
Abstract
HAp nanofibers (or whiskers) have been attracted considerable attention for their application as adsorbents and reinforcing fillers owing to their unique morphologies. However, fabrication of HAp nanofibers has been limited to high-temperature and/or long-term methods. Herein, we report that HAp nanofibers with more than 5 μm in length (aspect ratio >100) can be easily obtained by a simple wet precipitation method without additives at relatively low temperature (80 °C) under acidic conditions (initial pH of 6.5 and final pH of 3.9), without pH control during the precipitation.
You have full access to this open access chapter, Download conference paper PDF
Similar content being viewed by others


Keywords
1 Introduction
Hydroxyapatite (HAp) is recognized as a major inorganic component of human hard tissues (bones and teeth). Synthetic HAp, a type of bioceramics, exhibits biocompatibility (i.e., nontoxicity) (Lawton et al. 1989; Abdel-Gawad and Awwad 2010) and excellent cell adhesion properties (Dorozhkin 2010). Therefore, HAp and its composites with polymers or metals have been widely used in orthopedic and dental tissue engineering fields (Choi et al. 2010; Honda et al. 2010; Okada and Matsumoto 2015). Other important applications of HAp include their use as drug delivery carriers (Matsumoto et al. 2004; Bouladjine et al. 2009; Tomoda et al. 2010) and in liquid chromatographic packing materials (Kawasaki 1991) by utilizing the favorable adsorption capacity of the HAp surface for biomolecules, such as cell adhesion proteins (Kilpadi et al. 2001; Lee et al. 2010). HAp belongs to a hexagonal crystal system and possesses different properties on its a and c planes, and hence, its morphology strongly affects protein adsorption properties (Kilpadi et al. 2001; Lee et al. 2010). Therefore, the control of HAp morphology is of fundamental importance to improve its adsorption properties.
HAp nanofibers (or whiskers) have been attracted considerable attention, owing to their unique morphologies, for application as adsorbents and reinforcing components in biomedical composites (Qi et al. 2017). Nevertheless, previous reports have demonstrated the synthesis of HAp nanofibers only at high temperature, during long periods, or by adding additives, such as hydrothermal methods (e.g., 180–200 °C) (Sadat-Shojai et al. 2012; Chen and Zhu 2016), homogeneous precipitation methods (e.g., 72 h) (Aizawa et al. 2005; Zhan et al. 2005), and wet precipitation methods with surfactants (Liu et al. 2002; Chen and Zhu 2016). Therefore, synthesis of HAp nanofibers at mild conditions could reduce costs and enable more diverse applications of HAp. However, the synthesis of HAp nanofibers at low temperature without additives is still challenging.
Herein, we report that HAp nanofibers could be easily obtained by a simple wet precipitation method without additives at relatively low temperature (80 °C) under acidic conditions, without pH control during the precipitation. The formation process of the HAp nanofibers by the simple wet precipitation method was also evaluated.
2 Materials and Methods
Unless otherwise mentioned, all materials were of reagent grade and were purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). All materials were used as received. Milli-Q water (Millipore Corp., Bedford, MA, USA) with a specific resistance of 18.2 × 106 Ω·cm was used.
An aqueous solution of Ca(NO3)2·H2O (10 g/L = 42.3 mM; 160 mL) was poured into a 500-mL conical flask equipped with an inlet for nitrogen and a magnetic stirrer. After the reactor was heated at a predetermined temperature (30, 50, or 80 °C), initial pH of the solution was adjusted to 6.5 (i.e., acidic condition) or 10.0 (i.e., alkaline condition) by adding a 28% ammonia solution. After the temperature and the initial pH were equilibrated for 5 min, an aqueous solution of (NH4)2HPO4 (101.6 mM; 40 mL; pH 8.0) was added at a feed rate of 8.0 mL/h into the conical flask, and the resultant mixture was stirred for another 12 h at a constant temperature, with variations within 0.1 °C. During the reaction, although the pH was not controlled, the changes in pH were recorded with a pH meter (HM-31P; pH resolution, 0.01; DKK-TOA Corp., Tokyo, Japan) at a time interval of 2 min. The resulting product was then centrifugally washed three times with distilled water and then dried at room temperature under reduced pressure for 1 day.
The dispersed sample was dried on an aluminum stub and coated using an osmium coater Neoc-Pro (Meiwafosis Co. Ltd., Tokyo, Japan) before particle morphology observation by scanning electron microscopy (SEM) using a JSM-6701F microscope (JEOL Ltd., Tokyo, Japan) operated at 5 kV or the sample was dried on a collodion-coated grid before particle morphology observation and electron diffraction measurement by transmission electron microscopy (TEM) using a JEM-2100F microscope (JEOL Ltd.) operated at 200 kV. The number-averaged size (N = 50) was determined from SEM photographs with image analysis software (Image J; National Institutes of Health, Bethesda, MD, USA). Fourier-transform infrared (FTIR) spectra were obtained using an IRAffinity-1S system (Shimadzu Corp., Kyoto, Japan) with a KBr pellet method at a resolution of 4 cm−1 with 32 scans. Product identification was also conducted by X-ray diffraction (XRD) measurements (RINT2500HF; Rigaku Corp., Tokyo, Japan) equipped with a Cu-Kα radiation source.
3 Results and Discussion
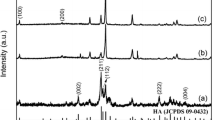
In the case of alkaline conditions (i.e., initial pH = 10.0), the final pH after the reaction decreased to 8.2–9.3, and HAp crystals were obtained as shown in Fig. 22.1a (i–iii). The crystal morphologies varied by changing the temperature (Fig. 22.2a–c); i.e., more elongated HAp crystals were obtained by increasing the reaction temperature, which is consistent with previous reports (Sadat-Shojai et al. 2013; Okada and Matsumoto 2015). Note that the size distributions of HAp crystals formed in the alkaline conditions were broad (i.e., polydispersed), which should be due to long particle nucleation stage. In other words, the saturated solution concentration of HAp is too small at alkaline conditions (Matsumoto et al. 2007), and hence the particle nuclei formed throughout the feeding period (5 h) of phosphate ion solution into the calcium ion solution.

(a) XRD patterns of the products synthesized by wet precipitation methods under different initial pH and temperature conditions: (i) pH 10.0, 30 °C; (ii) pH 10.0, 50 °C; (iii) pH 10.0, 80 °C; (iv) pH 6.5, 30 °C; (v) pH 6.5, 50 °C; (vi) pH 6.5, 80 °C. (b) A phase diagram of the products after hydrolysis of α-tricalcium phosphates at different pH and temperature conditions (Monma 1980; Monma et al. 1981) and the pH changes during wet precipitation methods at different initial pH and temperature conditions: (open squares) pH 6.5, 30 °C; (open triangles) pH 6.5, 50 °C; (open circles) pH 6.5, 80 °C. Abbreviations: HAp hydroxyapatite, OCP octacalcium phosphate, DCPD dicalcium phosphate dihydrate

SEM photographs of the products synthesized by wet precipitation methods under different initial pH and temperature conditions: (a) pH 10.0, 30 °C; (b) pH 10.0, 50 °C; (c) pH 10.0, 80 °C; (d) pH 6.5, 30 °C; (e) pH 6.5, 50 °C; (f) pH 6.5, 80 °C. The insets show magnified images
In the case of acidic conditions (i.e., initial pH = 6.5), the final pH significantly dropped to 5.5–3.9 after the reaction, as shown in Fig. 22.1b. Plate-like dicalcium phosphate dihydrate (DCPD) and/or octacalcium phosphate (OCP) were formed at lower temperatures of 30 °C and 50 °C (Figs. 22.1a (iv, v) and 22.2d–e), which is almost consistent with the phase diagram (Fig. 22.1b) reported from the data of the hydrolysis products of α-tricalcium phosphate (α-TCP) (Monma 1980; Monma et al. 1981). Interestingly, pure HAp crystals were obtained at 80 °C even in the acidic condition (final pH = 3.9), which is not consistent with the phase diagram for α-TCP hydrolysis (Fig. 22.1b). The HAp crystals formed under the acidic condition at 80 °C showed a fiber-like morphology, with the long axis being parallel to c axis of HAp (Fig. 22.3), and were longer than those formed under the alkaline condition at the same temperature.

(a) TEM image of the product synthesized by the wet precipitation method at initial pH 6.5 and 80 °C and (b) electron diffraction pattern of the area highlighted by a dotted circle in the image (a). The incident electron beam direction in the diffraction pattern was parallel to [1–10] zone of HAp crystal
In order to check the formation process of the HAp nanofibers under the acidic condition at 80 °C, a part of precipitation was collected during the reaction as shown in Fig. 22.4. From the XRD measurements (Fig. 22.4c), only HAp crystals were detected throughout the reaction even at the end of the feeding and ripening periods. From SEM observation (Fig. 22.4b), needle-like crystals of around 500 nm in length were formed at 30 min, and they elongated into fiber-like crystals with more than 10 μm in length and around 50–100 nm in width (i.e., aspect ratio >100) at 1300 min.

(a) A variation of pH during the wet precipitation method at initial pH of 6.5 and 80 °C. (b) SEM photographs, (c) XRD patterns, and (d) FTIR spectra of the part of precipitation collected during the wet precipitation method at 30, 60, 300 (end of feeding), and 1300 min
Taken together, these results indicate that needle-like HAp was firstly precipitated at the initial pH of 6.5 at 80 °C, which is consistent with the phase diagram (Fig. 22.1b). The firstly formed HAp acted as a seed crystal during the following feeding and ripening periods even at acidic conditions. The unexpected stability of enamel apatite (i.e., ribbon-like apatite crystal elongated extremely in its c-axis direction) at acidic conditions has been also reported; that is, enamel apatite did not transformed to DCPD or other calcium phosphates even at pH 4 for 2 months due to a decrease in the ion product of enamel apatite with the decrease in pH (Larsen and Jensen 1989). The acidic condition would be preferable for preventing both secondary nuclei formation (due to an increase in the saturated solubility of HAp (Matsumoto et al. 2007) and CO3 2− contamination (due to a decrease in the saturated solubility of CO2), which are known to inhibit HAp crystal growth (i.e., elongation).
The HAp nanofibers prepared by the simple wet precipitation method without additives at relatively low temperature (80 °C) have potential applications as adsorbents and reinforcing components in biomedical composites because of their unique morphologies (Qi et al. 2017). The preparation method described here accompanies with the crystal growth of HAp (without secondary nuclei formation, under appropriate conditions). Further optimization of this low-temperature wet precipitation method could enable (1) the preparation of more uniform size distribution of HAp nanocrystals for the pre-prepared seed crystals with controlled initial size and number of seeds (or by developing a modified wet precipitation method with a stepwise pH control from alkaline to acidic condition during the reaction) and (2) the development of brush-like HAp coating for the substrates pre-coated with HAp seed crystals. Note that brush-like HAp coating of titanium substrate showed superior osteoconductivity compared with other morphologies (i.e., needle-like, plate-like, net-like, and spherical) of HAp (Kuroda and Okido 2012). Nevertheless, brush-like HAp coating has been limited to some metallic substrates because the previously developed methods require electroconductive and thermally stable substrates due to their coating conditions [e.g., electrochemical and/or high temperature conditions such as above 140 °C (Kuroda and Okido 2012) for wet methods and above 300 °C (Teshima et al. 2012) for dry methods]. The fabrication of brush-like HAp coated substrate at low temperature and its application will be reported in the near future.
References
Abdel-Gawad EI, Awwad SA (2010) Biocompatibility of intravenous nano hydroxyapatite in male rats. Nat Sci 8:60–68
Aizawa M, Porter AE, Best SM, Bonfield W (2005) Ultrastructural observation of single-crystal apatite fibres. Biomaterials 26:3427–3433
Bouladjine A, Al-Kattan A, Dufour P, Drouet C (2009) New advances in nanocrystalline apatite colloids intended for cellular drug delivery. Langmuir 25:12256–12265
Chen F, Zhu YJ (2016) Large-scale automated production of highly ordered ultralong hydroxyapatite nanowires and construction of various fire-resistant flexible ordered architectures. ACS Nano 10:11483–11495
Choi S-W, Zhang Y, Thomopoulos S, Xia Y (2010) In vitro mineralization by preosteoblasts in poly(DL-lactide-co-glycolide) inverse opal scaffolds reinforced with hydroxyapatite nanoparticles. Langmuir 26:12126–12131
Dorozhkin SV (2010) Nanosized and nanocrystalline calcium orthophosphates. Acta Biomater 6:715–734
Honda M, Fujimi TJ, Izumi S, Izawa K, Aizawa M, Morisue H, Tsuchiya T, Kanzawa N (2010) Topographical analyses of proliferation and differentiation of osteoblasts in micro- and macropores of apatite-fiber scaffold. J Biomed Mater Res A 94:937–944
Kawasaki T (1991) Hydroxyapatite as a liquid chromatographic packing. J Chromatogr 544:147–184
Kilpadi KL, Chang PL, Bellis SL (2001) Hydroxylapatite binds more serum proteins, purified integrins, and osteoblast precursor cells than titanium or steel. J Biomed Mater Res 57:258–267
Kuroda K, Okido M (2012) Hydroxyapatite coating of titanium implants using hydroprocessing and evaluation of their osteoconductivity. Bioinorg Chem Appl 2012:730693 7 pages
Larsen MJ, Jensen SJ (1989) Stability and mutual conversion of enamel apatite and brushite at 20 °C as a function of pH of the aqueous phase. Arch Oral Biol 34:963–968
Lawton DM, Lamaletie MDJ, Gardner DL (1989) Biocompatibility of hydroxyapatite ceramic: response of chondrocytes in a test system using low temperature scanning electron microscopy. J Dent 17:21–27
Lee JS, Wagoner Johnson AJ, Murphy WL (2010) A modular, hydroxyapatite-binding version of vascular endothelial growth factor. Adv Mater 22:5494–5498
Liu Y, Wang W, Zhan Y, Zheng C, Wang G (2002) A simple route to hydroxyapatite nanofibers. Mater Lett 56:496–501
Matsumoto T, Okazaki M, Inoue M, Yamaguchi S, Kusunose T, Toyonaga T, Hamada Y, Takahashi J (2004) Hydroxyapatite particles as a controlled release carrier of protein. Biomaterials 25:3807–3812
Matsumoto T, Okazaki M, Nakahira A, Sasaki J, Egusa H, Sohmura T (2007) Modification of apatite materials for bone tissue engineering and drug delivery carriers. Curr Med Chem 14:2726–2733
Monma H (1980) Preparation of octacalcium phosphate by the hydrolysis of α-tricalcium phosphate. J Mater Sci 15:2428–2434
Monma H, Ueno S, Kanazawa T (1981) Properties of hydroxyapatite prepared by the hydrolysis of tricalcium phosphate. J Chem Technol Biotechnol 31:15–24
Okada M, Matsumoto T (2015) Synthesis and modification of apatite nanoparticles for use in dental and medical applications. Jpn Dent Sci Rev 51:85–95
Qi M-L, He K, Huang Z-N, Shahbazian-Yassar R, Xiao G-Y, Lu Y-P, Shokuhfar T (2017) Hydroxyapatite fibers: a review of synthesis methods. JOM 69:1354–1360
Sadat-Shojai M, Khorasani MT, Jamshidi A (2012) Hydrothermal processing of hydroxyapatite nanoparticles – a Taguchi experimental design approach. J Cryst Growth 361:73–84
Sadat-Shojai M, Khorasani MT, Dinpanah-Khoshdargi E, Jamshidi A (2013) Synthesis methods for nanosized hydroxyapatite with diverse structures. Acta Biomater 9:7591–7621
Teshima K, Wagata H, Sakurai K, Enomoto H, Mori S, Yubuta K, Shishido T, Oishi S (2012) High-quality ultralong hydroxyapatite nanowhiskers grown directly on titanium surfaces by novel low-temperature flux coating method. Cryst Growth Des 12:4890–4896
Tomoda K, Ariizumi H, Nakaji T, Makino K (2010) Hydroxyapatite particles as drug carriers for proteins. Colloids Surf B Biointerfaces 76:226–235
Zhan J, Tseng YH, Chan JCC, Mou CY (2005) Biomimetic formation of hydroxyapatite nanorods by a single-crystal-to- single-crystal transformation. Adv Funct Mater 15:2005–2010
Acknowledgments
This work was supported partly by the Japan Society for the Promotion of Science KAKENHI (grant numbers: JP16H05533, JP15K1572307, and JP25220912), the Matching Planner Program (MP27115663113) from Japan Science and Technology Agency, and the Cooperative Research Project of Research Center for Biomedical Engineering, Ministry of Education, Culture, Sports, Science and Technology of Japan.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2018 The Author(s)
About this paper

Cite this paper
Okada, M., Hara, E.S., Matsumoto, T. (2018). Fabrication of Hydroxyapatite Nanofibers with High Aspect Ratio via Low-Temperature Wet Precipitation Methods Under Acidic Conditions. In: Endo, K., Kogure, T., Nagasawa, H. (eds) Biomineralization. Springer, Singapore. https://doi.org/10.1007/978-981-13-1002-7_22
Download citation
DOI: https://doi.org/10.1007/978-981-13-1002-7_22
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-1001-0
Online ISBN: 978-981-13-1002-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)