish AG, Beverly PCL, Clapham PR et al (1984) The CD4 (T4) antigen is an essential component of the receptor for the AIDS retrovirus. Nature 312:763–767
Greve JM, Davis G, Meyer AM, Forte CP et al (1989) The major human rhinovirus receptor is ICAM-1. Cell 56:839–847
Bergelson JM, Shepley MP, Chan BMC et al (1992) Identification of the integrin VLA-2 as a receptor for echovirus 1. Science 255:1718–1720
Mendelsohn CL, Wimmer E, Racaniello VR (1989) Cellular receptor for poliovirus: molecular cloning, nucleotide sequence, and expression of a new member of the immunoglobulin superfamily. Cell 56:855–865
Yeager CL, Ashmun RA, Williams RK et al (1992) Human aminopeptidase N is a receptor for human coronavirus 229E. Nature 357:420–422
Raj VS, Mou H, Smits SL et al (2013) Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature 495:251–254
Li W, Moore MJ, Vasilieva N et al (2003) Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 426:450–454
Zeng Q, Langereis MA, van Vliet al et al (2008) Structure of coronavirus hemagglutinin-esterase offers insight into corona and influenza virus evolution. Proc Natl Acad Sci U S A 105:9065–9069
van Boheemen S, de Graaf M, Lauber C et al (2012) Genomic characterization of a newly discovered coronavirus associated with acute respiratory distress syndrome in humans. MBio 3, e00473-12
Acknowledgement
This work was supported by a grant from the Dutch Scientific Research (NWO; no. 40-00812-98-13066) granted to BJB and BLH. SLS is partly employed by Viroclinics Biosciences.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer Science+Business Media New York
About this protocol
Cite this protocol
Raj, V.S. et al. (2015). Identification of Protein Receptors for Coronaviruses by Mass Spectrometry. In: Maier, H., Bickerton, E., Britton, P. (eds) Coronaviruses. Methods in Molecular Biology, vol 1282. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-2438-7_15
Download citation
DOI: https://doi.org/10.1007/978-1-4939-2438-7_15
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-2437-0
Online ISBN: 978-1-4939-2438-7
eBook Packages: Springer Protocols