
Abstract
Heliconius wing patterns are an adaptive trait under strong selection in the wild. They are also amenable to genetic studies and have been the focus of evolutionary genetic analysis for many years. Early genetic studies characterised a large number of Mendelian loci with large effects on wing pattern elements in crossing experiments. The recent application of molecular genetic markers has consolidated these studies and led to recognition that a huge range of allelic variation at just a few major loci controls patterns across most of the Heliconius radiation. Some of these loci consist of tightly linked components that control different aspects of the phenotype and can be separated by occasional recombination. More recent quantitative analyses have also identified minor-effect loci that influence the expression of these major loci.
Studies of a single locus polymorphism in Heliconius numata provide an example of a ‘supergene’, in which a single major locus controls segregation of a variable phenotype. This supports ‘Turner’s Sieve’ hypothesis for the evolution of supergenes, whereby sequential linked mutations arise at the same locus. In addition, inversion polymorphisms are associated with wing pattern variation in wild populations, which reduce recombination across the supergene locus. This provides direct evidence that the architecture and organisation of genomes can be shaped by natural selection. There is also evidence that patterns of dominance of the alleles at this locus have also been shaped by natural selection. Mimicry therefore provides a case study of how natural selection shapes the genetic control of adaptive variation.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others

Keywords
A major research effort in evolutionary biology is devoted to determining the molecular changes in DNA sequences that control adaptive phenotypic changes. By identifying the number and identity of genes controlling traits, and the relative contribution of individual mutations to changes in the appearance of an organism, we can address a wealth of questions in evolutionary biology including some that were debated by early geneticists, such as the importance of large versus small mutations in evolution. Mimicry patterns in Heliconius butterflies have contributed significantly to our understanding of the genetic basis for adaptation over the past 40 years. Here I review what is known of the genetic basis for these bright colour patterns and some of the implications for our understanding of evolution.
1 Phenotypic Effects of Major Loci: The Red Locus Optix
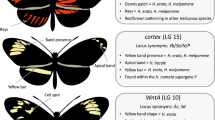
The most striking aspect of Heliconius wing pattern genetics is that a few major loci control large phenotypic changes (Fig. 9.1). This major locus control of adaptive traits is an emerging pattern in other organisms, but studies of butterflies provided some of the first clear examples (Nadeau and Jiggins 2010) and were already evident in early work (Sheppard et al. 1985). The locus that is best understood at a molecular level and has perhaps the largest phenotypic effect controls red patterns (Table 9.1). Alternate alleles represent regulatory switches controlling expression of the transcription factor optix. The most studied red patterns controlled by this locus can be divided into three main elements: the red forewing band, the red ray pattern on the hindwing and the basal patch on the forewing. The latter is known as the ‘Dennis’ patch, after an individual butterfly that William Beebe named ‘Dennis the Menace’. Once linked genetic markers were identified, it became clear that there is a remarkable degree of homology between species in the control of these elements (Baxter et al. 2008).

Phenotypes from a hybrid zone in Eastern Ecuador
There are three parental races that contribute variation to the hybrid zone, pictured here along the top row H. m. plesseni, H. m. malleti and H. m. ecuadorensis. Three major loci control the wing patterns, D controls red/orange pattern elements, Ac controls the shape of the forewing band (two spots or one) and Yb produces the yellow forewing band. These butterfly hybrids are all from the Neukirchen collection. Scale bar is 1 cm
This shows that convergent patterns in mimetic species are controlled by the same genetic mechanism. But what about other types of patterns? It turns out that a huge diversity of patterns are controlled by the same genetic loci. For example, this locus also controls orange patches in silvaniform butterflies, H. hecale and H. ismenius (Huber et al. 2015), and the brown forceps-shaped pattern on the ventral hindwing of H. cydno (Naisbit et al. 2003; Chamberlain et al. 2011). In fact, in every species so far investigated genetically, this locus has major phenotypic effects on red and orange pattern elements.
The optix locus actually consists of distinct, tightly linked elements. Direct estimation of recombination rates between these has proven difficult, but there are rare natural recombinants. For example in H. erato, a single individual with ray but not dennis was collected in a Peruvian hybrid zone, and similar individuals are known in H. melpomene (Mallet 1989). There are also established races that have recombinant genotypes, such as H. e. amalfreda and H. m. meriana that have dennis but not ray, while H. timareta timareta f. contigua is a form with ray but not dennis. Recent molecular analysis has confirmed that these phenotypes are indeed recombinants between tightly linked elements located in non-coding DNA near to optix (Wallbank et al. 2016). Thus, there are at least three very tightly linked elements that independently control different patches of red on the wing.
2 Phenotypic Effects of Major Loci: The Yellow Locus Cortex
This second major locus is similar in many ways to the red locus – it consists of tightly linked elements that similarly control different patches of yellow and white pattern. The cortex locus represents a cluster of tightly linked loci located on linkage group 15. These include effects known as Yb, Sb and N in H. melpomene and Cr in H. erato (Sheppard et al. 1985; Mallet 1986). Alleles that produce a yellow band are recessive to the absence of the band, although heterozygotes typically show an alteration in scale morphology in the band region that can be seen in altered reflectance in the otherwise black hindwing. Another allele at the same locus produces a band only on the underside of the hindwing and is present in the west Colombian race H. m. venustus. The same genomic region also controls a white hindwing margin found in the west Ecuador races H. e. cyrbia and H. m. cythera (Jiggins and McMillan 1997; Ferguson et al. 2010).
Many of the coloured patches on Heliconius wings are controlled in this very simple one-allele makes one-phenotype manner. However, there are also more complex interaction effects between loci. For example, in East Andean populations of H. erato, the yellow hindwing bar results from the joint effects of two loci, cortex and WntA. Thus, in Peruvian H. e. favorinus, recessive alleles at both loci are required for full expression of the hindwing bar (Mallet 1989) (although in Central American H. erato, a very similar bar results from a recessive allele at one locus). There is also evidence for rare recombination events between tightly linked loci at this locus. Thus, for example, Yb and Sb were mapped to within ~1 cM of one another, with two recombinant phenotypes identified in 175 individuals (Ferguson et al. 2010). Similar results are seen in crosses between H. melpomene rosina and H. c. chioneus (Naisbit et al. 2003).
In summary, these two loci both consist of a set of tightly linked genetic elements that control major phenotypic changes. Each locus controls pattern elements with broadly similar phenotypic effects: yellow and white patches in the case of cortex and red and orange patches in the case of optix. Patterns of dominance are also predictable, with alleles for red elements dominant, and those for yellow or white elements recessive, giving a dominance series of red > black > white > yellow. In both cases, loci most likely represent tightly linked cis-regulatory elements of the same protein-coding gene, with linkage a result of genetic architecture rather than being favoured by selection.
3 Phenotypic Effects of Major Loci: The Shape Locus WntA
The third major locus is located on linkage group 10 and primarily controls the shape of the forewing elements. For example, in crosses between H. melpomene rosina and H. cydno chioneus, a recessive allele ac places a triangle that forms a white hourglass shape in the main forewing cell of H. cydno (Naisbit et al. 2003). In the Ecuadorean H. m. plesseni, this locus produces the ‘split’ forewing band – the largely recessive H. m. plesseni allele expresses the more proximal of the two white patches of this form and also influences the shape of the more distal patch (Salazar 2012). This locus likely results from variation in expression of the gene WntA (Martin et al. 2012).
A wide variety of loci have previously been described (St, Sd and Ly) which all map to the same genomic location (Papa et al. 2013), corresponding to WntA. These loci influence the shape of forewing band elements. In some cases the phenotypic effects of this locus are extremely similar to those seen in H. melpomene; thus, for example, in H. e. notabilis, which is mimetic with H. m. plesseni, Sd also acts to generate the split forewing band phenotype (Salazar 2012). In Amazonian forms, the allele at this locus also generates the broken yellow forewing band (Sheppard et al. 1985; Papa et al. 2013).
4 Phenotypic Effects of Other Loci
A further locus, termed K, controls the colour change between yellow and white pigments in H. melpomene, H. cydno and H. pachinus. Most strikingly, this locus controls a polymorphism of yellow and white forms in H. cydno alithea in western Ecuador. The K locus is located on linkage group 1 and is linked with the gene wingless (Kronforst et al. 2006). This differs from other loci in that it influences solely colour, with no effect on pattern. There are also a number of minor-effect loci described in the older literature, but in most cases, these have been found to represent allelic effects of the major loci described above. Nonetheless, some of these loci are likely to be distinct. For example, a locus named Or described in both H. melpomene and H. erato controls the switch between red and orange colours (Sheppard et al. 1985). ‘Postman’ races typically have a bright red forewing band, while Amazonian forms have orange dennis and ray patterns. Another locus that has been better characterised is Ro, which generates a rounded forewing band phenotype such as that seen in H. e. notabilis (Salazar 2012; Papa et al. 2013; Nadeau et al. 2014). Some of the most beautiful but poorly characterised are the iridescent blue and green colours that result from structural variation in the wing scales. These traits vary continuously and are difficult to quantify (Jiggins and McMillan 1997). However, while most analysis of Heliconius genetics has relied on the scoring of presence/absence of major pattern elements, a better characterisation of these minor-effect loci is gained by a quantitative analysis of pattern segregation.
5 Quantitative Analysis
A comprehensive QTL analysis was carried out by Papa et al. using crosses between H. e. notabilis and H. himera (Papa et al. 2013). This confirmed the subjective finding from generations of earlier researchers that major loci control the segregation of most of the wing variation in crosses. For example, an additive model showed that the optix locus controlled 87% of variance in the amount of white versus yellow in the forewing, while the amount of red was best described by an epistatic model in which optix explained ~56% of the variation. The sizes of the two forewing spots showed a less skewed distribution of effect sizes and were controlled by several QTL of moderate effect (>5%), some as large in effect as the major locus WntA. For example, four QTL together explained 63% of the variance in the ‘big spot’, one of which was the WntA locus. This spot shape analysis therefore suggests a less skewed, more quantitative genetic architecture. Nonetheless, the overall variance explained across the complete set of H. erato crosses described in this paper is strongly dominated by large-effect loci.
These QTL analyses of specific wing pattern traits still fail to capture and quantify both segregation of the presence and absence of major pattern elements in the same analysis as quantitative variation in the expression of those traits. More recently, analytical methods have been developed that capture all of the variation in colour and pattern into a single PCA analysis (Huber et al. 2015; Le Poul et al. 2014), which was used to analyse broods of H. hecale and H. ismenius. All of the significant QTL identified corresponded to the existing major wing patterning loci. More minor QTL did not pass the significance threshold, although some of these additional loci would likely become significant with larger sample sizes. These quantitative analyses therefore support the conclusion that most variations are controlled by a handful of major-effect loci, although their expression is modified by minor-effect loci. In the future, there is a clear need for studies that combine large mapping families with objective methods for pattern analysis to better characterise the distribution of wing patterning variants.
6 Non-genetic Effects and Plasticity
There has been considerable interest recently in the role of phenotypic plasticity in evolution, and it has been proposed that plasticity can promote evolutionary novelty, for example, by allowing populations to explore new phenotypes without genetic change (Pfennig et al. 2010; Moczek et al. 2011). However, there is little evidence for phenotypic plasticity in the expression of Heliconius wing patterns. First, most of the variation in wing pattern among hybrid butterflies can be explained by genetic variation at just a handful of major loci. Second, in the wild there is very little phenotypic variation in wing pattern among individuals occurring across a wide range of altitudes and habitats – apart from genetically divergent wing pattern races. Some pigment colours do fade with age, or in stressed individuals, but this is not adaptive plasticity. In summary, while plasticity may play a role in many aspects of Heliconius biology, such as learning of behaviour, there is no evidence that it plays a role in wing pattern evolution.
7 A Distribution of Effect Sizes?
Early workers used major genes in butterfly mimicry as an argument for major mutations driving evolution, but Fisher countered that mutations with a large effect on the organism will virtually always be deleterious (Fisher 1930). More recently Orr has shown that during an adaptive walk, we expect an exponential distribution of mutational effect sizes (Orr 1998, 2005). Early in the process, there is a high likelihood of mutations that move the population a large distance relative to the optimum. Later on, smaller effect mutations are more probable, that act to ‘fine-tune’ the adaptation. To some extent this modern view therefore reconciles the two camps.
The theory developed by Orr and others hypothesised a population evolving towards a single adaptive peak. However, the frequency-dependent nature of mimicry and warning colour means that these traits have a different dynamic. If a population of butterflies has a bright warning colour pattern (hereafter the ‘mimic’), predators will learn this pattern, and the population will generally be well protected from predation. There may be other butterfly species locally that are perhaps more abundant or more toxic (the ‘model’) and therefore have a better-protected wing patterns, so the mimic species would gain in fitness by evolving mimicry of the model pattern. However, an individual ‘mimic’ that deviates from the rest of the population would be selected against, even if it becomes slightly more similar to the model. The two patterns would have to be very similar for predators to generalise between them, in order for gradual evolution towards the model to be possible (Turner 1981). Most current Heliconius patterns in different mimicry rings are sufficiently different from one another that gradual convergence seems unlikely. There is a valley of low fitness between the model and mimic which would seem to prevent gradual evolution of mimicry. This difficulty can be overcome if a single mutation causes a large change, sufficient to induce enough similarity to the model in one step that overall fitness is increased. This initial mutation is unlikely to produce a perfect mimic, so subsequent mutations will then be needed to perfect the phenotype. This argument was first outlined by Nicholson (1927) and termed the ‘Nicholson two-step model’ by John Turner (1977, 1984, 1987). Mimicry may therefore have a different genetic architecture to traits evolving under a single-peak-climbing model (Baxter et al. 2009).
The major locus control of Heliconius patterns seems to fit with the predictions of the ‘Nicholson two-step model’ (Huber et al. 2015; Papa et al. 2013; Turner 1981; Baxter et al. 2009), with a few major loci and additional modifiers of small effect. However, there are a number of reasons to be sceptical of this simple interpretation. First, many races within both H. erato and H. melpomene differ at several unlinked major-effect loci. For example, hybrid zones in both Peru and Ecuador between races of both H. melpomene and H. erato differ in at least two major loci (Mallet 1989; Salazar 2012; Nadeau et al. 2014). It is not clear whether a substitution at just one of these loci would be sufficient to gain enough mimetic similarity to provide protection, while the population ‘waited’ for a subsequent mutation at the second locus. Turner has acknowledged this difficulty but suggested either multiple rounds of ‘two-step’ evolution or that changes at just one of the loci would be sufficient to confer a fitness advantage (Turner 1977).
Another mismatch between the theory and empirical data is that the data from crossing experiments refers to the phenotypic effects of genetic loci, not separate mutations (Baxter et al. 2009). As pointed out by Fisher (1930), and more recently in dissection of major effect QTL in other organisms (Stam and Laurie 1996; Linnen et al. 2013), major-effect loci can result from accumulation of many mutations at a single locus. It seems likely that single large-effect genetic loci harbour many mutations corresponding to adaptive steps towards the peak. Testing the ‘two-step model’ therefore becomes a much more challenging problem of separating the order and effect size of individual mutations at a single locus. Nonetheless, mimicry can arise through hybridisation, in which an already well-adapted large-effect allele is acquired from a related species. This represents a clear case of single-step ‘major-effect’ evolution, so there certainly are at least some cases in which large changes are involved (The Heliconius Genome Consortium, 2012). Overall therefore, the ‘rugged’ adaptive landscape of mimicry likely favours adaptation via large steps as described under a two-step theory, and this might provide some part of the explanation for the major-effect loci involved in Heliconius mimicry.
8 Supergenes and Polymorphism
The broad picture of wing pattern genetics outlined above applies to most Heliconius that have been studied, but there is one species in the genus that has a very different pattern: H. numata. Mimicry patterns in Heliconius numata are polymorphic, with different morphs mimetic with different species mostly in the genus Melinaea. These dramatic differences are controlled by a single genetic locus, with several alternate alleles. Such loci are known as ‘supergenes’, which we have defined as ‘A genetic architecture involving multiple linked functional genetic elements that allows switching between discrete, complex phenotypes maintained in a stable local polymorphism’ (Thompson and Jiggins 2014). There are two major characteristics of the Heliconius numata supergene that maintain an integrated phenotype. First, a lack of recombination – all aspects of the phenotype are inherited as a single non-recombining locus – and second, dominance: alternate alleles show complete dominance relationships such that heterozygote genotypes develop the wing pattern of one or other parent.
The P supergene is genetically homologous to the region of the cortex locus in H. melpomene (Joron et al. 2006). The genetic architecture of 3–4 major loci is ancestral because it is shared by all other species in the genus that have been studied (Huber et al. 2015), so in H. numata this locus has ‘taken over’ control of all aspects of pattern variation (Jones et al. 2012). There are several hypotheses to explain the gradual evolution of tightly linked elements in a supergene. A long-standing hypothesis is that alleles located in different regions of the genome might be translocated into tight linkage (Turner 1967). However, there is no evidence for long-range movement of genes; the gene content of the region is similar in all Heliconius. The P locus has therefore evolved control of pattern variation normally influenced by genes on different chromosomes, rather than by moving those genes into linkage. The second hypothesis is that sequential mutations might arise in tight linkage with the polymorphic locus and be favoured by selection (Turner 1977; Charlesworth and Charlesworth 1976). Mutations that improve one mimetic form are likely to make things worse for other forms. However, if a new mutation is tightly linked at the P locus, then it will always be inherited with the alleles with which it is coadapted. This process has become known as ‘Turner’s sieve’, because it involves sieving of the genetic variation that arises in order to select only linked variants (Turner 1977; Charlesworth and Charlesworth 1976; Turner 1978). The fact that P consists of linked elements suggests that these must have arisen through multiple sequential mutations.
Once linked elements have arisen, theory predicts that selection can act to further reduce recombination between them (Turner 1967; Charlesworth and Charlesworth 1976; Kirkpatrick and Barton 2006). Mathieu Joron and his group have identified large genomic inversions (400 kb) that segregate in polymorphic populations around the P locus (Fig. 9.2). Alternate gene arrangements are fully associated with wing pattern phenotypes in natural populations and show strong linkage disequilibrium in natural populations. Effectively, there is a block of about 400 kb of DNA sequence that is inherited in complete association with different wing pattern forms (Joron et al. 2011). Similar inversions have been seen in complex polymorphisms in other species – notably a behavioural and plumage polymorphism in the white-throated sparrow, a social polymorphism in fire ants and a behavioural polymorphism in the ruff, a wading bird (Thompson and Jiggins 2014; Huynh et al. 2011; Wang et al. 2013; Küpper et al. 2015; Lamichhaney et al. 2015). In all cases, inversions lock together inheritance of a large part of one chromosome. Perhaps more similar to the Heliconius numata case is Papilio polytes, in which a very localised inversion around the Dsx gene controls a wing pattern mimicry polymorphism (Kunte et al. 2014; Nishikawa et al. 2015). These examples all suggest that the evolution of inversions to reduce recombination between coadapted alleles may be a common phenomenon.

Structural variation associated with the Heliconius numata supergene
At least two genetic inversions are associated with the H. numata supergene. The ancestral gene order, which matches that in H. melpomene and H. erato is shown on the left and is associated with ancestral phenotypes such as H. n. silvana. Two sequentially derived inversions are associated with dominant alleles and are shown in the middle and right. Redrawn from (Joron et al. 2011)
The second aspect of a supergene that ensures mimicry is a strong pattern of dominance (Llaurens et al. 2015). Alternate alleles show complete dominance, with an allelic series between morphs (Le Poul et al. 2014; Joron et al. 2011; Brown 1976). Remarkably, one heterozygote genotype is distinct but appears to have been stabilised because of its effective mimicry of a different species (Le Poul et al. 2014). In most Heliconius, there are predictable rules for dominance. Red/orange pattern elements are generally dominant over black, while yellow/white pattern elements are recessive. The complete dominance of alleles across the entire wing surface in H. numata therefore represents a derived state that apparently overturns typical ‘rules of inheritance’. Dominance has been optimised by natural selection.
These patterns of dominance could be controlled by mutations within the supergene itself or unlinked loci acting to control dominance at P. Although there is evidence for both of these processes, recent analysis provides strong evidence for evolution of dominance at the P locus itself. Patterns of dominance between derived and ancestral alleles show unusual patterns of dominance in which the typical dominance patterns are overruled. In contrast, among derived alleles, patterns of dominance follow the typical colour hierarchy seen in other Heliconius species (Le Poul et al. 2014). These patterns suggest that dominance is a property of the alleles themselves, rather than the genetic background. This will be a fascinating system in which to explore mechanisms underlying the evolution of dominance.
9 Conclusions
The extraordinary diversity of wing patterns among the Heliconius butterflies has provided insights into the diversification of animal form and its genetic control. An important discovery has been the repeated role of just a handful of loci in diversification of not just convergent mimetic patterns but also diverse and novel phenotypes. Nonetheless, there is still a need for better quantitative analysis of patterns that will reveal the distribution of loci controlling adaptation. These patterns parallel discoveries in other systems, for example, sticklebacks, where similarly there are a few loci with major effects on phenotype (Colosimo et al. 2005; Chan et al. 2010), but many traits are also influenced by more polygenic control (Peichel and Marques 2017).
I have reviewed our understanding of wing patterning based on genetic crossing experiments, but have not considered in detail the developmental basis for pattern diversity, which has recently been reviewed elsewhere (Jiggins et al. 2017).
References
Baxter SW, Johnston SE, Jiggins CD (2009) Butterfly speciation and the distribution of gene effect sizes fixed during adaptation. Heredity 102:57–65. doi:10.1038/hdy.2008.109
Baxter SW, Papa R, Chamberlain N, Humphray SJ, Joron M, Morrison C, ffrench-Constant RH, McMillan WO, Jiggins CD (2008) Convergent evolution in the genetic basis of Müllerian mimicry in Heliconius butterflies. Genetics 180:1567–1577. doi:10.1534/genetics.107.082982
Brown KS (1976) An illustrated key to the silvaniform Heliconius (Lepidoptera: Nymphalidae) with descriptions of new subspecies. Trans Am Entomol Soc 102:373–484
Chamberlain NL, Hill RI, Baxter SW, Jiggins CD, Kronforst MR (2011) Comparative population genetics of a mimicry locus among hybridizing Heliconius butterfly species. Heredity 107:200–204. doi:10.1038/hdy.2011.3
Chan YF et al (2010) Adaptive evolution of pelvic reduction in sticklebacks by recurrent deletion of a Pitx1 enhancer. Science 327:302–305. doi:10.1126/science.1182213
Charlesworth D, Charlesworth B (1976) Theoretical genetics of batesian mimicry II. Evolution of supergenes. J Theor Biol 55:305–324. doi:10.1016/S0022-5193(75)80082-8
Colosimo PF et al (2005) Widespread parallel evolution in sticklebacks by repeated fixation of ectodysplasin alleles. Science 307:1928–1933. doi:10.1126/science.1107239
Ferguson L et al (2010) Characterization of a hotspot for mimicry: assembly of a butterfly wing transcriptome to genomic sequence at the HmYb/Sb locus. Mol Ecol 19:240–254. doi:10.1111/j.1365-294X.2009.04475.x
Fisher RA (1930) The Genetical theory of natural selection, 1st edn. Oxford University Press, Oxford
Huber B et al (2015) Conservatism and novelty in the genetic architecture of adaptation in Heliconius butterflies. Heredity 114:515–524. doi:10.1038/hdy.2015.22
Huynh LY, Maney DL, Thomas JW (2011) Chromosome-wide linkage disequilibrium caused by an inversion polymorphism in the white-throated sparrow (Zonotrichia albicollis). Heredity 106:537–546. doi:10.1038/hdy.2010.85
Jiggins CD, McMillan WO (1997) The genetic basis of an adaptive radiation: warning colour in two Heliconius species. Proc R Soc Biol Sci 264:1167–1175. doi:10.1098/rspb.1997.0161
Jiggins CD, Wallbank RWR, Hanly JJ (2017) Waiting in the wings: what can we learn about gene co-option from the diversification of butterfly wing patterns? Philos Trans R Soc B 372:20150485. doi:10.1098/rstb.2015.0485
Jones RT, Salazar PA, ffrench-Constant RH, Jiggins CD, Joron M (2012) Evolution of a mimicry supergene from a multilocus architecture. Proc R Soc Lond B Biol Sci 279:316–325. doi:10.1098/rspb.2011.0882
Joron M et al (2006) A conserved supergene locus controls colour pattern diversity in Heliconius butterflies. PLoS Biol 4:1831–1840. doi:10.1371/journal.pbio.0040303
Joron M et al (2011) Chromosomal rearrangements maintain a polymorphic supergene controlling butterfly mimicry. Nature 477:203–206. doi:10.1038/nature10341
Kirkpatrick M, Barton N (2006) Chromosome inversions, local adaptation and speciation. Genetics 173:419–434. doi:10.1534/genetics.105.047985
Kronforst MR, Young LG, Kapan DD, McNeely C, O’Neill RJ, Gilbert LE (2006) Linkage of butterfly mate preference and wing color preference cue at the genomic location of wingless. Proc Natl Acad Sci U S A 103:6575–6580. doi:10.1073/Pnas.0509685103
Kunte K, Zhang W, Tenger-Trolander A, Palmer DH, Martin A, Reed RD, Mullen SP, Kronforst MR (2014) doublesex is a mimicry supergene. Nature 507:229–232. doi:10.1038/nature13112
Küpper C et al (2015) A supergene determines highly divergent male reproductive morphs in the ruff. Nat Genet 48:79–83. doi:10.1038/ng.3443
Lamichhaney S et al (2015) Structural genomic changes underlie alternative reproductive strategies in the ruff (Philomachus pugnax). Nat Genet 48:84–88. doi:10.1038/ng.3430
Le Poul Y, Whibley A, Chouteau M, Prunier F, Llaurens V, Joron M (2014) Evolution of dominance mechanisms at a butterfly mimicry supergene. Nat Commun 5:5644. doi:10.1038/ncomms6644
Linares M (1996) The genetics of the mimetic coloration in the butterfly Heliconius cydno weymeri. J Heredity 87(2):142–149
Linnen CR, Poh Y-P, Peterson BK, Barrett RDH, Larson JG, Jensen JD, Hoekstra HE (2013) Adaptive evolution of multiple traits through multiple mutations at a single gene. Science 339:1312–1316. doi:10.1126/science.1233213
Llaurens V, Joron M, Billiard S (2015) Molecular mechanisms of dominance evolution in Müllerian mimicry. Evolution 69:3097–3108. doi:10.1111/evo.12810
Mallet J (1986) Hybrid zones of Heliconius butterflies in Panama and the stability and movement of warning color clines. Heredity 56:191–202
Mallet J (1989) The genetics of warning color in Peruvian hybrid zones of Heliconius erato and Heliconius melpomene. Proc R Soc Lond Ser B-Biol Sci 236:163–185
Martin A et al (2012) Diversification of complex butterfly wing patterns by repeated regulatory evolution of a Wnt ligand. Proc Natl Acad Sci 109:12632–12637. doi:10.1073/pnas.1204800109
Mavarez J, Salazar CA, Bermingham E, Salcedo C, Jiggins CD, Linares M (2006) Speciation by hybridization in Heliconius butterflies. Nature 441(7095):868–871
Moczek AP, Sultan S, Foster S, Ledón-Rettig C, Dworkin I, Nijhout HF, Abouheif E, Pfennig DW (2011) The role of developmental plasticity in evolutionary innovation. Proc R Soc Lond B Biol Sci 278:2705–2713. doi:10.1098/rspb.2011.0971
Nadeau NJ, Jiggins CD (2010) A golden age for evolutionary genetics? Genomic studies of adaptation in natural populations. Trends Genet 26:484–492. doi:10.1016/j.tig.2010.08.004
Nadeau NJ et al (2014) Population genomics of parallel hybrid zones in the mimetic butterflies, H. melpomene and H. erato. Genome Res 24:1316–1333. doi:10.1101/gr.169292.113
Naisbit RE, Jiggins CD, Mallet J (2003) Mimicry: developmental genes that contribute to speciation. Evol Dev 5:269–280
Nijhout HF (1990) A comprehensive model for colour pattern formation in butterflies. Proc Royal Soc B Biol Sci 239(1294):81–113
Nicholson AJ (1927) A new theory of mimicry in insects. Aust Zool 5:10–104
Nishikawa H et al (2015) A genetic mechanism for female-limited Batesian mimicry in Papilio butterfly. Nat Genet 47:405–409. doi:10.1038/ng.3241
Orr HA (1998) The population genetics of adaptation: the distribution of factors fixed during adaptive evolution. Evolution 52:935–949. doi:10.2307/2411226
Orr HA (2005) The genetic theory of adaptation: a brief history. Nat Rev Genet 6:119–127. doi:10.1038/nrg1523
Papa R, Kapan DD, Counterman BA, Maldonado K, Lindstrom DP, Reed RD, Nijhout HF, Hrbek T, McMillan WO (2013) Multi-allelic major effect genes interact with minor effect QTLs to control adaptive color pattern variation in Heliconius erato. PLoS One 8:e57033. doi:10.1371/journal.pone.0057033
Peichel CL, Marques DA (2017) The genetic and molecular architecture of phenotypic diversity in sticklebacks. Philos Trans R Soc B 372:20150486. doi:10.1098/rstb.2015.0486
Pfennig DW, Wund MA, Snell-Rood EC, Cruickshank T, Schlichting CD, Moczek AP (2010) Phenotypic plasticity’s impacts on diversification and speciation. Trends Ecol Evol 25:459–467. doi:10.1016/j.tree.2010.05.006
Salazar PCA (2012) Hybridization and the genetics of wing colour-pattern diversity in Heliconius butterflies. Cambridge University, Cambridge
Sheppard PM, Turner JRG, Brown KS, Benson WW, Singer MC (1985) Genetics and the evolution of Muellerian mimicry in Heliconius butterflies. Philos Trans R Soc Lond Ser B Biol Sci 308:433–610. doi:10.2307/2398716
Stam LF, Laurie CC (1996) Molecular dissection of a major gene effect on a quantitative trait: the level of alcohol dehydrogenase expression in Drosophila melanogaster. Genetics 144:1559–1564
The Heliconius Genome Consortium (2012) Butterfly genome reveals promiscuous exchange of mimicry adaptations among species. Nature 487:94–98. doi:10.1038/nature11041
Thompson MJ, Jiggins CD (2014) Supergenes and their role in evolution. Heredity 113:1–8. doi:10.1038/hdy.2014.20
Turner JRG (1967) On supergenes. I. The evolution of supergenes. Am Nat 101:195–221
Turner JRG (1977) Butterfly mimicry – genetical evolution of an adaptation. In: Hecht MK, Steere WC, Wallace B (eds) Evolutionary biology. Plenum Press, New York, pp 163–206
Turner JRG (1978) Why male butterflies are non-mimetic: natural selection, sexual selection, group selection, modification and sieving. Biol J Linn Soc 10:385–432. doi:10.1111/j.1095-8312.1978.tb00023.x
Turner JRG (1981) Adaptation and evolution in Heliconius – a defense of Neodarwinism. Annu Rev Ecol Syst 12:99–121
Turner JRG (1984) Mimicry: the palatability spectrum and its consequences. In: Vane-Wright RI, Ackery PR (eds) The biology of butterflies. Academic Press, London, pp 141–161
Turner JRG (1987) The evolutionary dynamics of batesian and muellerian mimicry: similarities and differences. Ecol Entomol 12:81–95. doi:10.1111/j.1365-2311.1987.tb00987.x
Wallbank RWR et al (2016) Evolutionary novelty in a butterfly wing pattern through enhancer shuffling. PLoS Biol 14:e1002353. doi:10.1371/journal.pbio.1002353
Wang J, Wurm Y, Nipitwattanaphon M, Riba-Grognuz O, Huang Y-C, Shoemaker D, Keller L (2013) A Y-like social chromosome causes alternative colony organization in fire ants. Nature 493:664–668. doi:10.1038/nature11832
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
This chapter is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
Copyright information
© 2017 The Author(s)
About this chapter
Cite this chapter
Jiggins, C.D. (2017). What Can We Learn About Adaptation from the Wing Pattern Genetics of Heliconius Butterflies?. In: Sekimura, T., Nijhout, H. (eds) Diversity and Evolution of Butterfly Wing Patterns. Springer, Singapore. https://doi.org/10.1007/978-981-10-4956-9_9
Download citation
DOI: https://doi.org/10.1007/978-981-10-4956-9_9
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-4955-2
Online ISBN: 978-981-10-4956-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)