
Abstract
Mimicry is an important evolutionary trait involved in prey-predator interactions. In a swallowtail butterfly Papilio polytes, only mimetic-form females mimic the unpalatable butterfly, Pachliopta aristolochiae, but it remains unclear how this female-limited polymorphic Batesian mimicry is generated and maintained. To explore the molecular mechanisms, we determined two whole genome sequences of P. polytes and its related species P. xuthus for comparison. The genome projects revealed a single long-autosomal inversion outside doublesex (dsx) between mimetic (H) and non-mimetic (h) chromosomes (Chr25) in P. polytes. The inversion site was just same as the mimicry locus H identified by linkage mapping. The gene synteny around dsx among Lepidoptera suggests that H-chromosome originates from h-chromosome. The 130 kb inverted region includes three genes, doublesex (dsx), UXT, and U3X, all of which were expressed from H-chromosome, but rarely from h-chromosome, indicating that these genes in H-chromosome are involved in the mimetic trait as supergene. Amino acid sequences of Dsx were substituted at over 13 sites between H- and h-chromosomes. To certify the functional difference of Dsx, we performed electroporation-mediated knockdown and found that only female dsx from H-chromosome (dsx_H) induced mimetic patterns but simultaneously repressed non-mimetic patterns on female wings. We propose that dsx_H switches the coloration of predetermined patterns in female wings and that female-limited polymorphism is tightly kept by chromosomal inversion. In this chapter, I will introduce the above results and discuss about the molecular mechanism and evolutionary process underlying the female-limited Batesian mimicry in P. polytes.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others


Keywords
- Batesian mimicry
- Female-limited polymorphic mimicry
- Papilio polytes
- Whole genome sequence
- Doublesex
- Supergene
- Chromosomal inversion
- Electroporation-mediated functional analysis
- Wing coloration pattern
1 Research Background
One of the most essential problems in evolutionary biology is to elucidate the molecular basis of various and adaptive morphological phenotypes in living organisms. The morphological diversity plays an important role in adaptation to the surrounding environment in many cases (Darwin 1872). Insects at the bottom of the food chain have been continuously attacked by the predators and thus developed various defense strategies to avoid predation during evolution (Ruxton et al. 2005).
Among the various strategies used by butterflies to avoid predators, some butterflies have become unpalatable and inform predators to their toxicity by exhibiting the conspicuous wing patterns. Some unpalatable butterflies share similar wing patterns to provide mutualistic protection called Mullerian mimicry (Müller 1878). In contrast, some palatable butterflies have evolved Batesian mimicry, in which they resemble unpalatable model to protect them from predators (Bates 1862; Brower 1958; Uesugi 1996). Multiple loci are involved in the expression of Mullerian mimicry phenotypes in Heliconius butterflies (Jiggins et al. 2005; Kapan et al. 2006), whereas the phenotypes of Batesian mimicry species reported so far are determined by a single locus (Clarke and Sheppard 1959, 1962, 1972).
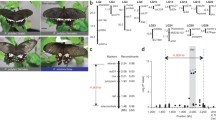
It is noteworthy that the Common Mormon butterfly, Papilio polytes, shows a female-limited Batesian mimicry (Clarke and Sheppard 1972). The females have two forms: non-mimetic female (also called cyrus) which wing patterns are almost identical to monomorphic males and mimetic female (also called polytes) which resembles wing patterns of the distasteful butterfly, the Common Rose, Pachliopta aristolochiae (Fig. 10.1). This female-limited dimorphism is controlled by a single autosomal locus H, and the mimetic phenotype (genotype: HH or Hh) is dominant (Clarke and Sheppard 1972). However, how the female-limited Batesian mimicry is generated or how the female dimorphism is maintained is largely unknown.

P. polytes and model butterfly, Pachliopta aristolochiae
There are two models for the H gene: a conceptual “supergene” that comprises a series of the neighboring genes tightly linked to each other (Clarke and Sheppard 1960, 1972) and a single regulatory gene that controls downstream, unlinked genes affecting the color pattern. It is hypothesized that a supergene unit is created by recombination events and fixed by inhibitory effects of a chromosomal inversion on recombination (Nijhout 2003; Joron et al. 2011), although the mechanism underlying this hypothesis has remained obscure.
Recently, we found that drastic changes of gene networks not only in red but also pale-yellow regions can switch wing color patterns between non-mimetic and mimetic female of P. polytes (Nishikawa et al. 2013). It is presumed that these pigmentation processes involved in Batesian mimicry of P. polytes should be downstream of the H gene. To elucidate the evolutionary processes of this mimicry comprehensively, it is important to clarify the H locus and its structure and function. More recently, Kunte et al. (2014) and our group have identified the H gene locus and revealed its structure, independently (Nishikawa et al. 2015).
2 Papilio Genome Projects Reveal the H Locus and Chromosomal Inversion Near dsx
To reveal the H locus and its flanking structure, we first determined the whole genome sequences of P. polytes and P. xuthus for comparison (Nishikawa et al. 2015). We have prepared the P. polytes genome DNA (Ishigakijima Island strain in Japan) from one inbred female (genotype, H/h) after four generations of laboratory inbreeding and the P. xuthus genome DNA from a male captured in the field near Tokyo, Japan. We used a whole genome shotgun approach with next-generation sequencing platform. Filtered paired-end reads (135.2 Gb pairs for P. polytes and 73.8 Gb pairs for P. xuthus) were assembled using Platanus (version 1.2.1) (Kajitani et al. 2014) with some mate-pair libraries sequenced by Illumina Hiseq2000 and Hiseq2500. Consequently, we obtained 3873 and 5572 scaffolds, with an N50 of 3.7 Mb and 6.2 Mb pairs, spanning 227 Mb and 244 Mb pairs of the genome sequences for P. polytes and for P. xuthus, respectively.
In validating resulting assembled scaffolds, we noticed that there were two independent scaffolds including dsx in P. polytes while only one scaffold including dsx in P. xuthus. Because these two scaffolds in P. polytes were significantly different in sequences and the genome DNA was prepared from one heterozygous (Hh) mimetic female, we assumed that each haplotype (H or h) was highly diverged around the dsx locus in the two independent scaffolds. To survey such heterozygous regions in the whole genome of P. polytes, we picked windows in which the coverage depth was ≤350, which is approximately half the homozygous peak of 600. After clustering overlapping windows, we found 15 highly diverse (identity of ≤90%) and long (≥100 kbp) heterozygous regions; 14 were mapped on heterozygous sex chromosome-1 (ZW) and one on chromosome-25 near dsx (Nishikawa et al. 2015). In the heterozygous region near dsx, we detected an approximately 130 kbp autosomal inversion (Fig. 10.2b). Strikingly, in the whole genome data of P. polytes, we could not find a long heterozygous region other than in the sex chromosomes (Z/W) which include many various repetitive sequences. Thus, the putative H locus region located on the chromosome-25 is thought to be only a long and unique heterozygous site among the whole autosomal chromosomes, which structure is maintained by reduced recombination due to the chromosomal inversion.

Schematic view of the H locus in P. polytes. (a) Phylogenetic tree of Lepidoptera based on Dsx amino acid sequences. (b) Gene structure around dsx for the h and H alleles and gene synteny among Lepidoptera. (c) Expression level of genes within inverted region of the H locus, at early pupal stage in hind wings of mimetic (red) and non-mimetic (black) females. (d) Sequence homology around the H locus between H and h alleles and between h and P. xuthus
3 Linkage Mapping of the H Locus
To identify the mimicry locus H, we also performed the linkage mapping using non-mimetic type of P. polytes in Minamidaitōjima Island in Japan and mimetic type of Papilio alphenor in the Philippines (Nishikawa et al. 2015) (this work was performed mainly by Dr. H. Hori). After analyzing 84 F2 backcrossed females with mimetic phenotype in heterozygote of H (Hh) and 69 of non-mimetic females (hh) using amplified fragment length polymorphism (AFLP) and restriction fragment length polymorphism (RFLP) markers, we mapped the mimicry locus in P. polytes within 800 kbp genomic region containing 41 genes between two markers designed in kinesin and intermediate on chromosome-25. The association between the region and mimicry phenotype in natural populations was further examined using single nucleotide polymorphisms (SNPs) in 54 wild-caught females (Nishikawa et al. 2015). Consequently, eight SNPs in dsx showed significantly higher association (chi-squared test of independence, P < 10−10) but none outside the gene. This is consistent with the result of the association study by K. Kunte et al. (2014) using laboratory-reared P. polytes alphenor. It is noteworthy that the H locus revealed by linkage mapping coincides completely with the long heterozygous region revealed by whole genome sequencing. This means that a genetic locus responsible for some polymorphic trait with a long heterozygous region can be identified only by genome sequencing without linkage mapping.
4 Detailed Structure of a Long Heterozygous Region Linked to the H Locus
Gene prediction and RNA-seq mapping showed that most of the inverted region of the H locus was occupied by dsx and the intron/exon structure was reversed in the h- and H-chromosomes, suggesting that a simple inversion occurred near both ends of dsx (Kunte et al. 2014; Nishikawa et al. 2015). Sequence comparison of the inverted region between H and h showed low-level homology not only directly but also in reverse (Fig. 10.2d). However, it is remarkable that some scattered regions including exons (shown by blue) for dsx were highly conserved (Fig. 10.2d).
To estimate the evolutionary process of the chromosomal inversion between H- and h- chromosomes, we further compared the gene synteny around dsx of P. polytes, with those of other Lepidoptera (Fig. 10.2a, b). We found that all tested genomes (Papilio species, Heliconius, Bombyx and Manduca) except Danaus have the same oriented synteny as the h-chromosome of P. polytes. Only in H-chromosome of P. polytes, dsx resides in the reverse orientation. These observations suggest that the H-chromosome may have originated from h-chromosome by a single inversion. Based on the gene synteny, we speculate that different types of inversion may have occurred near dsx in the Danaus genome independently. When comparing the inverted region (named hetero_130kbp) with a corresponding region of P. xuthus, the homology between h and P. xuthus was a little bit lower than that between h and H (Fig. 10.2d). This fact and phylogenetic tree suggest that the chromosomal inversion may have occurred after the branch of P. polytes and P. xuthus (Fig. 10.2a, d).
To clarify structural features of the inverted region of the H locus, we identified the exact place for the chromosomal inversion (Nishikawa et al. 2015). We have detected the sharp decline of the sequence conservation at both ends of inverted region between H- and h-chromosomes and considered these as putative breakpoints (Fig. 10.2d). The left breakpoint which closes on Prospero resides on about 700 bp downstream of the sixth exon of dsx in h-chromosome but on about 14.6 kbp upstream of the first exon of dsx in H-chromosome. The right breakpoint which closes on Sir2 resides on about 8.9 kb upstream of the first exon of dsx in h-chromosome but on about 1.1 kb downstream of dsx in H-chromosome. Compared with dsx in h-chromosome (dsx_h), dsx in H-chromosome (dsx_H) was longer in the second, fourth, fifth, and sixth introns and sixth exon. Just outside of both breakpoints, in contrast, more than 99% homology was carried on between the h- and H-sequences (Fig. 10.2d). These structures implied that many mutations and several insertion and deletion events may have accumulated in the inverted region for H after the inversion and were maintained by suppression of recombination between two chromosomes.
5 Dimorphic Dsx Structure Associated with the H and h Alleles
The fact that a complete dsx was encoded inside of the inversion region indicates a possible involvement of the gene on the mimetic phenotype. RNA-seq assembly from mimetic (HH) and non-mimetic female (hh) revealed three types of female isoforms of dsx (F1, F2, and F3) in wings (Fig. 10.3). Dsx isoforms are generated by alternative splicing between the third and the forth exon both on the h and H alleles. Translational stop codon appeared in the fourth exon for F1 and for F3 and in the third exon for F2. Amino acid differences among isoforms were restricted merely in the C-terminal region (4–23 amino acids); three isoforms shared the first 244 amino acids including dsx DNA-binding motif and oligomerization domain (Fig. 10.3). Although there were 13–15 amino acid changes in three dsx isoforms between H and h alleles (Fig. 10.3), most substitutions occurred around the DNA-binding motif and dimerization domain (An et al. 1996). The comparison of dsx sequences among Lepidoptera showed that only five amino acids were specifically changed in dsx_H of P. polytes (Fig. 10.3, *). Recently, we have revealed dimorphic structure of Dsx sequences in another polymorphic, female-limited Batesian mimic species P. memnon, which shows different sites of amino acid changes between mimetic and non-mimetic alleles, in comparison with P. polytes (Komata et al. 2016). This finding suggests parallel evolution of the mimicry locus in two Papilio species, and further researches are necessary to clarify the structural features of Dsx involved in the mimicry traits.

Amino acid sequences of Dsx for the H and h alleles. The N-terminal 244 amino acid sequence of Dsx which is common to female-specific isoforms (F1, F2, and F3) is shown on top. Each sequence of three isoforms for C-terminal region is shown at the bottom. The h allele sequence (Dsx_h) is shown. The amino acids substituted in Dsx_H are shown in blue below the sequence. * indicates an amino acid residue which is substituted only in Dsx_H among Lepidoptera
Respective differences of amino acid sequence for F1, F2, and F3 between dsx_H and dsx_h are 2, 0, and 1, respectively (Fig. 10.3). This indicates that at least the C-terminal region of F2 may not be involved in the specific function of dsx_H on the mimetic phenotype. Furthermore, sequence comparison of these isoforms with those in other Lepidoptera revealed highly conserved structure except the C-terminal amino acid in F1 isoform. These observations suggest that no special isoform of dsx_H seems to be involved in the mimetic wing coloration, although it needs further evidence to show this possibility.
In males which show merely non-mimetic phenotype, we found only one isoform of dsx which skips exons 3 and 4 included in all female isoforms, implying the importance of exons 3 and 4 for the mimicry. In these regions of three female isoforms, however, there was only one amino acid (the C-terminal end of F1) changed specifically in dsx_H, as described above. The male-specific isoform of dsx_H was scarcely expressed in prepupal to pupal wings, suggesting that male dsx_H is not involved in the mimetic phenotype (Fig. 10.4).

Hypothetical model of Dsx_H function on mimetic and non-mimetic wing coloration patterns. The expression patterns of dsx_H in wings of mimetic (red) and non-mimetic (blue) females and males (dotted green) during larval to pupal stages are shown below
6 Expression Profiles of Genes Around the Inverted Region of H Locus
To clarify the transcribed regions around the inverted regions, we mapped reads of RNA-seq to h and H alleles and found that three independent transcripts near left breakpoints, ubiquitously expressed transcript (UXT, transcriptional regulator) (Schroer et al. 1999), unknown-3-exons (U3X, long noncoding RNA emerged in H), and unknown transcript downstream of Prospero. These genes were highly expressed in wings of mimetic females (HH or Hh) compared with that in wings in non-mimetic females (hh) (Nishikawa et al. 2015) (Fig. 10.2c), suggesting a possible involvement of these genes in the mimicry. The 5′ untranslated region (UTR) structure and transcriptional start site for UXT were altered by the inversion event between H and h, while the open reading frame was the same. A newly emerged gene U3X was found merely in the heterozygous region of the H-chromosome in the whole genome of P. polytes. The downstream sequence of Prospero was differently expressed between h- and H-chromosomes while located outside of inverted regions. These facts demonstrate that the chromosomal inversion affects not only the genome structure but also the expression of neighboring genes drastically probably through changes of gene regulatory elements.
We found that there seemed no significant differences in expression level of each isoform (F1, F2, and F3) of dsx between mimetic and non-mimetic wings in P1–2, P4–5, and P10.5 stages (Nishikawa et al. 2015). However, the expression level of dsx_H in mimetic female wings was quite higher than in non-mimetic wings in early pupal stages, but dsx_h did not show such expression profiles. RNA-seq analyses support the results that dsx_H was dominantly expressed in Hh female wings (Nishikawa et al. 2015). Differential expression level between dsx_h and dsx_H becomes significant on female wings at P2 stage when the patterning of wing pigmentation may be determined (Nishikawa et al. 2013) (Fig. 10.4). In contrast to Hh male, dsx_H was scarcely expressed, while dsx_h was dominantly expressed both in wandering and early pupal stages. In the report of Kunte et al. (2014), however, the expression pattern of dsx_H was upregulated at late pupal stage, which was different with our result.
The comparison of promoter regions of dsx_H and dsx_h showed highly nucleotide conservation near the transcriptional start site, but the conservation gradually reduced in more upstream regions (Nishikawa et al. 2015). Some of the nucleotide differences in the regulatory regions or intron regions between dsx_H and dsx_h may be responsible for the specific regulation of dsx_H in the female wings. The above results suggest that not only amino acid substitution but also regulatory changes for female dsx_H are possibly involved in the mimetic phenotype.
7 Functional Analysis of dsx
To verify the dsx function on the mimetic wing pattern formation, we performed the functional analysis with electroporation-mediated siRNA incorporation optimized for pupal wings, which enables mosaic analysis by knocking down target genes (Ando and Fujiwara 2013; Yamaguchi et al. 2011; Fujiwara and Nishikawa 2016). First, to confirm the validity of this newly established method, we knocked down tyrosine hydroxylase (TH) that is involved in melanin synthesis and found that the black pigmentation in adult wings was clearly repressed in the siRNA incorporated region. When injecting Universal Negative Control siRNA which is used generally as a negative control, no phenotypic change was observed.
Using this method, we injected siRNA designed to knock down dsx_H but not dsx_h into the whole hind wings of mimetic female and applied electroporation. This treatment caused the mimetic wing pattern switching to non-mimetic wing patterns. Furthermore, electroporation of dsx_H siRNA into part of early pupal hind wings of mimetic females also resulted in severe repression of mimetic wing patterns in the siRNA incorporated region; the peripheral red spots became the small pale orange ones; the central white marking mostly disappeared in the mosaic area. By this treatment, the ectopic white patterns for non-mimetic females emerged in the predicted position (Nishikawa et al. 2015). These results indicated that dsx_H not only induces the mimetic wing patterns but also simultaneously represses the emergence of the non-mimetic wing patterns. In contrast, dsx_h siRNA in mimetic females did not influence wing phenotype. When knocking down both dsx_H and dsx_h expression by siRNA which was designed in the common region between H and h (dsx_H/h), the same phenotype was observed as dsx_H siRNA alone (Nishikawa et al. 2015). These results implied that dsx_h is not involved both in mimetic and non-mimetic wing pattern formation. This strongly suggests that only dsx_H is involved in the mimicry phenotypes. It is noteworthy that H/H homozygous individuals are viable. This means that dsx_H should have basic functions for sexual differentiation in addition to the wing coloration.
It is unexpected that dsx_H not only induces the formation of mimetic color patterns but also represses non-mimetic patterns (Fig. 10.4). We previously showed that white (pale-yellow in mimetic) regions on mimetic and non-mimetic female wings of P. polytes are composed of different pigments (Nishikawa et al. 2013). Additionally, kynurenine/N-beta-alanyldopamine (NBAD) synthesis and Toll signaling genes were upregulated in the red spots of mimetic wings of P. polytes (Nishikawa et al. 2013; Rembold and Umebachi 1984). In addition to these observations, in the mimetic female wings, the positions of white bands in non-mimetic females are altered to the central area, which pattern resembles that of the model species Pachliopta aristolochiae. Therefore, both region-specific patterns and the synthesis of pale-yellow and red pigments in the mimetic female wings should be switched by dsx_H. We observed that chemical properties of pale-yellow pigments in mimetic females are similar to the model butterfly. Thus, various changes controlled by dsx_H lead to the successful mimicry of mimetic females of P. polytes.
More noteworthy, the appearance of non-mimetic-type white bands in mimetic pupal wings by knockdown dsx_H suggests that the pigmentation pattern is preset at least in early pupal stages. We propose a possible model for the functional role of dsx_H on the color pattern formation (Fig. 10.4). We assume that dsx_H acts as the pigmentation selector for the mimetic pattern. In late larval to early pupal stages, both mimetic and non-mimetic color patterns are predetermined by pattern formation genes other than dsx. Moreover, dsx_H merely selects the pigmentation processes for the mimetic pattern and represses the non-mimetic pattern in the fate-determined wings. In heliconid butterflies, the black region at the center of the forewings is determined by WntA (Martin et al. 2012), and the forewing band pattern is determined by optix (Reed et al. 2011). In early pupal wings of Bicyclus anynana, the Distal-less (Dll) homeobox gene is involved in positive regulation of focal differentiation and eyespot signaling (Monteiro et al. 2006; Brakefield et al. 1996). In addition, the red spots in the margin of hind wings are often observed among many butterflies in Papilionidae, irrelevant to males or females. From these facts, we speculated that the wing patterns of P. polytes may be determined initially by other genes than dsx.
8 Evolution of Female-Limited Batesian Mimicry
We revealed fine structure of the chromosomal inversion linked to female-limited Batesian mimicry of Papilio polytes, which has been studied mainly by classical genetics or from ecological points of views to date. Female-limited Batesian mimicry is widely observed among Papilio species (Kunte 2009) and may be controlled by a similar dsx-mediated system used in P. polytes. The characteristic of female-limited mimicry of P. polytes is the intraspecies polymorphisms and mimetic and non-mimetic phenotypes, which molecular background is explained well by the existence of two differentiated chromosomes, h- and H-chromosomes. The structure and function of dsx_H (and maybe neighboring genes) specialized for mimetic phenotype may be tightly kept through reduced recombination due to chromosomal inversion just outside of dsx for a long time in natural populations.
A question to be answered is when the chromosomal inversion or the differentiation of dsx_H and dsx_h occurred. The comparison of gene synteny among Lepidoptera indicates that H-chromosome of P. polytes may be originated from the h-type chromosome which structure is common in many species. Therefore, the chromosomal inversion might have occurred after the branch of P. polytes and P. xuthus 40 million years ago (Zakharov et al. 2004) (Figs. 10.1 and 10.2a), and thereafter the sequence difference between dsx_H and dsx_h had been fixed and accumulated. Indeed, the rate of single nucleotide variations (SNVs) and phylogenetic analysis of dsx showed considerable rate of nucleotide substitutions in the inverted region, suggesting that dsx_H has a high evolutionary rate and may evolve a new function under positive selection pressure. On the other hand, we observed lower homology in the whole hetero_130 kbp region between the H- and h-chromosomes of P. polytes, which level seemed similar to that between the P. polytes and h-chromosomes (Fig 10.2d). This indicates the chromosomal inversion might have occurred in very ancient age, and dsx_H function was refined by repeated and accumulated mutations during evolution. This evolutionary scenario needs to be certified by further detailed analysis of genome structure in other closely related species.
References
An W, Cho S, Ishii H, Wensink PC (1996) Sex-specific and non-sex-specific oligomerization domains in both of the doublesex transcription factors from Drosophila melanogaster. Mol Cell Biol 16:3106–3111
Ando T, Fujiwara H (2013) Electroporation-mediated somatic transgenesis for rapid functional analysis in insects. Development 140:454–458. doi:10.1242/dev.085241
Bates HW (1862) Contributions to an insect fauna of the Amazon valley. Lepidoptera: Heliconidae Transactions of the Linnean Society of London 23:495–566
Brakefield PM, Gates J, Keys D, Kesbeke F, Wijngaarden PJ, Monteiro A, French V, Carroll SB (1996) Development, plasticity and evolution of butterfly eyespot patterns. Nature 384:236–242
Brower JVZ (1958) Experimental studies of mimicry in some North American butterflies. Part II. Battus philenor and Papilio troilus, P. polyxenes and P. glaucus. Evolution 12:123–136
Clarke CA, Sheppard PM (1959) The genetics of Papilio Dardanus, Brown I. Race cenea from South Africa. Genetics 44:1347–1358
Clarke CA, Sheppard PM (1960) Super-genes and mimicry. Heredity 14:175–185
Clarke CA, Sheppard PM (1962) The genetics of the mimetic butterfly Papilio glaucus. Ecology 43:159–161
Clarke CA, Sheppard PM (1972) The genetic of the mimetic butterfly Papilio polytes. Philos Trans R Soc Lond Ser B Biol Sci 263:431–458
Darwin CR (1872) The origin of species by means of natural selection, or the preservation of favoured races in the struggle for life, 6th edn. John Murray, London
Fujiwara H, Nishikawa H (2016) Functional analysis of genes involved in color pattern formation in Lepidoptera. Curr Opin Insect Sci 17:16–23. doi:10.1016/j.cois.2016.05.015
Jiggins CD, Mavarez J, Beltrán M (2005) A genetic linkage map of the mimetic butterfly Heliconius melpomene. Genetics 171:557–570
Joron M, Frezal L, Jones RT, Chamberlain NL, Lee SF, Haag CR, Whibley A, Becuwe M, Baxter SW, Ferguson L, Wilkinson PA, Salazar C, Davidson C, Clark R, Quail MA, Beasley H, Glithero R, Lloyd C, Sims S, Jones MC, Rogers J, Jiggins CD, French-Constant RH (2011) Chromosomal rearrangements maintain a polymorphic supergene controlling butterfly mimicry. Nature 477:203–206. doi:10.1038/nature10341
Kajitani R, Toshimoto K, Noguchi H, Toyoda A, Ogura Y, Okuno M, Yabana M, Harada M, Nagayasu E, Maruyama H, Kohara Y, Fujiyama A, Hayashi T, Itoh T (2014) Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res 24:1384–1395. doi:10.1101/gr.170720.113
Kapan DD, Flanagan NS, Tobler A, Papa R, Reed RD, Gonzalez JA, Restrepo MR, Martinez L, Maldonado K, Ritschoff C, Heckel DG, McMillan WO (2006) Localization of Mullerian mimicry genes on a dense linkage map of Heliconius erato. Genetics 173:735–757
Komata S, Lin CP, Iijima T, Fujiwara H, Sota T (2016) Identification of doublesex alleles associated with the female-limited Batesian mimicry polymorphism in Papilio memnon. Sci Report 6:34782. doi:10.1038/srep34782
Kunte K (2009) The diversity and evolution of batesian mimicry in Papilio swallowtail butterflies. Evolution 63:2707–2716. doi:10.1111/j.1558-5646.2009.00752.x
Kunte K, Zhang W, Tenger-Trolander A, Palmer DH, Martin A, Reed RD, Mullen SP, Kronforst MR (2014) Doublesex is a mimicry supergene. Nature 507:229–232. doi:10.1038/nature13112
Martin A, Papa R, Nadeau NJ, Hill RI, Counterman BA, Halder G, Jiggins CD, Kronforst MR, Long AD, McMillan WO, Reed RD (2012) Diversification of complex butterfly wing patterns by repeated regulatory evolution of a Wnt ligand. Proc Natl Acad Sci U S A 109:12632–12637. doi:10.1073/pnas.1204800109
Monteiro A, Glaser G, Stockslager S, Glansdorp N, Ramos D (2006) Comparative insights into questions of lepidopteran wing pattern homology. BMC Dev Biol 6:52
Müller F (1878) Über die Vortheile der Mimicry bei Schmetterlingen. Zool Anz 1:54–55
Nijhout HF (2003) Polymorphic mimicry in Papilio Dardanus: mosaic dominance, big effects, and origins. Evol Dev 5:579–592
Nishikawa H, Iga M, Yamaguchi J, Saito K, Kataoka H, Suzuki Y, Sugano S, Fujiwara H (2013) Molecular basis of wing coloration in a Batesian mimic butterfly, Papilio polytes. Sci Report 3:3184. doi:10.1038/srep03184
Nishikawa H, Iijima T, Kajitani R, Yamaguchi J, Ando T, Suzuki Y, Sugano S, Fujiyama A, Kosugi S, Hirakawa H, Tabata S, Ozaki K, Morimoto H, Ihara K, Obara M, Hori H, Itoh T, Fujiwara H (2015) A genetic mechanism for female-limited Batesian mimicry in Papilio butterfly. Nat Genet 47:405–409. doi:10.1038/ng.3241
Reed RD, Papa R, Martin A, Hines HM, Counterman BA, Pardo-Diaz C, Jiggins CD, Chamberlain NL, Kronforst MR, Chen R, Halder G, Nijhout HF, McMillan WO (2011) Optix drives the repeated convergent evolution of butterfly wing pattern mimicry. Science 333:1137–1141. doi:10.1126/science.1208227
Rembold H, Umebachi Y (1984) The structure of papiliochrome II, the yellow wing pigment of the Papilionid butterflies. In: Schlossberger HG, Kochen W, Linzen B, Steinhart H (eds) Progress in Tryptophan and Serotonin Research. Walter de Gruyter, Berlin, pp 743–746
Ruxton GD, Sherratt TN, Speed M (2005) Avoiding attack: the evolutionary ecology of crypsis, warning signals and mimicry. Oxford Univ. Press, Oxford
Schroer A, Schneider S, Ropers H, Nothwang H (1999) Cloning and characterization of UXT, a novel gene in human Xp11, which is widely and abundantly expressed in tumor tissue. Genomics 56:340–343
Uesugi K (1996) The adaptive significance of Batesian mimicry in the swallowtail butterfly, Papilio polytes (lnsecta, Papilionidae): associative learning in a predator. Ethology 102:762–775
Yamaguchi J, Mizoguchi T, Fujiwara H (2011) siRNAs induce efficient RNAi response in Bombyx mori embryos. PLoS One 6:e25469. doi:10.1371/journal.pone.0025469
Zakharov EV, Caterino MS, Sperling FA (2004) Molecular phylogeny, historical biogeography, and divergence time estimates for swallowtail butterflies of the genus Papilio (Lepidoptera: Papilionidae). Syst Biol 53:193–215
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
This chapter is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
Copyright information
© 2017 The Author(s)
About this chapter
Cite this chapter
Fujiwara, H. (2017). Molecular Mechanism and Evolutionary Process Underlying Female-Limited Batesian Mimicry in Papilio polytes . In: Sekimura, T., Nijhout, H. (eds) Diversity and Evolution of Butterfly Wing Patterns. Springer, Singapore. https://doi.org/10.1007/978-981-10-4956-9_10
Download citation
DOI: https://doi.org/10.1007/978-981-10-4956-9_10
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-4955-2
Online ISBN: 978-981-10-4956-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)